
Abstract
We developed a novel Designed Primer-based RNA-sequencing strategy (DP-seq) that uses a defined set of heptamer primers to amplify the majority of expressed transcripts from limiting amounts of mRNA, while preserving their relative abundance. Our strategy reproducibly yielded high levels of amplification from as low as 50 picograms of mRNA while offering a dynamic range of over five orders of magnitude in RNA concentrations. We also demonstrated the potential of DP-seq to selectively suppress the amplification of the highly expressing ribosomal transcripts by more than 70% in our sequencing library. Using lineage segregation in embryonic stem cell cultures as a model of early mammalian embryogenesis, DP-seq revealed novel sets of low abundant transcripts, some corresponding to the identity of cellular progeny before they arise, reflecting the specification of cell fate prior to actual germ layer segregation.
Similar content being viewed by others

Introduction
Next Generation Sequencing-based approaches for whole transcriptome analysis produce millions of sequencing reads, which represent the vast majority of the expressed transcripts. The high number of reads allows a digital estimation of transcript abundance, resulting in a large dynamic range and high sensitivity1,2,3,4. This is in contrast to microarray platforms, which rely on the hybridization efficiencies of transcript specific probes to their corresponding targets and thus result in analog expression profiles and a low dynamic range. With the recent dramatic increase in sequencing depth and the decrease in cost per base sequenced, high throughput sequencing technologies have emerged as preferred platforms for mRNA expression analysis of the complex mammalian transcriptome5,6.
A major limitation of the current gold standard RNA sequencing approach7 is the large amount of starting material (10–100 ng of mRNA) required to generate a sequencing library. This limits the potential of this protocol when it is difficult to obtain such large amounts of RNA such as in the fields of developmental biology or forensics or even for FACS sorted cell populations. Also, the standard RNA-seq protocol7 maintains the relative order of transcript expression with a few highly expressed transcripts occupying majority of the sequencing space. This results in a poor coverage of low abundant transcripts at current sequencing depths8,9. Reliable quantitation of the low abundant transcripts within large mammalian transcriptomes is further hampered by multireads and biases introduced by the transcript length10 and random hexamer primer hybridization11. A number of amplification-based protocols have been developed to address these issues such as “random priming” strategies12,13,14, which utilize the hybridization and extension potential of hexamer/heptamer primers to amplify the starting material (mRNA or cDNA). However, these random priming methods often result in a low yield of good quality reads, due to mis-hybridization of primers and/or primer dimerization. Furthermore, these methods do not discriminate the regions of the transcriptome to amplify, a feature also shared by other uniform amplification based strategies15,16,17,18,19.
Here we describe a novel quantitative cDNA expression profiling strategy, involving the amplification of a majority of the mouse transcriptome using a defined set of 44 heptamer primers. The amplification protocol allows an efficient amplification of the majority of the expressed transcripts from as low as 50 pg of mRNA and was optimized to reduce mis-hybridization of primers and primer dimerization. We further explored the potential of our primer design strategy to selectively suppress the amplification of the highly expressing transcripts such as ribosome encoding transcripts. Our sequencing data demonstrated a significant reduction in the representation of the ribosomal transcripts with multiple choices of primer sets. We compared our methodology with a full-length cDNA amplification strategy (Smart-seq)19 and observed comparable transcriptome coverage and similar technical noise. We implemented DP-seq on a model of embryological lineage segregation, achieved by graded activation of Activin A/TGFβ signaling in mouse embryonic stem cells (mESCs). The fold changes in transcript expression were in excellent agreement with quantitative RT-PCR and we observed a dynamic range spanning more than five orders of magnitude in RNA concentration with a reliable estimation of low abundance transcripts. Our transcriptome data identified key lineage markers, while the high sensitivity indicated that novel lineage specific transcripts anticipate the differentiation of specific cell types.
Results
Sequencing-library generation using heptamer primers based amplification
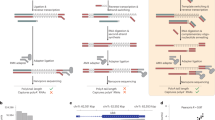
A novel cDNA sequencing-library generation methodology was developed to reliably represent the relative abundance of transcripts using limited amounts of mRNA. DP-seq consisted of three distinct phases ( Figure 1a ). In the first phase, we developed a primer design strategy that identified a defined set of 44 heptamer primers amplifying > 80% of the mouse transcriptome ( Figure 1a , green panel). This strategy incorporated known biases in PCR, namely the secondary structure of primer-binding sites in single stranded cDNA, GC content and the proximity to the 3′ end of the transcript to identify potential primer-binding sites. Of the 16384 input sequences of heptamer primers, we selected primers with annealing temperatures between 16–25°C. To minimize mis-priming, heptamer primers starting with adenines at the 5′ end and purine rich primers were filtered out. Next, an iterative randomized algorithm was implemented to identify 44 heptamer primers, which preferentially amplified unique regions of mouse transcripts (see Supplementary Fig. S1 online). The primers were split into multiple sets ensuring no two primers had a mutual interaction energy (Gibbs free energy) lesser than -5 kcal/mol in order to reduce primer dimerization. Of the 26566 known transcripts in the mouse NCBI RefSeq mRNA database, our heptamer primers covered 15072 (56.7%) transcripts uniquely.

Schematic representation of sequencing library preparation using heptamer primers based amplification, DP-seq.
(a) Step 1: Primer selection was based on identifying potential primer-binding sites that were less likely to form secondary structures and resided upstream to the unique regions on the mouse transcriptome. Step 2: targeted cDNA amplification. A Standard cDNA library was prepared and the primers selected from Step 1 were annealed to the single stranded cDNA library and were extended and amplified as indicated. Step 3: Library preparation. Illumina paired end adaptors were ligated to the ends of the amplicon library and the correct orientation of adaptors were selected. The library was further amplified using Illumina's paired end adaptor primers and were size selected for synthesis-based sequencing. (b) Expression profiles of genes responding to graded activation of the Activin A/TGFβ signaling pathway in mouse embryoid bodies at day 4. Quantitative RT-PCR data was normalized with respect to untreated serum-free media controls. (c) The fidelity of amplification of the cDNA library using heptamer primers. Fold changes observed in 11 genes (from part (b), Afp and Cer1) across different dosages of Activin A showed perfect agreement with quantitative RT-PCR performed on cDNA (R2 = 0.94; n = 45). (d) Distribution of reads on the mouse genome.
In the second phase of the methodology, we performed a targeted amplification of the mouse transcriptome using the defined set of heptamer primers ( Figure 1a , pink panel). This phase consisted of two components; (i) determination of the minimum length of the primer required to achieve efficient amplification and (ii) optimization of the amplification protocol to extend and amplify partially hybridized primers. We determined 14 bp (Tm ~ 45–50°C) as the optimal length of the primers required to efficiently amplify regions of interest in the mouse transcriptome. As such the heptamer primers were extended by addition of a universal 7 bp sequence (5′-CCGAATA′-3′) at the 5′ end of heptamer primers. Standard PCR protocols failed to amplify partially hybridized primers because of low annealing temperatures of the last 7 bp, resulting in significant distortions in the expression level of low abundance transcripts. We therefore developed a novel protocol that uses a combination of mesophilic (Klenow polymerase) and thermophilic polymerases (Taq polymerase) to efficiently amplify regions of interest on cDNA. Klenow polymerase, which retains its optimal extension activity at 37°C, extends our partially hybridized primers (last 7 bp) at this temperature. The extended primers withstand the high temperature required for a Taq polymerase extension at 72°C, resulting in the formation of a double stranded amplicon library. These amplicons possess complementary sequences of the entire 14 bp of our primers at its ends. Since our 14 bp primers have a high Tm (45–50°C), they efficiently hybridize to the template and allow amplification of these amplicons during the subsequent cycles of Taq polymerase PCR.
In the last phase of the sequencing library generation, the amplicon library was 5′ end phosphorylated and ligated to Illumina's adaptors ( Figure 1a , blue panel). Since only distinct adaptor orientation fragments can be sequenced in Illumina's platform, we used a biotin-streptavidin chemistry (see Supplementary Methods online) to select the correct orientations of the adaptors. The fragments were later PCR amplified using Illumina's adaptor specific PCR primers and size selected for synthesis-based sequencing. The selection of fragments with a correct orientation of the adaptors can be skipped by ligating standard Illumina Y-adaptors to the amplicon library and using a custom sequencing primer that contains the universal tail sequence (5′-CCGAATA′-3′) at its 3′ end.
Evaluation of heptamer amplification-based transcriptomics
We implemented DP-seq on an in vitro cell culture based model of primitive streak (PS) induction in mESCs20,21. Signaling by the TGFβ-family member Nodal through Activin receptor like kinase-4 is essential for mesoderm22,23,24 and endoderm25,26 formation and the dose-dependent induction of these tissues can be mimicked by treatment with Activin A. Various dosages of Activin A (3 ng/mL, AA3; 15 ng/mL, AA15; and 100 ng/mL, AA100) were therefore used to induce mesoderm and definitive endoderm, while its inhibition by a small molecule inhibitor, SB-431542 (SB)27, was used to induce neuro-ectoderm28.
As expected, small doses of Activin A substantially induced mesodermal markers (e.g., Kdr, Mesp1) while higher doses of Activin A were required for the induction of anterior lineages including definitive endoderm (e.g., Gsc, Foxa2) ( Figure 1b ). On the other hand, complete inhibition of Activin A/TGFβ signaling caused an up-regulation of neuro-ectoderm markers (e.g., Sox1)29. Moreover, direct target genes (e.g., Lefty1, Lefty2 and T also known as Brachyury)30,31 of the Activin A/TGFβ signaling pathway were regulated dose dependently. The differential expression of these low abundant genes in DP-seq showed excellent concordance with quantitative RT-PCR (R2 = 0.94, Figure 1c ) thus validating the DP-seq approach.
For a typical transcriptome measurement, we obtained ~30 million reads per lane of Illumina's genome analyzer flow cell ( Table 1 ). About 59% (18 million) reads uniquely mapped to more than 11000 transcripts with ≥10 reads. About 19% of the reads were non-uniquely mapped with a vast majority of them mapping to isoform groups. Another 18% of the reads (71% uniquely) mapped to genomic locations (excluding the open reading frames of known transcripts) and mitochondria transcripts ( Figure 1d ). Of these genomic reads, 72% mapped to intronic regions of transcripts while another 20% mapped within 5 Kb of the known transcripts. These reads most likely represent non-coding RNA, since we did not see a strong correlation between the fold changes in intronic reads with those from proximal exons.
The experimental data indicated expression of more than 100,000 different primer-binding sites representing ~18,000 known transcripts. This demonstrates the scale of massive multiplexing achieved by DP-seq. On average, we obtained expression of 10 different primer-binding sites for each expressed transcript. Notably, each site provided an independent measurement of relative abundance serving as technical replicates for the experiment (see Supplementary Fig. S2 online).
More than 50% of the uniquely mapped reads came from perfectly matched primer-binding sites while the rest were the product of mis–priming or single nucleotide polymorphisms (SNPs) in the primer-binding sites. Fold changes observed in predicted and mis-primed binding sites were highly correlated (R2 = 0.88) suggesting that mis-primed PCR products were able to conserve the relative abundance of transcripts (see Supplementary Fig. S2 online). Mis-primed products were mainly stabilized by a favorable interaction between the last three bases of the universal tail of the heptamer primers (5'-ATA-3') and the upstream regions of the primer-binding sites (tail interaction, see Supplementary Fig. S3 online). Finally, we observed no indication of primer – dimerization.
Analysis of the technical replicates revealed a strong correlation in quantitative transcript expression (R2 = 0.96, Figure 2a ). To assess the dynamic range, we spiked the untreated control (serum free media, SFM) with six artificial transcripts of the yeast POT1 promoter (~180 bp). The transcripts were flanked with different heptamer primer-binding sites and mixed in different dilutions, spanning six orders of magnitude in RNA concentration. The second most abundant transcript was similar in expression with the β-actin abundance in our biological samples. Our primers were able to effectively amplify all the six transcripts and maintained their relative abundance (R2 = 0.99, Figure 2b ). The distribution of fold changes (see Supplementary Fig. S2 online) observed in all possible pairwise comparisons of the samples was broad (2−8–210) suggesting a much higher dynamic range in comparison to microarray platforms (few hundred folds)4.

Performance of DP-seq.
(a) Comparison of two Activin A 15 ng/ml dosage replicates (R2 = 0.96). (b) Six in vitro synthesized transcripts derived from the yeast POT1 promoter with a length of 180 bp were added to untreated control cDNA at varying concentrations spanning six orders of magnitude. The reads obtained from the transcripts revealed a fold change of up to 105 (R2 = 0.99) in comparison to the lowest abundant transcript. (c) Sequencing libraries constructed from serial dilutions of mRNA exhibited high correlations within the technical replicates. Libraries constructed from at least 50 pg of mRNA showed high correlations (R2) in global expression measurements with the libraries made from 10 ng of mRNA. (d) Suppression of the ribosomal transcripts representation in the sequencing library generated from three different primer sets. The global transcriptome coverage remained high for all primer sets.
We next prepared serial dilutions of mRNA from 10 ng to 1 pg (10000 fold depth) of mRNA and constructed sequencing libraries to determine the lowest amount of mRNA required to prepare a reliable sequencing library. The number of amplification cycles was increased for lower dilutions to achieve appropriate amounts of DNA for the library construction. The transcript measurements from the technical replicates consistently showed high correlations for the libraries prepared from 10 ng–50 pg of mRNA ( Figure 2c ). The transcriptome coverage remained high even for libraries prepared from 1 pg of mRNA (~ 6000 transcripts; see Supplementary Table S1 online), although the noise in the quantification of the transcripts increased substantially (see Supplementary Fig. S4 online). We further investigated whether the transcript measurements were conserved within the dilution series of mRNA. Global transcript measurements of libraries constructed from at least 50 pg of mRNA showed high correlation with the libraries constructed from 10 ng of mRNA (see Supplementary Fig. S5 online). Sequencing libraries constructed from 1 pg of mRNA showed significant deviations in measurements of low copy number transcripts from 10 ng of libraries and a considerable amount of spurious PCR artifacts were observed.
Our methodology exhibited a few biases arising out of each stage of cDNA amplification (see Supplementary Fig. S3 online). The most dominant bias came from local secondary structures of the single stranded cDNA. Regions with stable secondary structures prevented the heptamer primer-binding sites from hybridizing with their corresponding heptamer primers, resulting in their poor representation in the sequencing library. There was also an inherent bias towards preferential amplification of fragments with shorter lengths and lower GC content, which are known to be associated with Taq Polymerase amplification and have been reported in other multiplexed PCR strategies32. Finally, we observed that the majority of the experimental heptamer primer-binding sites resided in proximity to the 3' end of the transcripts mainly because of the inability of the reverse transcriptase to produce full-length cDNA.
To determine whether DP-seq is capturing the majority of the expressed transcripts, we performed standard RNA-seq (Std. RNA-seq) on the AA3 sample using the protocol adopted from Mortazavi et al., 20087. We observed comparable transcriptome coverage with DP-seq libraries, representing > 80% of the expressed transcripts. Analysis of the technical replicates obtained from DP-seq and Std. RNA-seq revealed a similar noise structure (see Supplementary Fig. S6 online). The PCR biases observed in our methodology distorted the order of transcript expression within a biological sample (see Supplementary Fig. S6 online) resulting in a similar or enriched representation of the majority of low expressed transcripts (Reads Per Kilobase per Million mapped reads (RPKM) ≤10 in the Std. RNA-seq library). However, the relative abundance of the transcripts across different biological samples was not affected (shown in Figure 1c ) as these biases are expected to be similar for a given transcript across different biological samples. Furthermore, we observed an overlapping distribution of unique reads for the transcripts encoding transcription factors (http://genome.gsc.riken.jp/TFdb/) between the two protocols (see Supplementary Fig. S7 online).
We then investigated a novel aspect of our primer design strategy where we incorporated the PCR biases observed in our protocol to suppress the representation of highly expressed ribosomal transcripts, while maintaining the overall transcriptome coverage. Transcripts encoding 81 ribosomal proteins occupied about 9% of the sequencing space in the Std. RNA-seq library prepared from the AA3 sample. Detailed analysis of the PCR biases led us to propose heuristics on favorable amplification by our heptamer primers. Amplicons with heptamer primer-binding sites in open configuration (<−4 Kcal/mol); significant tail interaction (> = 2 bp interaction between the last four bases of the universal tail and the cDNA template); low GC content (< 0.55) and short fragment lengths (< 300 bp) were heavily penalized for the ribosomal transcripts. We designed three different primer sets and generated sequencing libraries from the AA3 sample. Our sequencing data revealed up to 70% reduction in the representation of the ribosomal transcripts while the global transcriptome coverage remained high for all primer sets ( Figure 2d ). Furthermore, the overall distribution of the reads coming from the transcription factor family also exhibited a similar distribution for a representative primer set (see Supplementary Fig. S7 online). This data demonstrates the potential of our designed primer based strategy to preferentially suppress the representation of the transcripts of interest (e.g. highly expressed transcripts) and distinguishes it from other uniform amplification based strategies.
Comparison with a different PCR-based RNA-Seq method
We performed a thorough comparison of our methodology with Smart-seq19, which performs full-length cDNA amplification from limiting amounts of mRNA. Sequencing libraries were generated from 50 picograms of mRNA derived from Activin A (3 ng/mL and 100 ng/mL) treated samples using DP-seq and Smart-seq. The same samples were also used to generate Std. RNA-seq libraries from 10 ng of mRNA. The libraries prepared from both methods were highly reproducible and displayed strong correlations in the expression measurements of the transcripts in the technical replicates (see Supplementary Fig. S8 online). Both DP-seq and Smart-seq exhibited similar transcriptome coverage (see Supplementary Table S1 online) and overlapping noise in the quantification of the transcripts ( Figure 3a ). However, the transcriptome coverage obtained in either method was significantly lower than that of Std. RNA-seq libraries with the majority of low expressed transcripts (average RPKM < 3 in the Std. RNA-seq library) showing stochastic loss. Consequently, the distribution of unique reads for the low expressed transcripts was shifted towards a low read number ( Figure 3b ). A similar observation was made for moderately expressed transcripts (average RPKM between 3 and 300 in the Std. RNA-seq library) with DP-seq and Smart-seq libraries displaying an overlapping distribution of unique reads (see Supplementary Fig. S9 online). Our mapping analysis revealed a significant length bias in Smart-seq sequencing libraries resulting from poor amplification of long cDNA species (> 4 Kb). This was not observed with DP-seq as it performs amplification of selected regions of cDNA irrespective of its length, thus resulting in higher representation of a vast majority of the long transcripts (> 77%; Figure 3c ). Expression measurements of differentiating mESCs treated with a higher dose of Activin A (100 ng/mL) showed comparable up-regulation of mesendoderm markers (Cer1, Lefty1, Lefty2, Foxa2, Gsc etc.) and down-regulation of mesoderm and ectoderm genes, implying the conservation of the biological context in the sequencing libraries prepared from 50 pg of mRNA with either method ( Figure 3d ).

Comparison of DP-seq with Smart-seq on Activin A treated samples (AA3 and AA100).
(a) MA plot of technical replicates obtained from AA100 sample showed similar technical noise in the two methods. (b) Distribution of unique reads for the low expressed transcripts (RPKM < 3 in Std. RNA-seq library prepared from AA100 sample) obtained in the three methods. The majority of the low expressed transcripts did not show expression in the libraries constructed from 50 pg of mRNA in DP-seq and Smart-seq. (c) A length bias in Smart-seq resulted in higher reads for the long cDNA species (> 4 Kb) in the DP-seq libraries. (d) Comparable fold changes were observed for the known lineage markers in the three methods between AA100 and AA3 samples. The amount of mRNA used for sequencing library generation is shown in parentheses.
We next sought to compare the differential gene expression observed in DP-seq, Smart-seq and Std. RNA-seq for the two Activin A dosages. Differentially expressed transcripts were identified by generating the null distribution from the technical replicates (see Supplementary Methods online). The null distribution for Std. RNA-seq libraries showed little technical variation; as such a large proportion of differentially expressed transcripts were identified. The majority of these transcripts were expressed at low copy number. Smart-seq and DP-seq identified a comparable number of differentially regulated transcripts (1414 and 1297 respectively), however only a small proportion of them were common between the two methods (see Supplementary Fig. S9 online). Pairwise comparison of these methods with Std. RNA-seq revealed 56% overlap of the differentially expressed transcripts. Only 191 differentially regulated transcripts (common set) were common in all three methods. We found however that the differentially regulated transcripts that were method-specific are low expressed and were prone to large noise as these transcripts showed lower RPKM distributions as compared to the common set (see Supplementary Fig. S9 online). Further analysis of the fold changes observed for the common set in DP-seq and Smart-seq libraries showed strong correlations; however, they were poorly correlated with the fold changes observed in Std. RNA-seq libraries (R2 = 0.6456 for DP-seq and R2 = 0.5740 for Smart-seq). This highlights the issues caused by the increased noise in the quantification of low copy number transcript measurements, which is further amplified when using low amounts of input material.
Graded activation of the Activin A/TGFβ signaling pathway in mESCs
Mouse ESCs were differentiated in serum-free conditions in the presence of varying doses of Activin A and SB and the mRNA was profiled at day 4 (equivalent to 6.5–7.5 dpc) using DP-seq ( Figure 4a , also see Supplementary Methods online). The differential gene expression analysis revealed a stepwise increase in the number of transcripts differentially regulated as mESCs responded to the gradient of Activin A. The most transcriptional diversity was observed between SB and AA100 samples corresponding to the two extreme states of pathway activation. By mapping those transcripts to known Activin A/TGFβ pathway components using Ingenuity pathway analysis (Ingenuity® Systems, www.ingenuity.com), we observed a substantial down-regulation of many of these genes in response to pathway inhibition via SB ( Figure 4b ) whereas Activin A up-regulated these genes.

Graded expression of putative target genes of the Activin A/TGFβ signaling pathway in day 4 mESCs.
(a) Schematic representation of the experimental setup. Mouse ESCs were differentiated in serum free conditions and different dosages of Activin A and SB-431542 were introduced to create a graded activation of the Activin A/TGFβ signaling pathway. Cells were harvested at day 4 for sequencing library generation. Differential gene expression analysis identified ~15–20% of expressed transcripts as differentially regulated in each sample in comparison with untreated controls (see Supplementary Methods online). (b) Regulation of Activin A pathway components in response to SB-431542 and Activin A. (c) Putative TGFβ target genes in differentiating mESCs at day 4. The heat map corresponds to fold changes observed for transcripts in comparison to untreated control. Putative target genes were classified as transcripts that followed opposite trends of regulation upon treatment with Activin A and SB. Fifty transcripts were successively up-regulated while 23 transcripts followed graded down-regulation with increasing dosages of Activin A. The majority of the TGFβ target genes (marked with *) had FoxH1 transcription factor binding sites separated by 30–200 bp (also called ASE) in 10 Kb upstream and downstream of the transcription start site. Known TGFβ target genes are highlighted in bold. Low copy number transcripts (RPKM < 3 in AA3 sample) are displayed in red font.
Graded activation of the Activin A/TGFβ signaling pathway allowed us to identify putative TGFβ regulated genes during early differentiation of mESCs ( Figure 4c ). Potential TGFβ target genes were predicted based on (i) the opposing modulations in SB and AA3 conditions (in comparison to untreated control) and (ii) the subsequent up/down regulation with higher dosages of Activin A (see Supplementary Methods online). We identified many of the expected TGFβ target genes, including Cer133, Lefty131, Lefty231, Foxa234 and T30 ( Figure 4c , bold). Not all expected genes were found because they either did not meet our stringent classification criteria (e.g. Nodal30, Nanog35) or they were not expressed in this cellular context. More importantly, we have identified transcripts that respond similarly to the graded Activin A/TGFβ pathway modulation, which have not been linked previously to the pathway. Promoter analysis of these transcripts revealed the presence of multiple FoxH1 binding sites36,37,38 (Asymmetric Elements, ASE) within 10 Kb upstream and downstream of the transcription start site supporting our hypothesis that the Activin A/TGFβ signaling pathway regulates the expression of these transcripts.
Lineage segregation is achieved by regulation of Activin A/TGFβ signaling
Our preliminary experiments with T-GFP mESCs (GFP driven by Brachyury/T promoter) showed negligible induction of GFP+ cells at day 4 of differentiation upon treatment with SB (described in Supplementary Methods ). The untreated control condition (SFM) naturally drives mESCs to neuro-ectoderm lineage with only 5–10% GFP+ cells. However, in presence of mesoderm inducing factors such as Activin A (3 ng/mL), > 60% of the cells were GFP+ demonstrating efficient induction of mesoderm (see Supplementary Fig. S10 online). Neuro-ectoderm associated transcripts were classified as transcripts significantly up-regulated in SB and SFM in comparison to AA15 (see Supplementary Dataset 1 online) and comprised of known neuro-ectoderm markers (Sox1, Sox2 and Pax6, Figure 5a ). We then performed GO term (biological process annotation) enrichment and KEGG pathway enrichment to validate our classification (http://david.abcc.ncifcrf.gov/). Biological processes associated with neuron differentiation and morphogenesis (see Supplementary Table S2 online) were enriched in the transcript list with the Wnt and Activin A/TGFβ pathway significantly represented (see Supplementary Table S3 online).

Lineage segregation between neuro-ectoderm and PS (mesoderm and definitive endoderm) achieved by modulation of Activin A/TGFβ signaling pathway.
(a) Schematic of the mouse embryo at embryonic day 6.5–7.5 with the gradient of Nodal expression (yellow) with the maximum expression observed in the anterior tissue. Through inhibition of TGFβ signaling pathway cells commit to the neuro-ectoderm lineage (blue). A heat map of the neuro-ectoderm associated genes is depicted (left of the embryo) with their fold changes in different samples in comparison to untreated control. The heat map on the right of the embryo depicts successive fold changes of the PS markers with varying dosages of the Activin A. The transcripts with the highest fold change in AA100 in comparison with AA15 are enriched for definitive endoderm and other anterior tissue markers. Other PS transcripts are expected to have diffused expression pattern all throughout the streak. Genes with known expression in Theiler stages 9–11 of mouse embryo are highlighted in bold (MGI database). Low copy number transcripts (RPKM < 3 in the AA3 sample) are displayed in red font. (b) Small molecule inhibition of Wnt signaling pathway (IWR-1) induced the neuro-ectoderm lineage. The fold changes are normalized to the AA3 sample. (c) BMP4 enhanced expression of posterior and extraembryonic mesoderm markers at the expense of anterior markers. Quantitative RT-PCR fold changes for two BMP4 dosages are normalized with respect to Activin A alone induction. Error bars represent the standard deviation in biological replicates (n = 3). Asterisks indicate p > 0.05 (Student's t-test) compared to controls.
To correlate some of the novel neuro-ectodermal transcripts with embryology, we searched the MGI gene expression database for the expression patterns of the identified transcripts throughout all stages of mouse embryonic development. Expression of the vast majority of the neuro-ectoderm associated transcripts were not reported in embryonic day 6.5–7.5 embryos, the stages that correspond to the studied mESC derived samples. A number of these transcripts, however, were expressed in neuro-ectoderm derivatives at later stages of development. To validate the early expression of these transcripts in the neuro-ectoderm lineage, we used Wnt pathway inhibition (IWR-1)39 as an alternative to induce neuro-ectoderm and confirmed the up-regulation of a number of these neuro-ectoderm associated transcripts ( Figure 5b and Supplementary Fig. S11 online). On the other hand, transcripts significantly up-regulated in AA15 in comparison to SB and SFM were designated as PS associated transcripts (see Supplementary Dataset 1 online). The list included a number of known mesoderm and endoderm markers (T, Mesp1, Foxa2 and Sox17). GO enrichment analysis (http://david.abcc.ncifcrf.gov/) revealed biological processes associated with gastrulation, tissue morphogenesis and tube development (see Supplementary Table S2 and S3 online).
Graded Activin A/TGFβ signaling has been shown to induce different mesoderm and endoderm tissues, correlating with the anteroposterior position of progenitors within the PS, with the highest levels of signaling corresponding to anterior most located progenitors40,41,42. Transcripts with a maximum fold change between AA100 and AA15 in comparison to other two fold changes (AA3/SFM and AA15/AA3) should mark anterior PS derivatives and in our experiments indeed comprised definitive endoderm markers. Conversely, the majority of the transcripts with maximum fold changes in AA3/SFM and AA15/AA3 were expected to have a diffused expression pattern throughout the PS ( Figure 5a ), which was confirmed by reported in-situ hybridizations for some of these transcripts43. To further validate our classification, we studied some of these new transcripts by posteriorizing Activin A induced-mesoderm with BMP444,45,46. Transcripts known to be expressed in the extra-embryonic mesoderm and the extreme posterior PS were indeed enriched and anterior PS transcripts were significantly down-regulated ( Figure 5c ). Pan-PS transcripts also exhibited down-regulation by BMP4 suggesting a dominant posteriorization effect of BMP4 signaling (see Supplementary Fig. S11 online).
Discussion
Sequencing library generation from low amounts of starting material has remained a challenge for most of the existing RNA – seq protocols. Random priming strategies amplify from low amounts of RNA, however, reliable quantitation of low abundant transcripts is not regularly obtained. In our initial experiments with a random priming strategy14, primer-dimerization and mismatches in the primer-binding sites resulted in majority of the reads mapping to multiple mRNA species. Only 18% of the reads mapped uniquely to the transcriptome and low abundant transcripts were significantly under-represented because of a poor dynamic range. The methodology presented in this work addresses these issues by facilitating generation of reliable sequencing libraries from as low as 50 pg of mRNA. The dynamic range of our protocol exceeded five orders of magnitude in RNA concentrations allowing a more reliable detection of the majority of the low expressed transcripts.
Primer design was a critical component of DP-seq. The ubiquitous presence of heptamer primer-binding sites on the mouse transcriptome was utilized to amplify more than 80% of known transcripts (see Supplementary Fig. S2 online) from a small set of 44 heptamer primers. We optimized PCR conditions for heptamer hybridization to achieve successful amplification of more than 50,000 different fragments representing ~18,000 transcripts in the mouse Refseq mRNA database. A number of considerations were made while determining the base composition of primers to reduce mis-priming and primer dimerization. As a result, majority of the reads (55%) came from perfect binding of the primers while another 38% had one mismatch in primer-binding site. This enabled us to use the entire read length for alignment to the mouse transcriptome.
Our transcriptome data demonstrated excellent reproducibility and sensitivity. We were able to reliably estimate up to a 216-fold change in transcript expression from limiting amounts of mRNA. Furthermore, fold changes observed in low abundant transcripts were in perfect agreement with quantitative RT-PCR. Technical replicate data revealed comparable noise in the quantification of transcript expression with respect to standard RNA-seq protocols. Furthermore, the global measurements of transcript expression of libraries constructed from at least 50 pg of mRNA showed high correlations with the library made from 10 ng of mRNA.
A standard RNA-seq approach7 requires at least 10–100 ng of mRNA for reliable library generation. To address this issue, a number of protocols15,18,19 were recently developed. DP-seq offers a cost effective way of generating reliable sequencing library from limiting amounts of mRNA. The cost of amplification only includes a one-time purchase of 44 primers (14 bp) that are sufficient to generate hundreds of sequencing libraries. Our protocol is compatible with regular first strand cDNA synthesis kits and the polymerases used in our protocol (Taq and Klenow polymerase) are cheap and readily available. The processing time required for the generation of a sequencing library is also short, as DP-seq library preparation does not require fragmentation of the cDNA library or poly-adenylation of the 3' end of the amplicon library. A direct comparison of DP-seq with Smart-seq revealed comparable transcriptome coverage and similar technical noise in the quantification of the low expressed transcripts. Furthermore, DP-seq does not suffer from length bias and provides a higher representation and hence better quantification of the long cDNA species in the sequencing library. DP-seq primers amplify select regions of the known transcriptome, as such the sequencing libraries are devoid of the information regarding RNA structure (exon usage, TSS, etc.) or uncharacterized transcripts.
Typical RNA-seq protocols do not discriminate against high abundant transcripts. Consequently, most of the sequencing effort is spent on a small number of highly abundant transcripts47. We exploited the PCR biases observed in our protocol to reduce the representation of ribosomal transcripts by designing primers that have less likelihood of hybridizing efficiently to these transcripts. Complete elimination of the ribosomal transcripts was not achieved because of the mis-priming of the heptamer primers. It would be desirable to reduce mis-priming seen in our approach and further refinements in the design strategy to address above issues are in progress.
The increased sensitivity of our methodology allowed us to detect known transcripts that had only been associated with later stages of germ layer segregation. These findings are of interest since it supports the view that low-level expression of lineage specific transcripts precedes overt manifestation of lineage phenotype, at least as traditionally assayed. This might not be surprising, since lineage commitment probably involves making chromatin of lineage specific transcripts accessible to transcriptional machinery and might result in low-level transcription. Indeed, recent work on the analysis of activation marks in the promoters of differentiation specific transcripts has demonstrated that promoter activity is detected well before established landmarks of differentiation are achieved48,49. It will be very interesting to explore this idea further, at the single cell level, to determine when and how this early transcriptional activation determines germ line specification.
Methods
Primer design
Heptamer primer-binding sites are ubiquitously present in the mouse transcriptome enabling the selection of a small set of heptamer primers to cover more than 80% of the mouse transcriptome. Moreover, while hexamer primers have a low range of annealing temperatures, heptamer primers hybridize with greater efficiency to allow Klenow polymerase to extend these primers and perform efficient amplification.
We first implemented a suffix array data structure to identify 32-mer unique regions in the mouse transcriptome. All suffixes in the suffix array were divided into disjoint segments using 32-mer sequences. For each segment, we then identified all related segments that possess up to 2 mismatches with the 32-mer sequence. If the segment and all of its related segments contained suffixes mapping to only one transcript, then the segment was designated as unique. Next, we predicted the local secondary structures of the known transcripts as stable secondary structures were expected to shield heptamer primers from hybridizing to their primer-binding site. For each transcript in the Mouse NCBI RefSeq mRNA database, we ran a window of 47 bp along the transcript length and determined its propensity to form stable secondary structure using UNAfold software50. Gibbs free energy (ΔG) was estimated at 37°C for standard PCR buffer conditions (2 mM MgCl2 and 50 mM NaCl). Regions with a ΔG ≥ −4 kcal/mol were considered to be available for heptamer primer hybridization (open configuration).
We combined the two datasets and identified all heptamer primer-binding sites, (i) flanking unique regions on mouse transcriptome and (ii) residing in open configuration. We then implemented an iterative randomized algorithm (see Supplementary Fig. S1 online ) to identify a defined set of heptamer primers forming valid amplicons for >80% of the mouse transcriptome. We defined a valid amplicon as follows:
-
1
It has a length between 50 and 300 bp.
-
2
Both, forward and reverse primer-binding sites are in open configuration.
-
3
At least one of the primer-binding sites must have a ΔG ≥−2 Kcal/mol.
-
4
A 32 bp unique region should follow one of the primer-binding sites.
-
5
The GC content of the amplicon should not exceed 58%.
-
6
The amplicon must be within 5 Kb of the 3' end.
Using this approach, we identified 44 unique primers, which were split into 3 sets to reduce primer-dimerization (see Supplementary Table S4 ). This configuration covered ~80% of transcripts with 57% of transcripts covered uniquely. More than 170000 valid amplicons were predicted from 201242 primer-binding sites.
The three primer sets used for suppressing the representation of the ribosomal transcripts are detailed in Supplementary Table S5 online .
Accession Code. Gene Expression Omnibus: GSE 45474 (sequencing read data).
References
Asmann, Y. W. et al. 3' tag digital gene expression profiling of human brain and universal reference RNA using Illumina Genome Analyzer. BMC Genomics 10, 531 (2009).
Marguerat, S. & Bahler, J. RNA-seq: from technology to biology. Cell Mol Life Sci 67, 569–579 (2010).
Marioni, J. C., Mason, C. E., Mane, S. M., Stephens, M. & Gilad, Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18, 1509–1517 (2008).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10, 57–63 (2009).
Metzker, M. L. Sequencing technologies - the next generation. Nat Rev Genet 11, 31–46 (2010).
Ozsolak, F. & Milos, P. M. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet 12, 87–98 (2011).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5, 621–628 (2008).
Fang, Z. & Cui, X. Design and validation issues in RNA-seq experiments. Brief Bioinform 12, 280–287 (2011).
Bloom, J. S., Khan, Z., Kruglyak, L., Singh, M. & Caudy, A. A. Measuring differential gene expression by short read sequencing: quantitative comparison to 2-channel gene expression microarrays. BMC Genomics 10, 221 (2009).
Oshlack, A. & Wakefield, M. J. Transcript length bias in RNA-seq data confounds systems biology. Biol Direct 4, 14 (2009).
Hansen, K. D., Brenner, S. E. & Dudoit, S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res 38, e131 (2010).
Adli, M., Zhu, J. & Bernstein, B. E. Genome-wide chromatin maps derived from limited numbers of hematopoietic progenitors. Nat Methods 7, 615–618 (2010).
Armour, C. D. et al. Digital transcriptome profiling using selective hexamer priming for cDNA synthesis. Nat Methods 6, 647–649 (2009).
Li, H. et al. Determination of tag density required for digital transcriptome analysis: application to an androgen-sensitive prostate cancer model. Proc Natl Acad Sci U S A 105, 20179–20184 (2008).
Hashimshony, T., Wagner, F., Sher, N. & Yanai, I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep 2, 666–673 (2012).
Hoeijmakers, W. A., Bartfai, R., Francoijs, K. J. & Stunnenberg, H. G. Linear amplification for deep sequencing. Nat Protoc 6, 1026–1036 (2011).
Tang, F. et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 6, 377–382 (2009).
Gertz, J. et al. Transposase mediated construction of RNA-seq libraries. Genome Res 22, 134–141 (2012).
Ramskold, D. et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 30, 777–782 (2012).
Gadue, P., Huber, T. L., Paddison, P. J. & Keller, G. M. Wnt and TGF-beta signaling are required for the induction of an in vitro model of primitive streak formation using embryonic stem cells. Proc Natl Acad Sci U S A 103, 16806–16811 (2006).
Willems, E. & Leyns, L. Patterning of mouse embryonic stem cell-derived pan-mesoderm by Activin A/Nodal and Bmp4 signaling requires Fibroblast Growth Factor activity. Differentiation 76, 745–759 (2008).
Armes, N. A. & Smith, J. C. The ALK-2 and ALK-4 activin receptors transduce distinct mesoderm-inducing signals during early Xenopus development but do not co-operate to establish thresholds. Development 124, 3797–3804 (1997).
Gurdon, J. B., Harger, P., Mitchell, A. & Lemaire, P. Activin signalling and response to a morphogen gradient. Nature 371, 487–492 (1994).
Jones, C. M., Kuehn, M. R., Hogan, B. L., Smith, J. C. & Wright, C. V. Nodal-related signals induce axial mesoderm and dorsalize mesoderm during gastrulation. Development 121, 3651–3662 (1995).
Sulzbacher, S., Schroeder, I. S., Truong, T. T. & Wobus, A. M. Activin A-induced differentiation of embryonic stem cells into endoderm and pancreatic progenitors-the influence of differentiation factors and culture conditions. Stem Cell Rev 5, 159–173 (2009).
Tam, P. P., Kanai-Azuma, M. & Kanai, Y. Early endoderm development in vertebrates: lineage differentiation and morphogenetic function. Curr Opin Genet Dev 13, 393–400 (2003).
Inman, G. J. et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5 and ALK7. Mol Pharmacol 62, 65–74 (2002).
Vallier, L. et al. Early cell fate decisions of human embryonic stem cells and mouse epiblast stem cells are controlled by the same signalling pathways. PLoS One 4, e6082 (2009).
Pevny, L. H., Sockanathan, S., Placzek, M. & Lovell-Badge, R. A role for SOX1 in neural determination. Development 125, 1967–1978 (1998).
Dahle, O., Kumar, A. & Kuehn, M. R. Nodal signaling recruits the histone demethylase Jmjd3 to counteract polycomb-mediated repression at target genes. Sci Signal 3, ra48 (2010).
Guzman-Ayala, M. et al. Graded Smad2/3 activation is converted directly into levels of target gene expression in embryonic stem cells. PLoS One 4, e4268 (2009).
Zajac, P., Oberg, C. & Ahmadian, A. Analysis of short tandem repeats by parallel DNA threading. PLoS One 4, e7823 (2009).
Katoh, M. CER1 is a common target of WNT and NODAL signaling pathways in human embryonic stem cells. Int J Mol Med 17, 795–799 (2006).
Zhang, Y. et al. High throughput determination of TGFbeta1/SMAD3 targets in A549 lung epithelial cells. PLoS One 6, e20319 (2011).
Vallier, L. et al. Activin/Nodal signalling maintains pluripotency by controlling Nanog expression. Development 136, 1339–1349 (2009).
Labbe, E., Silvestri, C., Hoodless, P. A., Wrana, J. L. & Attisano, L. Smad2 and Smad3 positively and negatively regulate TGF beta-dependent transcription through the forkhead DNA-binding protein FAST2. Mol Cell 2, 109–120 (1998).
Norris, D. P., Brennan, J., Bikoff, E. K. & Robertson, E. J. The Foxh1-dependent autoregulatory enhancer controls the level of Nodal signals in the mouse embryo. Development 129, 3455–3468 (2002).
Shiratori, H. et al. Two-step regulation of left-right asymmetric expression of Pitx2: initiation by nodal signaling and maintenance by Nkx2. Mol Cell 7, 137–149 (2001).
Chen, B. et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol 5, 100–107 (2009).
Hoodless, P. A. et al. FoxH1 (Fast) functions to specify the anterior primitive streak in the mouse. Genes Dev 15, 1257–1271 (2001).
Rossant, J. & Tam, P. P. Blastocyst lineage formation, early embryonic asymmetries and axis patterning in the mouse. Development 136, 701–713 (2009).
Yamamoto, M. et al. The transcription factor FoxH1 (FAST) mediates Nodal signaling during anterior-posterior patterning and node formation in the mouse. Genes Dev 15, 1242–1256 (2001).
Faust, C., Schumacher, A., Holdener, B. & Magnuson, T. The eed mutation disrupts anterior mesoderm production in mice. Development 121, 273–285 (1995).
Kattman, S. J. et al. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 8, 228–240 (2011).
Kishigami, S. & Mishina, Y. BMP signaling and early embryonic patterning. Cytokine Growth Factor Rev 16, 265–278 (2005).
Nostro, M. C., Cheng, X., Keller, G. M. & Gadue, P. Wnt, activin and BMP signaling regulate distinct stages in the developmental pathway from embryonic stem cells to blood. Cell Stem Cell 2, 60–71 (2008).
Labaj, P. P. et al. Characterization and improvement of RNA-Seq precision in quantitative transcript expression profiling. Bioinformatics 27, i383–391 (2011).
Wamstad, J. A. et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 151, 206–220 (2012).
Paige, S. L. et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell 151, 221–232 (2012).
Markham, N. R. & Zuker, M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol 453, 3–31 (2008).
Acknowledgements
This work was supported by National Institutes of Health (NIH), National Institute of Diabetes and Digestive and Kidney Diseases Grant P01-DK074868 (S.S.), National Heart, Lung and Blood Institute Grant 5 R33 HL087375-02 (S.S. and M.M.) and NIH/National Institute of General Medical Sciences Grant GM078005-05 (S.S.). V.B. was recipient of a California Institute for Regenerative Medicine (CIRM) Graduate Fellowship (T1-00003 and TG2-01154, Interdisciplinary Stem Cell Training Program at UCSD). E.W. was a fellow supported by a CIRM postdoctoral training grant T2-00004 at SBMRI and by an AHA postdoctoral fellowship. The authors especially thank Wenqing Cai for sample preparation, data analysis and useful discussions, Dr. Suresh Subramani, Dr. Brian Saunders, Harish Nagarajan, Shakti Gupta and Dr. Gaurav Agarwal for many helpful discussions and Dr. Juan Carlos Izpisúa Belmonte for providing T-GFP mESCs.
Author information
Authors and Affiliations
Contributions
S.S. and V.B. conceived the designed primer strategy, V.B. and P.K. developed the methodology for primers, V.B. developed and implemented the sample preparation protocol, E.W., V.B., M.M., S.S. conceived and designed the embryonic stem cell experiments and analyses. V.B. and E.W. carried out the stem cell preparation and cultured embryonic stem cells, V.B. wrote the first draft of the manuscript and S.S. and M.M. supervised the work and revised the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Supplementary Information
Dataset 1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Bhargava, V., Ko, P., Willems, E. et al. Quantitative Transcriptomics using Designed Primer-based Amplification. Sci Rep 3, 1740 (2013). https://doi.org/10.1038/srep01740
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01740
This article is cited by
-
Non-coding RNAs in cancer: platforms and strategies for investigating the genomic “dark matter”
Journal of Experimental & Clinical Cancer Research (2020)
-
Single-cell RNA sequencing to explore immune cell heterogeneity
Nature Reviews Immunology (2018)
-
Robust transcriptional signatures for low-input RNA samples based on relative expression orderings
BMC Genomics (2017)
-
Nuclear RNA-seq of single neurons reveals molecular signatures of activation
Nature Communications (2016)
-
Technical Variations in Low-Input RNA-seq Methodologies
Scientific Reports (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
