
Abstract
Airway epithelial injury is the hallmark of various respiratory diseases, but its mechanisms remain poorly understood. While 13-S-hydroxyoctadecadienoic acid (13-S-HODE) is produced in high concentration during mitochondrial degradation in reticulocytes little is known about its role in asthma pathogenesis. Here, we show that extracellular 13-S-HODE induces mitochondrial dysfunction and airway epithelial apoptosis. This is associated with features of severe airway obstruction, lung remodeling, increase in epithelial stress related proinflammatory cytokines and drastic airway neutrophilia in mouse. Further, 13-S-HODE induced features are attenuated by inhibiting Transient Receptor Potential Cation Channel, Vanilloid-type 1 (TRPV1) both in mouse model and human bronchial epithelial cells. These findings are relevant to human asthma, as 13-S-HODE levels are increased in human asthmatic airways. Blocking of 13-S-HODE activity or disruption of TRPV1 activity attenuated airway injury and asthma mimicking features in murine allergic airway inflammation. These findings indicate that 13-S-HODE induces mitochondrial dysfunction and airway epithelial injury.
Similar content being viewed by others


Introduction
Asthma is a very common chronic lower airway disease. Allergic asthma is characterized by nonspecific airway hyperresponsiveness, reversible airway obstruction, airway inflammation including airway eosinophilia, mucus hypersecretion and structural changes of the airway. Most of the asthma features are thought to be mediated by Th2 response1. However, recent reports have emphasized the importance of airway epithelia in respiratory diseases including asthma in contrast to earlier concepts of dominant role of inflammation2,3. The airway epithela maintain airway homeostasis by controlling inflammatory and healing responses to foreign antigens and pollutants and airway epithelial injury is considered as one of the crucial events in various respiratory diseases, including asthma2,3. Indeed stressed epithelia release crucial cytokines such as IL-33 which have proinflammatory properties by inducing Th2 polarization4,5,6. Current research and therapeutic strategies for asthma focus on regulation of inflammatory cell function and preventing release of potentially toxic mediators. This is at least partly related to the fact that the mechanisms underlying the epithelial injury are not clear, thereby limiting downstream opportunities to protect against epithelial injury. Mitochondrial dysfunction is now recognized to be an important aspect of respiratory disease pathogenesis7,8,9,10. Whether and how mitochondrial dysfunction occurs in asthma and whether this is a critical aspect of epithelial injury is not known.
Various lipid metabolites have been implicated in different inflammatory diseases11. Although much is known about leukotrienes12, much less attention has been given to other lipid metabolites. 13-S-hydroxyoctadecadienoic acid (13-S-HODE) is a lipid metabolite derived from linoleic acid, a major polyunsaturated fatty acid, through various enzymatic sources like 15-lipoxygenase, cyclooxygenase and nonenzymatic mechanisms like auto-oxidation13,14,15. 13-S-HODE is produced in large quantities during mitochondrial degradation steps in RBC maturation16 whereas 13-hydroxy-linoleic acid, a related molecule, has been linked to airway hyperresponsiveness17. We studied 13-S-HODE as a potential link between mitochondrial dysfunction, epithelial injury and airway disease because of increasing evidence of the role of mitochondrial dysfunction7,8,9. In a recent report, the transfer of mitochondria from stem cells to alveolar epithelial cells reversed acute lung injury in sepsis, indicating the important role that mitochondrial health could play in lung disease18.
In this context, understanding the effects of 13-S-HODE on airway epithelium is essential. In this study, we demonstrate for the first time that 13-S-HODE causes disturbance in calcium homeostasis, mitochondrial structural alterations and bronchial epithelial injury. Exogenous administration of 13-S-HODE to naïve mouse leads to features of severe asthma such as severe airway obstruction, increasing airway neutrophilia, lung remodeling, increase in epithelial stress related proinflammatory cytokines such as IL-25 along with the features of mitochondrial dysfunction and bronchial epithelial injury. Further, 13-S-HODE induced features are reversible with inhibition of Transient Receptor Potential Cation Channel, Vanilloid-type 1 (TRPV1) both in human bronchial epithelia and mouse. While earlier study had demonstrated that TRPV1 inhibition didn't alleviate asthma features in C57BL/6 mice, our study revealed that TRPV1 levels were not affected in allergic C57BL/6 mice whereas TRPV1 levels were upregulated in BALB/c mice. Disruption of TRPV1 activity by genetic or pharmacological approaches attenuated airway injury and asthma features in murine models of allergic airway inflammation. These findings are relevant to human disease, as shown by increased in 13-S-HODE in extracellular fluids of human asthmatics and increase in TRPV1 expression in human asthmatic lungs. In addition, neutralization of 13-S-HODE attenuated asthma features in mice with allergic airway inflammation. These findings indicate extracellular 13-S-HODE causes mitochondrial dysfunction, airway injury and leads to severe asthma phenotype and thus represent potential therapeutic targets in inflammatory airway diseases.
Results
13-S-HODE causes mitochondrial structural alterations and injury in bronchial epithelium
It is known that 13-S-HODE is produced in high quantities during mitochondrial degradation steps in reticulocyte in the process of RBC maturation16. We found that 13-S-HODE is increased in allergized mouse lungs7 and we and others have shown that mitochondrial structural alterations and mitochondrial dysfunction in asthmatic bronchial epithelia7,8,9. So to determine the effect of 13-S-HODE on airway epithelia, we incubated bronchial epithelia with 13-S-HODE. Induction of human bronchial epithelial cells with 13-S-HODE caused epithelial injury evidenced by mitochondrial structural alterations such as dilatation of cristae, loss of tubular shape with swelling, increase in Annexin V positive cells, shrinkage of cells, loss of mitochondrial membrane potential with increase in JC-1 monomers and release of cytochrome c into cytosol to initiate apoptosis (Fig. 1a–g).

13-S-HODE leads to mitochondrial structural alterations and injury in human bronchial epithelium.
(a) TEM images of mitochondria of human bronchial epithelium that were induced either by vehicle (ethanol) or by 13-S-HODE for 48 hours. Scale bars, 0.5 μM. (b) Staining of 13-S-HODE induced human bronchial epithelium with MitoTracker Green. Images are at 100× magnification. Arrows indicate abnormal mitochondria. (c) Graphs of flow cytometric data of human bronchial epithelial cells that were induced with 13-S-HODE and stained with Annexin V to depict apoptotic cells. (d) Forward and side scatter graphs show the alteration in size of human bronchial epithelial cells with 13-S-HODE stimulation. (e) Graphs of flow cytometric data of human bronchial epithelial cells that were induced with 13-S-HODE and stained with JC-1 to depict apoptotic cells. (f) Histograms of JC-1 monomers (directly proportionate to cells that have collapsed mitochondria) after 13-S-HODE induction. (g) Cytochrome c in cytosol of human bronchial epithelium after 48 hours of 13-S-HODE induction.
13-S-HODE increased intramitochondrial calcium overload in bronchial epithelium
Since mitochondrial swelling can be associated with mitochondrial calcium influx, the mitochondrial calcium level was determined with mitochondrial matrix–targeted green fluorescent protein (GFP)-tagged inverse-pericam (i-pericam, a calcium-sensitive mitochondrial protein the fluorescence of which is inversely proportional to that of intramitochondrial calcium). There was a gradual increase in mitochondrial calcium levels with 13-S-HODE (Fig. 2a–b). As increase in calcium leads to activate calpain and calpain 10 overexpression has been shown to cause mitochondrial swelling and apoptosis19, we determined the calpain activity in the cytosol and the calpain 10 activity in the mitochondria. Expectedly, we found an increase in cytosolic calpain and mitochondrial calpain 10 activities (Fig. 2c–d) confirmed the increased intramitochondrial calcium overload and indicate the increase in intracellular calcium. It seemed that 13-S-HODE could cause loss of filamentous shape owing to mitochondrial calcium overload, depolarization and disturbance in osmotic balance between mitochondria and cytosol.

13-S-HODE increases intramitochondrial calcium overload in human bronchial epithelium.
(a) Selected confocal images of human bronchial epithelium that was transfected with mitochondrial matrix–targeted GFP-tagged i-pericam, stimulated with 13-S-HODE, stained with MitoTracker Red and recorded for 3 minutes. Upper row: i-pericam GFP expression control; middle row: MitoTracker Red staining; and lower row: merged images. Images are at 100× magnification. (b) The change in fluorescence was quantitated for 3 minutes. Selected mitochondrial regions were analyzed for their i-pericam fluorescence intensity and values are averaged (mean ± s.e.m.). (c, d) Fluorometry to estimate cytosolic calpain and calpain 10 activity in mitochondria. MITO, mitochondria Data representative of three independent experiments, results shown as mean ± s.e.m., significance determined with unpaired Student t test (***P < 0.05).
13-S-HODE causes severe airway dysfunction, airway neutrophilia, mitochondrial dysfunction and epithelial injury in naïve mouse
To determine whether the extracellular increase in 13-S-HODE was relevant to asthma pathogenesis, we instilled 13-S-HODE in murine lungs (Supplementary Fig. 1). 13-S-HODE instillation was associated with airway inflammation, marked epithelial injury and difficulty in breathing (Fig. 3a and Supplementary videos S1 and S2), which are characteristic features of human obstructive airway diseases such as asthma compared to vehicle (50% ethanol) instillation which didn't cause any alveolar pathology or alteration in lung function (Supplementary Fig. 2a, b). Unlike BALB/c mice, C57BL/6 mice didn't show the inflammatory foci in the airways and visible severe difficulty in breathing though they showed the pulmonary congestion and epithelial injury upon 13-S-HODE administration. There was a significant increase in lung compliance and decrease in lung elastance at basal levels with 13-S-HODE treatment in BALB/c mice (Fig. 3b, Supplementary Fig. 2c) similar to nonemphysematous reduction in lung elastance in patients with severe asthma20,21,22. Though 13-S-HODE administration didn't cause any alteration in airway resistance (data not shown), it caused a significant increase in enhanced pause (Penh), a unit-less index of bronchoconstriction (Fig. 3c). BALB/c mice, to which 13-S-HODE was administered, developed features of mitochondrial dysfunction such as reduction in mitochondrial membrane potential, reduction in complex IV activity in lung mitochondria and increase in the levels of cytochrome c in lung cytosol (Fig. 3d–f). Though similar features were observed with C57BL/6 mice (Fig. 3g–h) the severity of mitochondrial dysfunction was less compared to BALB/c mice. 13-S-HODE treatment led to airway neutrophilia along with an increase in IL-4 levels in the lung without affecting IL-5 and IL-13 (Fig. 3i–j).

Effects of exogenous 13-S-HODE on airway inflammation, airway physiology and mitochondrial functions in healthy BALB/c mice.
(a) Photomicrographs of hematoxylin and eosin–stained lung sections of mice that received different concentrations of 13-S-HODE (0-0.6 mg per mouse in BALB/c mice or 0.6 mg per mouse in C57BL/6 mice) or vehicle intranasally once a day for 3 consecutive days followed by sacrifice 6 hours after the last dose (n = 4–6 mice per group). Images are at 20× magnification. Invasive and noninvasive measurements to determine baseline lung compliance (b) and enhanced pause (c), respectively in BALB/c mice. Mitochondrial membrane potential (spectrofluorometry) (d), cytochrome c levels in lung cytosol (e) and complex IV activitiy (spectrophotometry) in lung mitochondria (f) were determined in BALB/c mice. Mitochondrial complex I activity (g) and cytosolic cytochrome c levels (h) in lung were also determined in C57BL/6 mice (n = 6 each group). (i) Cytokines in lung homogenates in BALB/c mice. (j) BAL fluid differential count in BALB/c mice. Macro, macrophage; Mono, mononuclear agranulocytes; and Neutro, neutrophil. Data representative of two independent experiments, results shown as mean ± s.e.m., significance determined with unpaired Student t test or Mann-Whitney test (**P < 0.001; ***P < 0.05).
Neutralization of 13-S-HODE in allergized BALB/c mice reduced asthma features
To determine whether extracellular 13-S-HODE was a critical modulator of asthma pathogenesis in vivo, we neutralized 13-S-HODE with intraperitoneal injection of 13-S-HODE–specific polyclonal antibody in BALB/c mice with allergic airway inflammation (Supplementary Fig. 1). Treatment of allergized mice with 13-S-HODE–specific polyclonal antibody which neutralized 13-S-HODE (approximately 85%), but not the isotype control, was associated with marked attenuation of airway inflammation, BAL fluid eosinophilia, goblet cell metaplasia, subepithelial fibrosis and airway hyperresponsiveness (Fig. 4 a–e). This attenuation of asthma features were associated with decrease in the levels of IL-25, an epithelial cytokine which is released upon epithelial stress and reversal of mitochondrial dysfunction with increase in mitochondrial complex I activity and decrease in cytosolic cytochrome c (Fig. 4 f–h). The reduction in IL-25 indicated that 13-S-HODE antibody reduced the epithelial stress6.

Neutralization of endogenous 13-S-HODE in allergized BALB/c mice attenuated asthma features.
Representative photomicrographs of bronchovascular regions of different groups of mice stained with H & E (a), PAS (b) and MT (c) stainings (n = 5–6 mice per group) and airway inflammation score (a, iv), BAL fluid differential count (b, iv) and morphometric analysis epithelial mucin content and subepithelial fibrosis (c, iv). Macro, macrophage; Mono, mononuclear agranulocytes; Eosino, eosinophil; and Neutro, neutrophil. Images are at 40× (H & E and PAS) and 20× (MT) magnification. (d) 13-S-HODE in BAL fluid supernatant was assayed. (e) Airway resistance in response to increasing concentrations of methacholine as the percent baseline airway resistance assuming saline aerosol-derived values as baseline (n = 4–6 each group) was determined. (f) IL-25 levels in lung. Lung mitochondrial complex I activity (g) and lung cytosolic cytochrome c (h) were assayed, n = 4–6 each group. Data are representative of two independent experiments, results are shown as mean ± s.e.m., significance is determined with unpaired Student t test or Mann-Whitney test (*P < 0.0001; **P < 0.001; ***P < 0.05).
Mechanisms for 13-S-HODE induced airway injury: TRPV1 activation and disturbance in calcium homeostasis in 13-S-HODE and allergic murine models
Since 13-S-HODE mediated epithelial injury was associated with increase in intracellular calcium, we suspected that 13-S-HODE mediated effects are linked with calcium related receptors which may also induce apoptosis. Among various possible receptors on which 13-S-HODE can act as putative ligands such as vanilloid receptors and peroxisome proliferator-activated receptors23,24, we have selected former as they are not only potent calcium channels but also can induce cell injury25,26. It is known that among 28 reported mammalian members of TRP channels, the expression of vanilloid receptor type 1 (TRPV1) is widely expressed in the airways including airway epithelia27,28,29 and plethora of literature is available to explore the role of TRPV1 in cough29. We performed immunohistochemical analysis in allergized lungs of both mouse and human to determine the TRPV1 expression. TRPV1 was expressed in bronchial epithelial cells and various connective tissue cells and its expression was increased in lungs of both allergized mouse and human asthmatics consistent with a putative target for 13-S-HODE (Fig. 5a). Further, TRPV1 levels also found to upregulated in allergized lungs of BALB/c mice. In contrast to BALB/c mice, C57BL/6 mice did not show any increase in TRPV1 levels in allergized lungs (Fig. 5b) indicate the strain specific variation in TRPV1 levels. To determine whether 13-S-HODE–mediated epithelial injury is through activation of TRPV1, we determined TRPV1 levels. However, there was no increase in TRPV1 protein levels with 13-S-HODE induction both in bronchial epithelia and mouse lungs (Fig. 5c–d). Suspecting 13-S-HODE could functionally activate TRPV1 without affecting protein levels, we measured calpain activity in lung cytosols of 13-S-HODE–treated mice as we found TRPV1 activation leads to increase in calpain activity in human bronchial epithelia (Fig. 2c, d). 13-S-HODE increased cytosolic calpain activity only in BALB/c mouse lungs (Fig. 5e–f). Thus the role of TRPV1 was complicated by variability across murine strains as TRPV1 and cytosolic calpain activity were increased in lungs of BALB/c mice but not C57BL/6 mice. However, there was an increase in the mitochondrial calpain 10 activity in both strains (Fig. 5g–i). Calpain 10 expression was found to be expressed in bronchial epithelia of allergized lungs (Fig. 5j). It is plausible that 13-S-HODE causes mitochondrial dysfunction in C57BL/6 mice by interacting with other receptor(s) which remains to be elucidated. So we have taken BALB/c mice for further studies.

Mouse strain specific variations in TRPV1 levels and calpain.
(a) Photomicrographs depicting immunohistochemical staining for TRPV1 in lungs of SHAM and OVA mice or lung sections from control participants and patients with asthma. Brown indicates positive expression of TRPV1; images are shown at 40× (upper panel) and 10 × (lower panel) magnifications. Graphs of data from ELISA of TRPV1 in lung cytosol in SHAM and OVA mice from either the BALB/c or C57BL/6 strain (b), in lung cytosol in mice in which 13-S-HODE was administered (c) and in Beas-2B cells induced with 13-S-HODE (d). Graphs of data from cytosolic calpain activity in healthy BALB/c mice in which 13-S-HODE was administered (e) and in healthy C57BL/6 mice in which 13-S-HODE was administered (f). Graphs of data from mitochondrial calpain activity in healthy BALB/c mice in which 13-S-HODE was administered (g) and in healthy C57BL/6 mice in which 13-S-HODE was administered (h). (i) Schematic diagram to illustrate the strain specific variations between BALB/c and C57BL/6 mice in TRPV1 levels and calpain activities. Red color indicate either increased in the levels or activity. (j) Photomicrographs depicting immunohistochemical staining for calpain 10 in lungs of SHAM and OVA mice; images are at 20× magnifications. Results are shown as mean ± s.e.m., significance determined with unpaired Student t test or Mann-Whitney test (**P < 0.001; ***P < 0.05).
Next, we determined the effects of TRPV1 inhibition (capsazepine, CPZ) in 13-S-HODE–treated human bronchial epithelial cells and mice. In bronchial epithelial cells, cytochrome c release and loss of mitochondrial tubular shape were restored by CPZ (Fig. 6a–b). CPZ also reduced mitochondrial calcium overload in 13-S-HODE–stimulated bronchial epithelial cells and CPZ might have affected both induced and basal levels of mitochondrial calcium (Fig. 6c). In mice, CPZ was able to prevent 13-S-HODE–mediated airway inflammation, abnormal lung function including difficulty in breathing, reduction in lung elastance, BAL fluid neutrophilia, increase in epithelial derived proinflammatory cytokines such as IL-25 and IL-33, mitochondrial dysfunction, lipid peroxidation and increased cytochrome c in lung cytosol (Fig. 6d–k, Supplementary Fig. 3a–b and Supplementary Video 3). CPZ also reduced 13-S-HODE–induced epithelial injury and ultrastructural changes of mitochondria (Fig. 6l–m and Supplementary Fig. 3c). Similar to pharmacological antagonism of TRPV1, siRNA inhibition of TRPV1 also reduced difficulty in breathing, reduced airway inflammation, reduced BAL fluid neutrophilia, increased complex I and complex IV activity in lung mitochondria and reduced lipid peroxidation in 13-S-HODE administered mice (Fig. 6n–q, Supplementary Fig. 3d–f and Supplementary Video 4).

A Mechanism for 13-S-HODE induced airway injury: 13-S-HODE–induced epithelial injury both in healthy BALB/c mice and human bronchial epithelium is through TRPV1.
(a) Cytochrome c in cytosols of 13-S-HODE–induced human bronchial epithelium treated with or without CPZ (capsazepine, a pharmacological antagonist of TRPV1) was determined (n = 3). (b) Mitochondria of human bronchial epithelium were visualized by MitoTracker Green–staining after 13-S-HODE induction with or without pretreatments of CPZ; images are at 100× magnification. (c) Intramitochondrial calcium in human bronchial epithelium that were stimulated with 13-S-HODE and pretreated with either vehicle or capsazepine (CPZ, 5 μM, antagonist of Transient Receptor Potential Cation Channel, Vanilloid-type 1 (TRPV1) was determined after transfection with i-pericam [i-pericam, a calcium-sensitive mitochondrial protein the fluorescence of which is inversely proportional to that of intramitochondrial calcium47]. Data representative of two or more independent experiments, results shown as mean ± s.e.m., significance determined with unpaired Student t test or Mann-Whitney test (***P < 0.05). Representative photomicrographs of hematoxylin and eosin–stained lung sections (d, 20× magnification), elastance of lung (e), BAL fluid differential count (f), IL-25 levels in lung (g), IL-33 levels in lung (h), complex I activity (i), complex IV activity (j) and cytochrome c (k) of 13-S-HODE–treated mice that were administered either CPZ or VEH (n = 5–6 mice per group) are shown. MITO, mitochondria. Macro, macrophage; Mono, mononuclear agranulocytes; and Neutro, neutrophil. TUNEL apoptotic assay (l, 60× magnification) and TEM analysis (m, 15000× magnification) of lung sections of 13-S-HODE–treated mice treated with CPZ were performed. Representative photomicrographs of hematoxylin and eosin–stained lung sections (n, 20× magnification), TRPV1 in lung (o), activities of mitochondrial complexes I (p) and IV (q) from 13-S-HODE–treated mice that were administered either TRPV1 or scrambled siRNA (n = 5–6 mice per group) are shown. Data are representative of two independent experiments, results are shown as mean ± s.e.m., significance is determined with unpaired Student t test or Mann-Whitney test (**P < 0.001; ***P < 0.05).
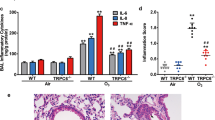
To determine the role of TRPV1 in asthma pathogenesis, we administered TRPV1 siRNA or CPZ to BALB/c mice with allergic airway inflammation (AAI), in which we had found upregulation of TRPV1. TRPV1 siRNA treatment of AAI mice reduced airway inflammation, goblet cell metaplasia, BAL fluid eosinophilia, subepithelial fibrosis and airway hyperresponsiveness along with a significant reduction in TRPV1 levels in the lungs (Fig. 7a–e). This attenuation was associated with a reduction in cytosolic calpain activity, mitochondrial calpain 10 activity, cytochrome c levels and caspase 3 activity in lung cytosol and epithelial apoptosis and with an increase in activities of complex I and complex IV in lung mitochondria (Fig. 7f–l). Pharmacological antagonism of TRPV1 with CPZ also reduced airway inflammation and airway hyperresponsiveness and restored mitochondrial function in lung (Fig. 7m–n, Supplementary Fig. 4a–e).

Knockdowns of TRPV1 restore mitochondrial function and reduce epithelial injury in allergic mice.
(a–c) Photomicrographs of bronchovascular regions of different groups of mice stained with H & E (a), PAS (b) and MT (c) stainings (n = 5–6 mice per group) and airway inflammation score (a, iv), BAL fluid differential count (b, iv) and morphometric analysis of epithelial mucin content and subepithelial fibrosis (c, iv). Macro, macrophage; Mono, mononuclear agranulocytes; Eosino, eosinophil; and Neutro, neutrophil. Images are at 40× (H & E and PAS) and 20× (MT) magnifications. (d) Airway hyperresponsiveness as airway resistance (n = 4–6 each group). ELISA was performed to determine TRPV1 (e) and activity assays were performed to determine calpain activity in cytosol (f) and mitochondrial calpain 10 activity in lung (g). Assays were performed to determine the activities of mitochondrial complexes I and IV (h, i). ELISA was performed to measure lung cytosolic cytochrome c (j) and caspase 3 activity was assessed (k) (n = 5–6 each group). MITO, mitochondria. (l) TUNEL calorimetry in lung with apoptotic index as the percentage TUNEL positive bronchial epithelia. All images are at 60× magnification. (m) Photomicrographs of hematoxylin and eosin–stained bronchovascular regions (10× magnification). (n) Airway hyperresponsiveness as airway resistance (n = 5–6 each group). Data representative of two independent experiments, results shown as mean ± s.e.m., significance determined with unpaired Student t test or Mann-Whitney test (*P < 0.0001; **P < 0.001; ***P < 0.05).
13-S-HODE is increased in extracellular fluids of human atopic asthmatics
To determine the relevance of these findings in human we measured its levels in the serum, sputum supernatant and bronchoalveolar lavage (BAL) fluid supernatant of study participants with atopic asthma and compared them with those of healthy individuals. We observed a marked increase in 13-S-HODE in these extracellular biological fluids in patient samples (Fig. 8a).

13-S-HODE levels are increased in extracellular fluids of patients with atopic asthma and Schematic diagram to depict the possible mechanisms for 13-S-HODE induced airway injury.
ELISA of 13-S-HODE in serum, sputum supernatant and BAL fluid supernatant (a1–a3) of healthy controls (Controls) or patients with atopic asthma (Atopic asthmatics). Results shown as mean ± s.e.m., significance determined with unpaired Student t test (*P < 0.0001; ***P < 0.05). N = 40, 5 and 3 for serum, sputum and BAL fluid supernatant, respectively, for controls and N = 80, 5 and 3 for serum, sputum and BAL fluid supernatant, respectively, for patients. (b) Schematic diagram to illustrate the possible mechanism of 13-S-HODE–induced airway epithelial injury. Linoleic acid is converted to 13-S-HODE by various sources and 13-S-HODE activates TRPV1 present in the epithelial membrane to alter intracellular calcium homeostasis, which leads to increased mitochondrial calcium and further mitochondrial dysfunction and initiates apoptosis.
Discussion
In this study, to our best knowledge, we report for the first time that 13-S-HODE, a linoleic acid metabolite, causes mitochondrial dysfunction and bronchial epithelial injury. Although much is known about leukotrienes in asthma12, much less attention has been given to other lipid metabolites. We studied 13-S-HODE because of increasing evidence of the role of mitochondrial dysfunction in asthma7,8,9 and high concentrations of 13-S-HODE are found in reticulocytes during degeneration of mitochondria of reticulocytes16. On the other hand, mitochondrial dysfunction seems to be crucial in the genesis of epithelial injury and asthma pathogenesis in mice7,8,9. Similarly dysfunctional mitochondria have been found in human asthmatic bronchial epithelia30. Transfer of mitochondria from stem cells to alveolar epithelial cells reversed acute lung injury in sepsis, indicating the crucial role that mitochondrial health in lung diseases17. In this context, understanding the effects of 13-S-HODE on airway epithelium is essential because we found its levels to be high in the airway secretions and extracellular fluids. Also it is practically difficult to reduce the levels of 13-S-hydroxyoctadecadienoic acid (13-S-HODE) as there are many sources for its synthesis13,14,15.
We showed how exogenous induction of 13-S-HODE can lead to mitochondrial dysfunction in cultured human bronchial epithelium. We found that 13-S-HODE leads to mitochondrial structural alterations such as loss of cristae, swelling and increase in intramitochondrial calcium. As increase in calcium levels activate calpain we measured calpain activity not only in mitochondria but also in cytosol. Increase in cytosolic calpain activity with 13-S-HODE administration indicates that 13-S-HODE might have increased the levels of intracellular calcium. Increase in mitochondrial calpain activity with 13-S-HODE is interesting in the context of mitochondrial structural alterations as calpain 10 overexpression is known to cause mitochondrial fragmentation and apoptosis in neurons31. Calpain 10, mitochondrial dominant calpain, is one of the recently reported candidate molecules in other chronic inflammatory diseases such as diabetes mellitus19,31 though there is no such report in asthma. Similarly we also found that increased calpain 10 activity in allergized lungs and importantly it was found to be increased in bronchial epithelia of allergized lungs.
Next we wanted to determine the effect of 13-S-HODE in in vivo conditions. To determine this, we instilled 13-S-HODE intranasally mimicking the increase in its levels in asthmatic conditions. 13-S-HODE not only causes mitochondrial dysfunction and injury in bronchial epithelia but also causes similar features in naïve mouse. These features were associated with increase in the levels of cytokines such as IL-25 and IL-33 which are the indicators of epithelial cell stress5,6. These cytokines are known to have proinflammatory properties as they induce the polarization of Th2 cells5,6. Evidently we found an increase in the levels of IL-4. In addition, we found an increase in the levels of IL-17 in C57BL/6 mice though IL-17 was not upregulated in BALB/c mouse with 13-S-HODE administration (data not shown). These 13-S-HODE induced features were associated with severe difficulty in breathing. To understand the reasons for this difficulty in breathing, we determined the lung function. We found a significant reduction in elastance and increase in compliance. In addition, 13-S-HODE administration to allergic mice leads to worsen the airway hyperresponsiveness (data not shown). 13-S-HODE induced features in mouse such as airway neutrophilia, reduction in elastance and increase in epithelial stress related cytokines and IL-17 levels are interesting and these features are the indicators of severe asthma and emphysema20,21,22,32. We found an increase in 13-S-HODE in airways of human asthmatics and 13-S-HODE neutralization attenuates asthma mimicking features in mice, so 13-S-HODE could be targeted for asthma therapeutics. Increases in the levels of IL-17 cytokines, airway neutrophilia are also the features of steroid resistant conditions. Our preliminary results indicate that steroid administration to 13-S-HODE given allergic mice leads to no reduction in airway neutrophilia though it reduced airway eosinophilia (data not shown). However, the involvement of 13-S-HODE in emphysema and steroid resistance requires further detailed investigation.
13-S-HODE administration also leads to alter calcium homeostasis both in bronchial epithelia and mouse lungs. Quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) array profiles of 13-S-HODE–instilled lung showed a significant upregulation of the genes involved in calcium-dependent mitochondrial transporters (aralar and citrin) and carnitine shuttle from cytosol to mitochondria (carnitine palmitoyltransferase 1b and solute carrier slc25a20) (Supplementary Table 1) though 13-S-HODE did not affect various tested OXPHOS genes except cox5a, atp5a1, and atp5d (Supplementary Table 2). Furthermore, we were able to show, by using genetic and pharmacological strategies, that the effects of 13-S-HODE were due to extracellular activation of TRPV1 channels, resulting in calcium influx and calpain activation. Though TRP receptors are well studied in airway in the aspect of neuronal cells scarce studies are available in non-neural tissues. Earlier study indicated that TRPV1 has no effect in asthma pathogenesis33. This may be true in C57BL/6 mice as we found no upregulation of TRPV1 in C57BL/6 mouse whereas we found that TRPV1 levels were upregulated in allergic BALB/c mouse. In support to our findings, differential distribution of TRPV1 has been reported in airway dorsal root ganglion neurons as BALB/c mice have more capsaicin-sensitive receptors (TRPV1) than do those of C57BL/6 mice34. This difference also could be one of the reasons that asthma mimicking features were not alleviated in C57BL/6 mice in an earlier study33. However, the reasons for increased TRPV1 expression in allergized lungs are not clear. Transport of TRPV1 to the cell membrane by nerve growth factor has been reported in neurons and nerve growth factor levels are increased in allergic inflammatory conditions35,36. Accordingly, we found its expression on the surface of the bronchial epithelium in allergized mice. Although the contribution of TRPV1-mediated cell death to the pathogenesis of diseases such as diabetes mellitus and neurodegenerative diseases is known31, such effects have not been studied in allergic airway inflammation. Although we did not focus on other types of vanilloid receptors, one cannot rule out the possibility of their roles in some 13-S-HODE–mediated effects. Together, these data indicated that 13-S-HODE concentrations were increased in extracellular fluids of asthmatic patients and extracellular receptor-mediated effects of 13-S-HODE are relevant to asthma pathogenesis. Thus, extracellular 13-S-HODE present in airways may activate TRPV1 present in the bronchial epithelial membrane to alter intracellular calcium homeostasis, thereby resulting in increased mitochondrial calcium and initiation of apoptosis (Fig. 8b). It is also possible that 13-S-HODE may have receptor independent effects that need further investigations.
In conclusion, our findings indicate a novel role of extracellular 13-S-HODE, a lipid metabolite derived from linoleic acid, in allergic responses and airway epithelial injury. We further identify TRPV1 as one of its downstream targets. Since 13-S-HODE leads to key features of steroid refractory conditions such as neutrophilia and increased in IL-17 cytokines, 13-S-HODE should be explored further as a potential therapeutic target.
Methods
In vitro experiments
Human bronchial epithelial cells (Beas-2B) were obtained (ATCC, Manassas, VA, USA), maintained, stimulated with vehicle (ethanol) or 13-S-HODE (15–35 μM, Cayman, Ann Arbor, Michigan, USA) for 6 to 48 hours and harvested for further experiments.
Intramitochondrial calcium imaging
Beas-2B cells were cultured on glass coverslips, transfected for 24 hours with 5 μg of mitochondrial matrix–targeted GFP-tagged i-pericam plasmid (gift of Dr. Mike Forte, Vollum Institute, Oregon, USA through Dr. Soumya Sinha Roy, Institute of Genomics and Integrative Biology (IGIB), Delhi) by using Lipofectamine (Invitrogen, CA, USA). Then those cells were induced with 13-S-HODE for 6 hours and stained with MitoTracker Red and 3-minute confocal recording was performed at room temperature in a Leica microscope system (Leica SP5). Imaging was performed with 63x/1.4 objective with laser lines for both i-pericam (Ex = 488, Em = 510/20) and MitoTracker (Ex = 561, Em = 590/30). Leica built-in software was used for data collection and snapshots were taken at different time points and exported as tiff files. Fluorescence intensity recorded at 0 second from 180 seconds of total recoding duration was considered as 1 and change in fluorescence intensity was plotted for 180 seconds.
Animal models and Mice grouping
BALB/c mice (6-8 weeks, obtained from National Institute of Nutrition, Hyderabad or Central Drug Research Institute, Lucknow and maintained in IGIB, Delhi) and C57BL/6 mice (obtained from Jackson Laboratories, USA and maintained in Johns Hopkins University, Baltimore, MD, USA) were used. The respective institutional animal ethics committees approved all mice experiments. Most of the mice were housed in the specific pathogen-free facility at the Johns Hopkins University or in the individual ventilated cages at IGIB.
There were two different murine models (Supplemental Figure 1) as detailed below:
13-S-HODE model (Supplemental Fig. 1a): There were four sets of mice: A, B, C and D. A (BALB/c) had four groups: 0 mg 13-S-HODE, 0.075 mg 13-S-HODE, 0.15 mg 13-S-HODE and 0.6 mg 13-S-HODE, n = 4–6 each, B (C57BL/6) had two groups: 0 mg 13-S-HODE and 0.6 mg 13-S-HODE, n = 8–10 each, C (BALB/c) had three groups: VEH/VEH (vehicle controls, VEH, vehicle), HODE/VEH (13-S-HODE administered mice treated with vehicle) and HODE/CPZ (13-S-HODE administered mice treated with capsazepine), n = 5–6 each) and D (BALB/c) had three groups: VEH/VEH, HODE/scrambled siRNA (13-S-HODE administered mice treated with scrambled siRNA) and HODE/TRPV1 siRNA (13-S-HODE administered mice treated with TRPV1 siRNA, n = 5–6 each.
OVA model (Supplemental Fig. 1b): There were three sets: E, F and G (all were BALB/c). E had three groups: SHAM/PBS/VEH (normal controls, VEH, vehicle), OVA/OVA/scrambled siRNA (allergic controls treated with scrambled siRNA) and OVA/OVA/TRPV1 siRNA (allergic mice treated with TRPV1 siRNA). F had three groups: SHAM/PBS/VEH, OVA/OVA/VEH (allergic controls treated with vehicle) and OVA/OVA/CPZ (allergic mice treated with capsazepine). G had three groups: SHAM/PBS/VEH, OVA/OVA/Iso Ab (allergic controls treated with isotypic control antibody) and OVA/OVA/13-S-HODE Ab (allergic controls treated with 13-S-HODE antibody).
Immunization and challenge of mice
Mice were immunized and challenged as previously described37,38,39 but with small modifications. Briefly, mice were immunized by three intraperitoneal (i.p.) injections of 50 μg OVA (OVA, chicken egg ovalbumin, Grade V, Sigma Chemical Co, USA) with 4 mg alum or 4 mg alum only on days 0, 7 and 14. After a week, mice were challenged with 3% OVA aerosol or PBS half-an-hour day for 7 days consecutively with a nebulizer (flow rate was 9 L/min, OMRON CX3 Model, Japan).
Administration of 13-S-HODE, capsazepine, siRNAs, 13-S-HODE antibody
We instilled 30 μl of 13-S-HODE (Cayman, USA) or VEH (50% ethanol) into the nasal openings in each isoflurane-anesthetized mouse. We used the maximum dose of intranasal 0.6 mg of 13-S-HODE per mouse compared with the oral 8 mg per mouse used in an earlier study40. We used 50 μg of TRPV1 or scrambled siRNA (Sigma, St. Louis, MO, USA) per mouse. Mice under isoflurane anesthesia were administered TRPV1 siRNA, or scrambled siRNA intranasally because intranasal delivery has 1,000-fold efficiency41. Results from our earlier study39 indicated that siRNAs (Sigma, St. Louis, MO, USA) did not induce either a type I interferon response or a toll-like receptor 3 response. We administered 2.5 mg/kg CPZ (Cayman, Ann Arbor, Michigan, USA) by means of intraperitoneal injection twice a day for 2 days. Selection of the particular dose for CPZ was based on our pilot experiments. CPZ was dissolved in normal saline containing 10% ethanol and 10% Tween 20. CPZ (2.5 mg/kg) or VEH was administered intraperitoneally in mice from day 21 to day 28 twice a day in a 100 μl volume per dose. 13-S-HODE–specific polyclonal antibody was not cross-reactive with any other lipid metabolites.
Airway hyperresponsiveness measurement
Airway hyperresponsiveness was measured with invasive flexiVent mice as previously described37. Briefly, every anesthetized mouse was intubated, ventilated and airway resistance was estimated with administration of increasing doses of methacholine with ultrasonic nebulizer (Scireq, Canada). Airway hyperreactivity to methacholine in conscious mice was estimated using single-chamber plethysmography (Buxco Electronics), as described earlier7. The results are expressed as enhanced pause which is a unit-less value that estimates the alteration in the amplitude of pressure wave and expiration time42.
Bronchoalveolar lavage (BAL) and absolute cell counts
PBS was instilled into the trachea to obtain BAL fluid and absolute cell counts were determined after estimating total cell counts and differential cell counts as described earlier37.
Lung histopathology
Formalin-fixed lung sections were stained with Haematoxylin & Eosin (H & E), Periodic acid-Schiff and Masson's Trichrome stainings and morphometric analysis was performed as described earlier37.
Transmission electron microscopy
Cells were fixed, pelleted and processed for TEM7. For lungs, in situ perfusion fixation was performed, fixed lungs were processed and stained and bronchial epithelium was selected for capturing images7.
TUNEL calorimetry
TUNEL assay was performed as previously described43.
Mitohchondrial isolation
Mitochondrial and cytosolic fractions were prepared by using a mitochondrial isolation kit (Sigma, St. Louis, MO, USA). We used a mitochondrial membrane potential assay (Sigma, St. Louis, MO, USA) in isolated mitochondria and we assessed activities of mitochondrial complexes I (Mitosciences, USA) and IV (Sigma, USA) according to manufacturer instructions.
ELISAs
Cytochrome c, 8-isoprostane, TRPV1, 13-S-HODE, IL-25 and IL-33 in serum or lung cytosol were estimated according to manufacturer instructions.
Calpain activity
Calpain activity in cytosol was assessed by using a calpain activity assay kit (Biovision, Inc, Mountain View, CA). Calpain 10 activity in mitochondria was assessed with components of a calpain activity assay kit (Biovision, Inc) by using Suc-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin substrate (50 μM, Sigma, St. Louis, MO, USA). Caspase 3 activity (Biovision, Inc) was assessed as per instructions.
Immunohstochemistry
Immunohistochemical analysis for TRPV1 (Novus Biologicals, CO, USA) was performed37 with respective secondary antibodies (Sigma, St. Louis, MO, USA).
Human participants
We used the serum samples of patients with atopic asthma and healthy participants recruited for our earlier studies44,45. The Review Board of the IGIB, Delhi, India, approved the studies and we also obtained written informed consent from both the patients with atopic asthma and the healthy controls. A subset of participants was recruited for the collection of BAL fluid and sputum46 after we obtained ethical approval from the Review Board of the All India Institute of Medical Sciences, New Delhi.
Real-time qRT-PCR arrays
Lungs were harvested and immersed in RNAlater solution (Invitrogen, CA, USA). Total RNAs were prepared by using buffer containing β-mercaptoethanol and qRT-PCR was performed according to manufacturer instructions (Qiagen, West Sussex, England and Roche Real-Time PCR System, IN, USA). Briefly, 1 μg of total RNA was used for first-strand complementary DNA synthesis (RT2 First Strand Kit) and the complementary DNA was used for performing qRT-PCR, which had 84 mitochondrial function–related genes and five housekeeping genes. Web-based analysis (Qiagen, West Sussex, England) and pairwise comparisons were performed.
Statistical analysis
All results are shown as mean ± s.e.m. Statistical significance of the differences between paired groups was determined with a two-tailed Student's t test. One-way analysis of variance was used to compare multiple groups by using JMP software and was evaluated further with a nonparametric Mann-Whitney rank-sum test wherever appropriate.
References
Ngoc, P. L., Gold, D. R., Tzianabos, A. O., Weiss, S. T. & Celedón, J. C. Cytokines, allergy and asthma. Curr. Opin. Allergy Clin. Immunol. 5, 161–166 (2005).
Lambrecht, B. N. & Hammad, H. The airway epithelium in asthma. Nat. Med. 18, 684–692 (2012).
Swamy, M., Jamora, C., Havran, W. & Hayday, A. Epithelial decision makers: in search of the ‘epiimmunome.’. Nat. Immunol. 11, 656–665 (2010).
He, R. & Geha, R. S. Thymic stromal lymphopoietin. Ann. N. Y. Acad. Sci. 1183, 13–24 (2010).
Borish, L. & Steinke, J. W. Interleukin-33 in asthma: how big of a role does it play? Curr. Allergy Asthma Rep. 11, 7–11 (2011).
Suzukawa, M. et al. Epithelial Cell-Derived IL-25, but Not Th17 Cell-Derived IL-17 or IL-17F, Is Crucial for MurineAsthma. J. Immunol. In Press (2012).
Mabalirajan, U. et al. Mitochondrial structural changes and dysfunction are associated with experimental allergic asthma. J. Immunol. 181, 3540–3548 (2008).
Aguilera-Aguirre, L. et al. Mitochondrial dysfunction increases allergic airway inflammation. J. Immunol. 183, 5379–5387 (2009).
Thomas, B. et al. Ciliary dysfunction and ultrastructural abnormalities are features of severe asthma. J. Allergy Clin. Immunol. 126, 722–729 (2010).
Rabinovich, R. A. et al. Mitochondrial dysfunction in COPD patients with low body mass index. Eur. Respir. J. 29, 643–650 (2012).
Wang, D. & Dubois, R. N. Eicosanoids and cancer. Nat. Rev. Cancer 10, 181–193 (2010).
Peters-Golden, M. & Peters-Golden, M. & Henderson, W. R., Jr Leukotrienes. N. Engl. J. Med. 357, 1841–1854 (2007).
Camacho, M., Godessart, N., Antón, R., García, M. & Vila, L. Interleukin-1 enhances the ability of cultured human umbilical vein endothelial cells to oxidize linoleic acid. J. Biol. Chem. 270, 17279–17286 (1995).
Baer, A. N., Costello, P. B. & Green, F. A. Free and esterified 13(R,S)-hydroxyoctadecadienoic acids: principal oxygenase products inpsoriatic skin scales. J. Lipid. Res. 31, 125–130 (1990).
Ogawa, E. et al. Epidermal FABP (FABP5) regulates keratinocyte differentiation by 13(S)-HODE-mediated activation of the NF-κB signaling pathway. J. Invest. Dermatol. 131, 604–612 (2011).
Kühn, H. & Brash, A. R. Occurrence of lipoxygenase products in membranes of rabbit reticulocytes. Evidence for a role of the reticulocyte lipoxygenase in the maturation of red cells. J. Biol. Chem. 265, 1454–1458 (1990).
Henricks, P. A., Engels, F., van der Linde, H. J., Garssen, J. & Nijkamp, F. P. 13-Hydroxy-linoleic acid induces airway hyperresponsiveness to histamine and methacholine in guinea pigs in vivo. J. Allergy Clin. Immunol. 96, 36–43 (1995).
Islam, M. N. et al. Mitochondrial transfer from bone-marrow derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 18, 759–765 (2012).
Arrington, D. D., Van Vleet, T. R. & Schnellmann, R. G. Calpain 10: a mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. Am. J. Physiol. Cell Physiol. 291, C1159–1171 (2006).
Wenzel, S. Severe asthma in adults. Am. J. Respir. Crit. Care Med. 172, 149–160 (2005).
Seow, C. Y., Schellenberg, R. R. & Paré, P. D. Structural and functional changes in the airway smooth muscle of asthmatic subjects. Am. J. Respir. Crit. Care Med. 158, S179–S186 (1998).
Bramley, A. M., Thomson, R. J., Roberts, C. R. & Schellenberg, R. R. Hypothesis: excessive bronchoconstriction in asthma is due to decreased airway elastance. Eur. Respir. J. 7, 337–341 (1994).
Patwardhan, A. M. et al. Heat generates oxidized linoleic acid metabolites that activate TRPV1 and produce pain in rodents. J. Clin. Invest. 120, 1617–1626 (2010).
Shureiqi, I. et al. The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-delta to induce apoptosis in colorectal cancer cells. Proc. Natl. Acad. Sci. U S A 100, 9968–9973 (2003).
Gees, M., Colsoul, B. & Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2, a003962 (2010).
Kim, S. R., Kim, S. U., Oh, U. & Jin, B. K. Transient receptor potential vanilloid subtype 1 mediates microglial cell death in vivo and in vitrovia Ca2+-mediated mitochondrial damage and cytochrome c release. J. Immunol. 177, 4322–4329 (2006).
Geppetti, P., Materazzi, S. & Nicoletti P. . The transient receptor potential vanilloid 1: role in airway inflammation and disease. Eur. J. Pharmacol. 533, 207–214 (2006).
Bessac, B. F. & Jordt, S. E. Breathtaking TRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology (Bethesda). 23, 360–370 (2008).
McLeod, R. L., Correll, C. C., Jia, Y. & Anthes, J. C. TRPV1 antagonists as potential antitussive agents. Lung 186 Suppl 1, S59–65 (2008).
Xu, W., Comhair, S. A. A., Janocha, A. J., Mavrakis, L. A. & Erzurum, S. C. Alteration of nitric oxide synthesis related to abnormal cellular bioenergetics in asthmatic airway epithelium. Am. J. Respir. Crit. Care Med. 181, A1436 (2010).
Hong, S., Agresta, L., Guo, C. & Wiley, J. W. The TRPV1 receptor is associated with preferential stress in large dorsal ganglion neurons in early diabetic sensory neuropathy. J Neurochem 105, 1212–1222 (2008).
Alcorn, J. F., Crowe, C. R. & Kolls, J. K. TH17 cells in asthma and COPD. Annu. Rev. Physiol. 72, 495–516 (2010).
Caceres, A. I. et al. A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proc. Natl. Acad. Sci. U S A 106, 9099–9104 (2009).
Veronesi, B. et al. Vanilloid (capsaicin) receptors influence inflammatory sensitivity in response to particulate matter. Toxicol. Appl. Pharmacol. 169, 66–76 (2000).
Zhang, X., Huang, J. & McNaughton, P. A. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 24, 4211–4223 (2005).
Bonini, S. et al. Circulating nerve growth factor levels are increased in humans with allergic diseases and asthma. Proc. Natl. Acad. Sci. U S A 93, 10955–10960 (1996).
Mabalirajan, U. et al. Beneficial effects of high dose of L-arginine on airway hyperresponsiveness and airway inflammation in a murine model of asthma. J. Allergy Clin. Immunol. 125, 626–635 (2010).
Kumar, M. et al. Let-7 microRNA-mediated regulation of IL-13 and allergic airway inflammation. J. Allergy Clin. Immunol. 128, 1077–1085 (2011).
Aich, J., Mabalirajan, U., Ahmad, T., Agrawal, A. & Ghosh, B. Loss of function of inositol polyphosphate 4 phosphatase reversibly increases the severity of allergic airway inflammation. Nat. Commun. 3, 877 (2012).
Khan-Merchant, N., Penumetcha, M., Meilhac, O. & Parthasarathy, S. Oxidized fatty acids promote atherosclerosis only in the presence of dietary cholesterol in low-density lipoprotein receptor knockout mice. J. Nutr. 132, 3256–3262 (2002).
Zhang, X. et al. Small interfering RNA targeting heme oxygenase-1 enhances ischemia-reperfusion-induced lung apoptosis. J. Biol. Chem. 279, 10677–10684 (2004).
Kuperman, D. A. et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 8, 885–889 (2002).
Mabalirajan, U. et al. L-arginine reduces mitochondrial dysfunction and epithelial injury in murine allergic airway inflammation. Int. Immunopharmacol. 10, 1514–1519 (2010).
Sharma, M. et al. A genetic variation in inositol polyphosphate 4 phosphatase a enhances susceptibility to asthma. Am. J. Respir. Crit. Care Med. 177, 712–719 (2008).
Kumar, A. & Ghosh, B. A single nucleotide polymorphism (A --> G) in intron 3 of IFN gamma gene is associated with asthma. Genes Immun. 9, 294–301 (2008).
Fireman, E. Assessment of hazardous dust exposure by BAL and induced sputum. Chest 115, 1720–1728 (2009).
Nagai, T., Sawano, A., Park, E. S. & Miyawaki, A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. U. S. A. 98, 3197–3202 (2001).
Acknowledgements
This work was supported by grants NWP 0033 and MLP 5502 from the Council of Scientific and Industrial Research, Government of India. We thank specially Professor Surendra Kumar Sharma (All India Institute of Medical Sciences, Delhi) for providing the human BAL and sputum samples, Dr. Soumya Sinha Roy, Dr. Satish Kumar Devarapu, Dr. Arjun Ram, Mr. Younus Ahmad Butt, Ms. Lipsa Panda, Mr. Bijay Ranjan Pattnaik, Mr. Sandeep Srivastava, Mr. Manish Kumar and Ms. Shravani Mukherjee for their help. All the authors declare no competing financial interests.
Author information
Authors and Affiliations
Contributions
U.M. designed, conducted, supervised and interpreted experiments and wrote the manuscript; R.R. performed animal experiments, biochemical assays, enzyme-linked immunosorbent assay (ELISA) and qRT-PCR; T.A. performed airway physiology and imaging experiments; S.K. performed animal (C57BL/6) experiments; S.S. performed in vitro experiments; G.D.L. performed TEM experiments; J.A., M.K., K.K. and V.P.S. provided support in animal experiments; A.K.D. supervised TEM experiments; S.B. supervised genetic knockdown experiments; S.B., A.A. and B.G. provided conceptual support, interpreted experiments and critically reviewed the manuscript; all authors reviewed the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Mabalirajan, U., Rehman, R., Ahmad, T. et al. Linoleic acid metabolite drives severe asthma by causing airway epithelial injury. Sci Rep 3, 1349 (2013). https://doi.org/10.1038/srep01349
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01349
This article is cited by
-
Neural Mechanisms Underlying the Coughing Reflex
Neuroscience Bulletin (2023)
-
Plasma lipid biomarkers in relation to BMI, lung function, and airway inflammation in pediatric asthma
Metabolomics (2021)
-
Up-regulation of 15-lipoxygenase enzymes and products in functional and non-functional pituitary adenomas
Lipids in Health and Disease (2019)
-
Delayed gut microbiota development in high-risk for asthma infants is temporarily modifiable by Lactobacillus supplementation
Nature Communications (2018)
-
Mediating Role of TRPV1 Ion Channels in the Co-exposure to PM2.5 and Formaldehyde of Balb/c Mice Asthma Model
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
