
Abstract
Long-term strain improvements through repeated mutagenesis and screening have generated a hyper-producer of cellulases and hemicellulases from Penicillium decumbens 114 which was isolated 30 years ago. Here, the genome of the hyper-producer P. decumbens JU-A10-T was sequenced and compared with that of the wild-type strain 114-2. Further, the transcriptomes and secretomes were compared between the strains. Selective hyper-production of cellulases and hemicellulases but not all the secreted proteins was observed in the mutant, making it a more specific producer of lignocellulolytic enzymes. Functional analysis identified that changes in several transcriptional regulatory elements played crucial roles in the cellulase hyper-producing characteristics of the mutant. Additionally, the mutant showed enhanced supply of amino acids and decreased synthesis of secondary metabolites compared with the wild-type. The results clearly point out that we can target gene regulators and promoters with minimal alterations of the genetic content but maximal effects in genetic engineering.
Similar content being viewed by others
Introduction
The rapid increase in crude oil prices and decrease in crude oil reserves have significantly stimulated the search and use of renewable resources and energy. Lignocellulosic materials from agriculture and forest by-products are relatively abundant renewable resources. The use of lignocellulosic biomass for the production of liquid fuels and chemicals has long been considered to be an important alternative for sustaining the human economy and society1,2. In one of the most common biorefinery schemes, lignocellulosic materials are partially treated by physical and chemical methods to release cellulose and hemicellulose which then are hydrolyzed to fermentable sugars by a lignocellulolytic enzyme system consisting of multiple cellulases and hemicellulases. The sugars can be converted to ethanol or other products2,3.
Although recombinant protein expression technology has been applied to the production of most industrial enzymes, lignocellulolytic enzyme systems are primarily produced by fungi that are often enhanced through strain improvement4,5. The most widely used commercial lignocellulolytic enzymes are produced by Trichoderma reesei strains that have been improved through repeated mutagenesis and screening6. Mutations in three genes, carbon repressor gene cre1 (ref. 7), β-glucosidase regulator gene bglR8 and glucosidase IIα subunit gene gls2α9, contribute to the hyper-production of cellulolytic enzymes in T. reesei mutant strains. Mutations in cre1 and bglR lead to reduced carbon catabolite repression and the mutated gls2α results in altered N-glycan patterns on secreted proteins. Although high numbers of genetic mutations have been found in several T. reesei mutants through comparative genomics analysis10,11, additional mechanisms accounting for the hyper-producing phenotype remain enigmatic.
Some Penicillium species have been reported to produce more balanced native lignocellulolytic enzyme systems than T. reesei12 and our previous study indicated that there were more diverse genes encoding cellulose binding domain-containing proteins and hemicellulases in the genome of cellulolytic fungus Penicillium decumbens than those in T. reesei13. The original P. decumbens isolate 114 was obtained from decayed straw-covered soil in 1979 (ref. 14) and has been improved through a series of mutagenesis and screening over the years (Fig. 1A). Carbon catabolite repression (CCR)-resistant strain UV11 was first obtained by selecting a mutant forming a clear halo zone on a holocellulose-glucose agar plate14. Multiple rounds of further mutagenesis and selection generated JU-A10 (ref. 15), followed by JU-A10-T, which has been used for industrial-scale cellulase production in China for 17 years with a productivity of 160 IU L−1 h−1 (ref. 16). The activities of the lignocellulolytic enzyme system of JU-A10-T increased several times compared to those of the sequenced strain 114-2. However, the molecular mechanisms behind the strain improvement for higher cellulase production are poorly understood.

P. decumbens mutagenesis and screening.
(A) Genealogy of strains used. The two strains used for genome sequencing are labeled in red. Strains shown in gray have been lost over the years. (B) Production of lignocellulolytic enzymes by 114-2 and JU-A10-T. The two strains were grown in the cellulose-wheat bran medium for 4 days. FPase stands for filter paper enzyme activity. Error bars represent standard deviations of triplicate independent cultures. (C) Colony morphology of 114-2 and JU-A10-T on cellulose plates. Strain JU-A10-T generated larger cellulolytic halo than strain 114-2 did.
In this study, comparative and functional genomics studies of P. decumbens mutant JU-A10-T and wild-type strain 114-2 were performed to decipher how strain improvement has significantly improved the production of the lignocellulolytic enzyme system. Mutations in gene regulators and promoter regions partially accounted for the increased production of cellulases and hemicellulases. Transcriptome analysis further revealed clues for the hyper production of lignocellulolytic enzymes in the mutant. Collectively, the results offer a multi-level understanding of enzyme hyper-production by P. decumbens strain JU-A10-T and the findings will provide guidance for targeted genetic modification to enhance the production of lignocellulolytic enzymes.
Results
The different phenotypes between wild-type strain 114-2 and mutant JU-A10-T
The mutant JU-A10-T produces ~9-fold higher cellulase activity (measured as filter paper enzyme, FPase), ~8-fold higher xylanase (a major type of hemicellulase) activity and ~4-fold higher total secreted proteins than wild-type strain 114-2 in cellulose-wheat bran (CW) medium, a typical cellulase producing medium for P. decumbens17 (Fig. 1B). Thus, the strain improvement has resulted in a mutant that produces more secreted proteins, especially cellulases and hemicellulases. In addition, strain 114-2 produces dark green conidia, while JU-A10-T produces much fewer and whitish-pink conidia on solid agar plates (Fig. 1C).
Global comparison of genomes of 114-2 and JU-A10-T
The genome of strain JU-A10-T was sequenced and assembled independently of that of 114-2 (Supplementary Table S1). The genome was assembled into 482 contigs totaling 30.69 Mb with 96.3% mapped onto the 8 chromosomes and the mitochondrial genome of 114-2 (Supplementary Fig. S1). The aligned DNA had an average of 1.4 single variations per kilobase (SNVs/kb; Supplementary Fig. S2A), less dense than that (7.8 SNVs/kb) between two Aspergillus niger strains18. Gene curation using transcriptomic data yielded 10,473 gene models in JU-A10-T (Supplementary Table S2). Strains 114-2 and JU-A10-T shared 9,599 proteins with an average amino acid identity of 99.55%, including 6,699 proteins with 100% identity over the full length (Supplementary Fig. S2B). The average protein identity was slightly lower than that (99.96%) between two P. digitatum isolates19. Proteins with sequence differences between 114-2 and JU-A10-T were enriched for those with transcription factor activities (P-value = 2.77 × 10−5). Manual check of sequence alignments suggested that 140 of the 476 predicted transcription factors contained amino acid sequence differences between the strains (Supplementary Table S3). Strain-specific genes were mainly found in a few genomic regions, encoding primarily proteins of unknown function, transposases, vegetative incompatibility proteins and proteins involved in biosynthesis of secondary metabolites (Supplementary Table S4). The features of strain-specific genes20 indicate that the parent isolate 114 was likely a heterokaryon and current strains 114-2 and JU-A10-T have been purified by subsequent transferring or screening processes (Fig. 1A).
Comparison of genes encoding lignocellulolytic enzymes
We have previously annotated 18 cellulases and 51 hemicellulases in 114-2 (ref. 13). JU-A10-T carried 66 of these enzymes and it did not contain three adjacent hemicellulase genes in 114-2 scaffold 8 (PDE_08036, PDE_08037 and PDE_08038). The shared proteins were mostly identical. Only 26 amino acid differences were identified in 16 cellulases or hemicellulases between the strains, all occurring at non-conserved positions (Supplementary Table S5) that are not expected to affect enzyme activities21.
JU-A10-T increased secretion of lignocellulolytic enzymes and decreased secretion of amylases and proteases
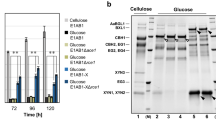
Activity assays showed that extracellular cellulase and hemicellulase activities were both elevated, while the β-glucosidase activity was slightly reduced in JU-A10-T relative to those in 114-2 (Fig. 1B). The difference indicated possible altered secretome composition between the strains. When the secretomes of 114-2 and JU-A10-T were analyzed using liquid chromatography-tandem mass spectrometry (LC-MS/MS), 90 proteins were identified (Supplementary Table S6). Proteins involved in the degradation of cellulose, hemicellulose, starch and proteins were detected in the secretomes of both strains. However, the proportion of cellulases and hemicellulases increased, while the production of amylases, proteases and other proteins decreased markedly in the JU-A10-T secretome, according to peptide spectral counts of LC-MS/MS22 (Fig. 2A). The result was confirmed by comparing the two-dimensional electrophoresis map of JU-A10-T secretome (Supplementary Fig. S3A) with that of 114-2 (ref. 13). Nearly all the protein spots on gel for JU-A10-T were identified as lignocellulolytic enzymes. Consistently, extracellular amylase activity and protease activity of JU-A10-T were significantly lower than those of 114-2, respectively (Supplementary Fig. S3B). To further assess the changes in secretome composition, transcripts of the strains were analyzed (Supplementary Table S7). Transcripts of the 34 experimentally detected cellulases and hemicellulases accounted for 1.07% of total transcripts in strain 114-2 and the percentage increased to 9.03% in JU-A10-T (Fig. 2B and Tables S6). On the flip side, transcription levels of amylase and protease genes were lower in JU-A10-T. The expression of the principal β-glucosidase BGLI gene was essentially unchanged in JU-A10-T.

Comparison of expression levels of secreted enzymes between strains 114-2 and JU-A10-T.
(A) Secretome comparison. Protein abundance ratios are calculated according to peptide spectral counts in LC-MS/MS. Proteins with lower abundances in JU-A10-T are shown in gray background. The 18 major proteins were at least 1% in at least one of the two secretomes. Glycoside hydrolases (GHs) were named according to the CAZy families they were classified, except BGLI which belongs to GH family 3. Chi18A, a chitinase; Mur25A, a muramidase; HP1, a hypothetical protein. (B) Transcription level comparison. All the proteins detected in LC-MS/MS and 2DE-MS/MS were included. Detailed protein abundance ratios and transcription levels are listed in Supplementary Table S6.
Genetic and transcriptional changes of transcription factors altered secretome composition in JU-A10-T
Like the first obtained mutant UV11, JU-A10-T showed CCR-resistant expression of cellulases and hemicellulases (e.g. higher expression in glucose medium, Supplementary Fig. S4A). The gene creA (PDE_03168) in P. decumbens, a homolog of T. reesei cre1 (ref. 7), encoding the key carbon catabolite repressor, had a frameshift mutation at the C-terminus in JU-A10-T (Supplementary Fig. S4B). The effect of CreA on the expression of extracellular hydrolytic enzymes was investigated by deleting the creA gene in strain 114-2. As expected, deletion of creA resulted in CCR-resistant expression of cellulases (Supplementary Fig. S4C). In addition, the ΔcreA strain showed ~1.7-fold higher FPase activity and ~3.4-fold higher amylase activity than 114-2 in cellulose medium (Fig. 3), confirming the negative effect of CreA on cellulase expression under both repressing and inducing conditions23.

Effect of deleting creA or amyR in strain 114-2 on the production of extracellular enzymes.
Mycelia from 2% (w/v) glucose medium were grown in 1% (w/v) cellulose medium for another 48 h. Enzyme activities (U/ml) and protein concentrations were determined in the culture supernatant and expressed as relative values with those of 114-2 set at 1. Error bars represent standard deviations of triplicate independent cultures. Statistical significance of the difference between 114-2 and each gene deletion mutant is shown. *, P < 0.05; **, P < 0.005.
Promoter regions (1.5-kb upstream region) of 11 cellulase genes contained 106 nucleotide differences between the strains (Supplementary Table S8). When the nucleotide differences were analyzed for their effects on putative binding motifs of CreA, one SNV in the promoter of a cellobiohydrolase gene cel7A-1 was found to affect a putative CreA-binding site (two adjacent 5′-SYGGRG-3′ motifs24,25; Supplementary Fig. S5). Further, Cel7A-1 had the highest fold change among the cellulases up-regulated in JU-A10-T (Supplementary Table S6). To investigate the mutation on transcription efficiency, the EGFP (enhanced green fluorescent protein) gene was fused to cel7A-1 promoters from 114-2 and JU-A10-T, respectively and then transformed into 114-2. Real-time quantitative RT-PCR analysis and microscopy analysis showed that the cel7A-1 promoter from JU-A10-T had a ~2.6-fold higher transcriptional efficiency than the promoter from 114-2 (Fig. 4).

Transcription efficiencies of cel7A-1 promoters from strains 114-2 and JU-A10-T.
Strains were grown in 1% (w/v) glucose medium or 1% (w/v) cellulose medium for 48 h. Transcription efficiency was analyzed by real-time quantitative RT-PCR of the reporter eGFP gene. Transcription levels of eGFP gene were normalized to 100-fold of those of β-actin. Error bars represent standard deviations of triplicate measurements. FM, fluorescence microscopy; LM, light microscopy. White bar = 10 μm.
In Aspergillus species, transcriptions of amylase genes are coordinately activated by the transcription factor AmyR26. The AmyR homologs in P. decumbens strains 114-2 and JU-A10-T (PDE_03964 and PDT_04193, respectively) were 100% identical, with a much lower gene transcription level in JU-A10-T than that in 114-2 (> 15-fold, Supplementary Dataset S1). Deletion of amyR in 114-2 dramatically decreased amylase activity by 94.2% (Fig. 3). Notably, the deletion also resulted in about 1.9-fold higher FPase activity, suggesting a negative role of AmyR in the expression of cellulase genes in strain 114-2.
Comparative transcriptome analysis revealed reorganized metabolism in JU-A10-T
The gene expression profiling data covered ~80% of the whole predicted genes (Supplementary Table S7), which allowed the study of genome-wide gene expression changes between the strains. Notably, JU-A10-T had remarkably more genes transcribed at low levels (< 10 transcripts per million clean tags) than 114-2 did (Fig. 5A). This indicated that some biological processes might be less active in JU-A10-T than those in 114-2. Correspondingly, 567 genes were significantly up-regulated, while up to 1,447 genes were significantly down-regulated in JU-A10-T (Supplementary Dataset S1).

Genome-wide gene expression changes between strains 114-2 and JU-A10-T.
(A) Distribution of gene transcription levels. TPM, transcripts per million clean tags. Note that the percent of genes with very high transcription levels (TPM > 2,000) in JU-A10-T is higher than that in 114-2. (B) Gene Ontology (GO) enrichment of genes of significantly differential expression between JU-A10-T and 114-2. For up-regulated genes, only the top 10 significantly enriched GO terms are shown. The full results of gene function enrichment including gene IDs are listed in Supplementary Table S9.
Compared with 114-2, genes significantly up-regulated in JU-A10-T predominantly coded extracellular polysaccharide hydrolases (Fig. 5B and Supplementary Table S9). Genes involved in glycolysis and the citric acid cycle had minimal variations in expression, but the expression levels of genes for the pentose phosphate pathway (PPP) were up-regulated in JU-A10-T, such as the putative glucose-6-phosphate dehydrogenase (PDE_01924) gene which was up-regulated ~9 folds (Supplementary Table S10). The data suggest increased flux through PPP, which provides NADPH and precursors for amino acids biosynthesis. Interestingly, the genes responsible for channeling xylose and arabinose, the hydrolytic products of hemicellulose, into PPP for consumption were also over-expressed in JU-10A-T (Supplementary Table S10). For amino acid biosynthesis, the genes of the lysine and cysteine biosynthesis were up-regulated by 2 to 7 folds (Supplementary Table S11), which may facilitate the synthesis of the four major (hemi-)cellulolytic enzymes rich in lysine and/or cysteine (Supplementary Fig. S6). Ribosomal protein genes were significantly enriched in the up-regulated gene set (FDR = 7.31 × 10−5, Supplementary Table S9). In addition, five genes involved in protein folding were significantly up-regulated in JU-A10-T (Supplementary Table S12). Putative protein disulfide-isomerase PDE_06215 and molecular chaperone PDE_08980 genes were up-regulated by 6.0 and 2.7 folds, respectively, which may aid the hyper-production of secreted proteins in JU-A10-T.
The down-regulated gene set in JU-A10-T was significantly enriched for oxidoreductases involved in metabolism (Fig. 5B and Supplementary Table S9). Of the 173 down-regulated oxidoreductases, 141 were mapped to KEGG pathways27, including 35 enzymes involved in degradation of amino acids (especially aromatic amino acids; Supplementary Table S13) and 29 genes responsible for biosynthesis of secondary metabolites. Further examination found that 104 out of 314 genes in predicted secondary metabolism gene clusters were significantly down-regulated, while only 11 were significantly up-regulated in JU-A10-T (Supplementary Table S14).
Discussion
Sequence analysis shows that the increased lignocellulolytic enzyme activities in mutant JU-A10-T are not due to the mutations in catalytic amino acid residues. This is not a surprise as the same conclusion has been reached with T. reesei mutant strains that over-produce cellulolytic enzymes10,11. Our sequence analysis also points to the enriched mutations in transcription regulators. Through functional experiments in wild-type 114-2 as the reference strain, some of the transcription factors are shown to be involved in the hyper-producing characteristics of JU-A10-T. First, global transcription factor CreA has a frame-shift mutation at the C-terminus, changing the sequence of the last 16 amino acid residues. The cre1 in T. reesei hyper-producing mutant Rut-C30 has a nonsense mutation that also enhances cellulase production7. This coincidence suggests that creA is a key target in genetic engineering of fungal strains for higher production of cellulolytic enzymes. Considering that glucose-resistant expression of cellulases has been observed in strain UV11 (Fig. 1A), the frameshift mutation in creA might occurr in the early phase of mutagenesis and screening14.
Secondly, down-regulation of AmyR expression contributes to the hyper-production of cellulolytic enzymes in JU-A10-T. AmyR was previously regarded as an amylase-specific regulator and was recently shown to also regulate the expression of β-glucosidases and galactosidases in A. niger28. In A. oryzae, mutation in amyR causes down-regulation of amylases and up-regulation of cellulolytic enzymes only in the presence of starch and a CCR-involved mechanism is proposed to account for the increased cellulolytic enzymes29. A CCR-involved mechanism is logic as AmyR activates the synthesis of amylases that convert starch to glucose and the latter inhibits the production of cellulolytic enzymes through CCR. However, AmyR may also directly repress the transcription of cellulolytic enzyme genes, as shown in our medium without starch (Fig. 3). To the best of our knowledge, this is the first report of a regulatory effect of AmyR on cellulose-induced cellulase expression in fungi.
Comparative analysis of N. crassa25 and T. reesei30 strains with different production levels of cellulolytic enzymes have shown the up-regulation of protein synthesis and processing-related proteins in fungal strains that over-produce cellulolytic enzymes. Our global gene expression analysis show similar changes between JU-A10-T and 114-2. In addition, we have identified elevated transcription of genes involved in lysine and cysteine biosynthesis required for the production of major lignocellulolytic enzymes in JU-A10-T. In summary, mutations in the key gene expression regulators and promoter regions, together with other functional mutations to be identified, made strain JU-A10-T a hyper-yield and “specific” producer of lignocellulolytic enzymes compared with the versatile wild-type strain 114-2. Activities facilitating the production of lignocellulolytic enzymes are enhanced and other activities, such as the synthesis of secondary metabolites, are decreased. The results clearly demonstrate that we may target gene regulators and promoters with minimal alterations of the genetic content but maximal effects in genetic engineering.
Methods
Strains and culture conditions
P. decumbens strains 114-2 (CGMCC 5302) and JU-A10-T were used in this study. All culture media were based on modified Mandels' salt solution (MMS), containing (g L−1): KH2PO4 3.0, NaNO3 2.6, MgSO4·7H2O 0.5, CaCl2 0.5, FeSO4·7H2O 0.0075, MnSO4·H2O 0.0025, ZnSO4·7H2O 0.0036 and CoCl2·6H2O 0.0037. Conidial suspensions were inoculated at a final concentration of 106 per ml and strains were grown at 30°C, 200 rpm (for liquid culture). For secretome analysis, digital gene expression profiling and comparison of extracellular enzyme activities between 114-2 and JU-A10-T, strains were grown in MMS supplemented with 0.1% (w/v) peptone and 1% (w/v) microcrystalline cellulose plus 1% (w/v) wheat bran (CW medium) for indicated time periods. MMS supplemented with 0.1% (w/v) peptone and 2% (w/v) glucose (G medium) was also used in comparison of extracellular enzyme activities. For comparison of extracellular enzyme activities between 114-2 and transcription factor gene deletion mutants, conidia were pre-cultured in G medium for 24 h. Then, mycelia were collected by vacuum filtration, washed with sterile water and re-suspended in MMS supplemented with 1% (w/v) microcrystalline cellulose for further growth.
Protein concentration and enzyme assays
Protein concentration and extracellular hydrolase activities were assayed as previously described17. For FPase assay, dilutions of supernatant (0.5 ml) were incubated with Whatman No.1 filter paper (1 × 6 cm, 50 mg) in 1.0 ml of 0.2 M acetate buffer (pH 4.8) at 50°C for 60 min. Statistical significance tests were carried out using Microsoft Excel program with a one-tailed equal variance t-test.
Genome sequencing, assembly and annotation
The genome of JU-A10-T was sequenced with 454 GS FLX Titanium platform and the Illumina Genome Analyzer. Genome assembly was performed with the standard 454 assembler Newbler31 version 2.3 to generate contigs. Illumina mate-paired reads of strain JU-A10-T were mapped to contigs from 454 assembler to generate scaffolds. Gene models were predicted independently with a set of gene finders as performed in strain 114-2 (ref. 13) and then manually curated with 454-sequenced transcriptome sequences (NCBI Sequence Read Archive database accession no. SRA048522) and nr BLAST32 hits to justify intron-exon boundaries and UTR regions. Blast2GO33 was used for Gene Ontology (GO)34 and InterPro35 annotation. Transcription factors were annotated according to the InterPro IDs in Fungal Transcription Factor Database36. Secondary metabolism gene clusters were predicted with SMURF37. Protein sequences from the eukaryotic genomes in KEGG GENES database27 (Version 57) were compiled as BLAST library and KO (KEGG Ortholog) identifiers were assigned with KOBAS38 algorithm with a combination of thresholds (e-value ≤ 1e-5 and rank < 5).
Genetic variation analysis
The genomes of 114-2 and JU-A10-T were aligned using mauve39,40 and Mummer41. In addition, sequencing reads of one strain were mapped to the genome of the other strain using BWA42,43, Bowtie44, SHRiMP45, gsMapper31 and MOSAIK-ALIGNER (Version1.1.0021, http://code.google.com/p/mosaik-aligner/). SNVs were called by Samtools and VarScan46 via the resulted SAM or BAM files from the aligners. The SNVs were then filtered by composition complexity, where those with more than five homo-polymers in 21 bp-windows were discarded. SNVs supposed by both reads level and genome level were retrieved as the final SNV set. Strain-specific genes were manually checked to remove false positive results caused by sequencing gaps or mistaken gene predictions.
Secretome analysis
Culture broths of strains grown in CW medium for 48 h were centrifuged at 12,000 × g at 4°C for 20 min and the supernatants were analyzed by reversed-phase (RP) LC-MS/MS and two-dimensional electrophoresis (2DE)-MS/MS techniques. RP-LC-MS/MS and data analysis were performed as previously described22. The spectral counting method was used to determine the abundance of proteins in the secretomes47. The secretome of JU-A10-T was also analyzed by 2DE using Ettan DIGE System (GE Healthcare) in dark as done in 114-2 (ref. 13). Spots of interest were manually excised, washed and trypsin-digested for MALDI-TOF/TOF mass spectrometry on 4700 Proteomics Analyzer (Applied Biosystems) in the positive ion reflector mode. An in-house P. decumbens protein sequence database was used for protein identification.
Digital gene expression profiling
Strains grown in CW medium for 41 h were used for total RNA extraction using the RNAisoTM reagent (TaKaRa, Japan) according to the manufacturer's protocol. Digital gene expression profiling experiment based on Solexa sequencing48 was performed by Beijing Genomics Institute (BGI) at Shenzhen, China. All clean tags obtained after raw data processing were mapped to the reference tag database (predicted transcripts plus downstream 300-nucleotide sequences) of the corresponding strain. Only sense expression was considered and no mismatch was allowed. The copy number of unambiguous tags (tags mapped to one single gene) for each gene was normalized to TPM (number of transcripts per million clean tags). Genes of significantly differential expressions were identified by a significance test49 with combined thresholds (FDR50 ≤ 0.001 and fold change ≥ 2). Balst2GO33 was used for function enrichment analysis of gene sets with the threshold of FDR ≤ 0.05.
Gene targeting of creA and amyR
The gene creA was deleted using ΔcreA::hph targeting cassette carrying hygromycin B phosphotransferase (hph) expression cassette flanked by 5′ and 3′ sequence of creA gene51. Retransformation of ΔcreA strain was performed using creA::ptrA targeting cassette carrying pyrithiamine resistance gene (ptrA)52 expression cassette flanked by creA expression cassette and downstream sequence of creA gene. The gene amyR was deleted in 114-2 using ΔamyR::ptrA targeting cassette carrying ptrA expression cassette flanked by 5′ and 3′ sequence of amyR gene. Construction of targeting cassettes and transformation of P. decumbens were performed as previously described51 and successful targeting was confirmed by Southern blot.
Promoter efficiency analysis
The reporter gene eGFP was introduced into the vector pDHt/SK under control of cel7A-1 promoters from 114-2 and JU-A10-T, respectively. The expression vectors were transformed into the recipient 114-2 via Agrobacterium-mediated fungal transformation. Transcription levels of eGFP and β-actin genes were analyzed by real-time quantitative PCR (Eppendorf, Germany), in which the primer pair 5′-AGTGCTTCAGCCGCTACCC-3′ and 5′-GATGCCGTTCTTCTGCTTGTC-3′ were used for amplification of eGFP gene and 5′-TGAGGCTCCCATCAACCC-3′ and 5′-CACCAGAGTCCAGCACGATA-3′ were used for amplification of β-actin gene (PDE_01092).
Accession numbers
The Whole Genome Shotgun project of strain JU-A10-T has been deposited in DDBJ/EMBL/GenBank under the accession number AGII00000000. The digital gene expression profiling data have been deposited in NCBI's Gene Expression Omnibus under the GEO Series accession number GSE34288.
References
Somerville, C. The billion-ton biofuels vision. Science 312, 1277 (2006).
Fernando, S., Adhikari, S., Chandrapal, C. & Murali, N. Biorefineries: current status, challenges and future direction. Energy & Fuels 20, 1727–1737 (2006).
Sanchez, C. Lignocellulosic residues: biodegradation and bioconversion by fungi. Biotechnol Adv 27, 185–194 (2009).
Cherry, J. R. & Fidantsef, A. L. Directed evolution of industrial enzymes: an update. Curr Opin Biotechnol 14, 438–443 (2003).
Gusakov, A. V. Alternatives to Trichoderma reesei in biofuel production. Trends Biotechnol 29, 419–425 (2011).
Esterbauer, H., Steiner, W., Labudova, I., Hermann, A. & Hayn, M. Production of Trichoderma cellulase in laboratory and pilot scale. Bioresour Technol 36, 51–65 (1991).
Ilmen, M., Thrane, C. & Penttila, M. The glucose repressor gene cre1 of Trichoderma: Isolation and expression of a full-length and a truncated mutant form. Mol Gen Genet 251, 451–460 (1996).
Nitta, M. et al. A new Zn(II)(2)Cys(6)-type transcription factor BglR regulates beta-glucosidase expression in Trichoderma reesei. Fungal Genet Biol 49, 388–397 (2012).
Geysens, S. et al. Cloning and characterization of the glucosidase II alpha subunit gene of Trichoderma reesei: a frameshift mutation results in the aberrant glycosylation profile of the hypercellulolytic strain Rut-C30. Appl Environ Microbiol 71, 2910–2924 (2005).
Le Crom, S. et al. Tracking the roots of cellulase hyperproduction by the fungus Trichoderma reesei using massively parallel DNA sequencing. Proc Natl Acad Sci U S A 106, 16151–16156 (2009).
Vitikainen, M. et al. Array comparative genomic hybridization analysis of Trichoderma reesei strains with enhanced cellulase production properties. BMC Genomics 11, 441 (2010).
Gusakov, A. V. & Sinitsyn, A. P. Cellulases from Penicillium species for producing fuels from biomass. Biofuels 3, 463–477 (2012).
Liu, G. et al. Genomic and secretomic analyses reveal unique features of the lignocellulolytic enzyme system of Penicillium decumbens. PLoS One 8, e55185 (2013).
Qu, Y., Gao, P. & Wang, Z. Screening of catabolite repression-resistant mutants of cellulase producing Penicillium spp. Acta Mycol Sinica 3, 238–243 (1984).
Qu, Y., Zhao, X., Gao, P. & Wang, Z. Cellulase production from spent sulfite liquor and paper-mill waste fiber. Appl Biochem Biotechnol 28-29, 363–368 (1991).
Fang, X., Shen, Y., Zhao, J., Bao, X. & Qu, Y. Status and prospect of lignocellulosic bioethanol production in China. Bioresour Technol 101, 4814–4819 (2010).
Sun, X., Liu, Z., Qu, Y. & Li, X. The effects of wheat bran composition on the production of biomass-hydrolyzing enzymes by Penicillium decumbens. Appl Biochem Biotechnol 146, 119–128 (2008).
Andersen, M. R. et al. Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res 21, 885–897 (2011).
Marcet-Houben, M. et al. Genome sequence of the necrotrophic fungus Penicillium digitatum, the main postharvest pathogen of citrus. BMC Genomics 13, 646 (2012).
Fedorova, N. D. et al. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genetics 4, e1000046 (2008).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081 (2009).
Li, R. X. et al. Localized-statistical quantification of human serum proteome associated with type 2 diabetes. PLoS One 3, e3224 (2008).
Aro, N., Pakula, T. & Penttila, M. Transcriptional regulation of plant cell wall degradation by filamentous fungi. FEMS Microbiol Rev 29, 719–739 (2005).
Cubero, B. & Scazzocchio, C. Two different, adjacent and divergent zinc finger binding sites are necessary for CREA-mediated carbon catabolite repression in the proline gene cluster of Aspergillus nidulans. EMBO J 13, 407–415 (1994).
Sun, J. & Glass, N. L. Identification of the CRE-1 cellulolytic regulon in Neurospora crassa. PLoS One 6, e25654 (2011).
Petersen, K. L., Lehmbeck, J. & Christensen, T. A new transcriptional activator for amylase genes in Aspergillus. Mol Gen Genet 262, 668–676 (1999).
Kanehisa, M., Goto, S., Furumichi, M., Tanabe, M. & Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 38, D355–360 (2010).
vanKuyk, P. A., Benen, J. A., Wosten, H. A., Visser, J. & de Vries, R. P. A broader role for AmyR in Aspergillus niger: regulation of the utilisation of D-glucose or D-galactose containing oligo- and polysaccharides. Appl Microbiol Biotechnol. 93, 285–293 (2012).
Watanabe, J. et al. Loss of Aspergillus oryzae amyR function indirectly affects hemicellulolytic and cellulolytic enzyme production. J Biosci Bioeng 111, 408–413 (2011).
Foreman, P. K. et al. Transcriptional regulation of biomass-degrading enzymes in the filamentous fungus Trichoderma reesei. J Biol Chem 278, 31988–31997 (2003).
Margulies, M. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol 215, 403–410 (1990).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat Genet 25, 25–29 (2000).
Hunter, S. et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 40, D306–312 (2012).
Park, J. et al. FTFD: an informatics pipeline supporting phylogenomic analysis of fungal transcription factors. Bioinformatics 24, 1024–1025 (2008).
Khaldi, N. et al. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol 47, 736–741 (2010).
Mao, X., Cai, T., Olyarchuk, J. G. & Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793 (2005).
Darling, A. C., Mau, B., Blattner, F. R. & Perna, N. T. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14, 1394–1403 (2004).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5, e11147 (2010).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biol 5, R12 (2004).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009).
Rumble, S. M. et al. SHRiMP: accurate mapping of short color-space reads. PLoS Comput Biol 5, e1000386 (2009).
Koboldt, D. C. et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283–2285 (2009).
Chundawat, S. P. et al. Proteomics-based compositional analysis of complex cellulase-hemicellulase mixtures. J Proteome Res 10, 4365–4372 (2011).
AC't Hoen, P. et al. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res 36, e141–e141 (2008).
Audic, S. & Claverie, J. M. The significance of digital gene expression profiles. Genome Res 7, 986–995 (1997).
Benjamini, Y. & Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann Stat, 1165–1188 (2001).
Li, Z., Du, C., Zhong, Y. & Wang, T. Development of a highly efficient gene targeting system allowing rapid genetic manipulations in Penicillium decumbens. Appl Microbiol Biotechnol 87, 1065–1076 (2010).
Kubodera, T., Yamashita, N. & Nishimura, A. Transformation of Aspergillus sp. and Trichoderma reesei using the pyrithiamine resistance gene (ptrA) of Aspergillus oryzae. Biosci Biotechnol Biochem 66, 404–406 (2002).
Acknowledgements
This study was supported by grants from National Basic Research Program of China (Grant no. 2011CB707403), National Natural Sciences Foundation of China (Grant no. 31030001) and International S&T Cooperation Program of China (2010DFA32560). We thank Bingqiang Liu and Yang Li for kind help in differential gene expression analysis.
Author information
Authors and Affiliations
Contributions
Y. Qu, Z.Z., T.W. and G.-P.Z. supervised the study and designed main experiments. G.Z., M.C., K.Z. and G.L. isolated DNA and RNA for sequencing. J.L., H.Z. and S.W. performed genome sequencing. L.Z. performed genome assembling and gene annotation. L.Z., G.Z., G.L., Y. Qin and L.M. participated in accurate adjustment of gene models. X.Y., J.Z. and L.C. prepared secreted protein samples for LC-MS/MS. G.Z. and R.L. performed efficiency analysis of promoters. X.W. and G.L. performed enzyme assays and 2DE analysis. Z.L. performed targeted gene deletion. G.L., L.Z. and G.Z. analyzed data and drafted the manuscript. H.X., X.B., X.F., L.W., Y.Z. and W.L. commented on the manuscript. Z.Z., L.X., C.W., G.-P.Z. and Y. Qu revised the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemental information
Supplementary Information
Dataset S1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Liu, G., Zhang, L., Qin, Y. et al. Long-term strain improvements accumulate mutations in regulatory elements responsible for hyper-production of cellulolytic enzymes. Sci Rep 3, 1569 (2013). https://doi.org/10.1038/srep01569
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01569
This article is cited by
-
Regulation of genes encoding polysaccharide-degrading enzymes in Penicillium
Applied Microbiology and Biotechnology (2024)
-
Preparation of cellulose nanocrystals from commercial dissolving pulp using an engineered cellulase system
Bioresources and Bioprocessing (2023)
-
Regulation of nutrient utilization in filamentous fungi
Applied Microbiology and Biotechnology (2023)
-
Construction of a novel filamentous fungal protein expression system based on redesigning of regulatory elements
Applied Microbiology and Biotechnology (2022)
-
Current perspective on improved fermentative production and purification of fungal cellulases for successful biorefinery applications: a brief review
Biomass Conversion and Biorefinery (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
