
Abstract
The N≡N bond (225 kcal mol−1) in dinitrogen is one of the strongest bonds in chemistry therefore artificial synthesis of ammonia under mild conditions is a significant challenge. Based on current knowledge, only bacteria and some plants can synthesise ammonia from air and water at ambient temperature and pressure. Here, for the first time, we report artificial ammonia synthesis bypassing N2 separation and H2 production stages. A maximum ammonia production rate of 1.14 × 10−5 mol m−2 s−1 has been achieved when a voltage of 1.6 V was applied. Potentially this can provide an alternative route for the mass production of the basic chemical ammonia under mild conditions. Considering climate change and the depletion of fossil fuels used for synthesis of ammonia by conventional methods, this is a renewable and sustainable chemical synthesis process for future.
Similar content being viewed by others


Introduction
Given the need to feed a growing world population whilst simultaneously reducing global carbon emissions, it is desired to break the link between industrial production of agricultural fertilisers based on ammonia and the use of fossil fuels. On the other hand, energy storage is a big challenge for renewable electricity. To synthesis basic chemicals such as ammonia from renewable electricity through electrochemical processes is a good option to save on carbon emissions and to reduce the pressure on renewable energy storage1.
Globally 131 million tons of ammonia were produced in 20102. The dominant ammonia production process is the Haber-Bosch process invented in 1904 which requires high temperature (~500°C) and high pressure (150–300 bar), in addition to efficient catalysts3,4. Natural gas or coal is used as the energy source of the ammonia industry. 1.87 tons of CO2 is released per ton of ammonia produced5. Globally 245 million tons of CO2 were released by the ammonia industry in 2010 equivalent to about 50% of the UK CO2 emissions (495.8 million tons) in that year6. In the Haber-Bosch process, the presence of ppm level oxygen may poison the commonly used Fe-based catalysts. In industry, extensive purification of N2 and H2 is needed and this remarkably increases the overall cost of the process7,8. Therefore researchers have been seeking a simpler way for synthesis of ammonia from nitrogen separated from air. To the best of our knowledge, the first report on synthesis of ammonia from nitrogen at room temperature is through the reduction of ligating molecular nitrogen9. Following this pioneering work, there are several key reports on synthesis of ammonia under mild conditions through complex intermediates3,10,11,12,13,14,15. On the other hand, ammonia can be synthesised at room temperature through electrochemical synthesis. In 1985, for the first time, Pickett et al. reported the electrochemical synthesis of ammonia at room temperature through protolysis of cis-[W(N2)2(PMe2Ph)4]16. There were reports on electrochemical synthesis of ammonia from N2 and H2 using Na2SO4 aqueous solution as the electrolyte but the current was quite small17,18. This could be related to the low proton conductivity of Na2SO4 solution. It is expected that the current density and ammonia production rate would be much higher if a conductive electrolyte is applied.
Proton conductors are important electrolytes for electrochemical devices19,20. Some perovskite oxides exhibit high proton conductivity and have been used in solid oxide fuel cells19,21,22. Stoukides reported the electrochemical synthesis of ammonia from N2 and H2 at 570°C based on a solid proton-conducting oxide SrCe0.95Yb0.05O3−δ23. The authors also further reported the synthesis of ammonia directly from N2 and H2O bypassing the process of H2 production24. There are other reports on electrochemical synthesis of ammonia from N2 and H2O in molten salts at a temperature ~300°C25,26. Recently we reported electrochemical synthesis of ammonia at ~500°C based on oxide-carbonate composite electrolytes4,27. Ammonia tends to decompose at ~500°C28 therefore low temperature synthesis is necessary to avoid ammonia decomposition; however, most good low temperature proton conducting materials are based on acidic materials29. Ammonia is a weak base and readily reacts with an acidic membrane reducing the proton conductivity. Sulfonated Nafion has been demonstrated as the best proton-conducting polymer which has been widely used in proton exchange membrane fuel cells (PEMFCs)29. In 2000, Kordali et al. reported the synthesis of ammonia from N2 and H2O based on a Ru/C cathode, Pt anode, 2 M KOH aqueous solution as the electrolyte using Nafion as a separation membrane (not electrolyte) and an ammonia formation rate of ~170 ng h−1 cm−2 (2.78 × 10−8 mol m−2 s−1) was achieved at 20°C30. Although ammonia was synthesised from N2 and H2O at room temperature using 2 M KOH solution as electrolyte, N2 cannot be replaced by air as CO2 in air may react with KOH to form K2CO3. It was reported that ammonia has been synthesised from N2 and H2 based on acidic H+-form Nafion membrane, with Ni-SDC (Sm-doped CeO2) as anode, SmBaCuMO5+δ as cathode with maximum formation rate of 4.1 × 10−9 mol cm−2 s−1 at 25°C31; however, the chemical compatibility of active metal Ni with the strongly acidic Nafion membrane is concerning. The reaction between active Ni and H+-form Nafion may form Ni2+-form Nafion and thus lose proton conduction therefore the reaction would not be sustainable32. In addition, to synthesise ammonia directly from air without the N2 separation stage would be a better choice.
It is well known that some higher plants can synthesise ammonia or its derivatives directly from air and water at room temperature33,34. The ammonia produced by plants is normally directly used as fertiliser by the plants. To the best of our knowledge, there is no report on artificial synthesis of ammonia direct from air and water. It has been a dream for researchers who can imitate this natural process to synthesise ammonia under similar conditions. In this report , for the first time, we demonstrated that ammonia can be synthesised directly from air (instead of N2) and H2O (instead of H2) under a mild condition (room temperature, one atmosphere) with supplied electricity which can be obtained from renewable resources such as solar, wind or marine.
Results
Proton conduction of mixed NH4+/H+ conducting Nafion 211 membrane
Ammonia is a base, the acidic Nafion membrane readily reacts with ammonia to form NH4+-form Nafion. In our experiments, for the first time, it was demonstrated that NH4+-form Nafion exhibits proton conduction through concentration cell measurements. A mixed NH4+/H+ conducting Nafion 211 membrane was used as solid electrolyte for electrochemical synthesis of ammonia. Conventional H+-form Nafion membrane will be converted to NH4+-form in the presence of ammonia. It is necessary to maintain the stability of the membrane electrolyte under the synthesis conditions35. It has been reported that the ionic conductivity of NH4+-form Nafion is very much dependent on the humidity and reached ~0.05 S/cm at 80°C with a relative humidity 100%35. The high ionic conductivity is believed due to NH4+ ions but proton conduction cannot be ruled out. It has been reported that some inorganic ammonium salts such as (NH4)3H(SeO4)2 exhibit proton conduction36,37. Therefore the NH4+-form Nafion may exhibit a certain level of proton conduction which can be used for continuous synthesis of ammonia.
The membrane electrode assembly (MEA) for synthesis of ammonia was fabricated by a process described in the experimental part. The H+-form Nafion 211 membrane was converted into NH4+-form Nafion through the reaction between 35 wt% ammonia aqueous solution and the H+-form membrane in the MEA. The MEA was then washed by de-ionised water for a week by pumping water through both sides of the cell until no ammonia can be detected at the outlets of the cell. A potential of 40 mV was applied to the MEA for 4 hours to activate the cell prior to concentration cell measurements. When wet H2 (ambient temperature humidification) was introduced to the anode, wet air (also ambient temperature humidification) was used at the cathode, an OCV of ~475 mV was obtained, indicating the membrane exhibit H+ or/and O2− conduction. When the air was replaced by 5%H2/Ar to form a hydrogen concentration cell, the OCV of the cell gradually decreased and stabilised at ~32.8 mV (Fig. 1A). The I–V curve of this hydrogen concentration cell is shown in Fig. 1B. A maximum current density of 7 mA cm−2 was observed indicating migration of protons through the membrane. The proton conduction of the thus treated membrane was thus demonstrated by a hydrogen concentration cell. The theoretical OCV of the wet H2/5%H2-Ar concentration cell is 38.47 mV estimated from the Nernst Equation. The proton transfer number is therefore ~85% assuming no leakage or crossover of gases. The other 15% ionic conductivity is possibly attributed to the NH4+ ions although NH4+ ions are proton carriers too. Therefore, the thus treated membrane is a mixed NH4+/H+ conductor and can be used for electrochemical synthesis of ammonia because it is chemically compatible with ammonia.

(A) The recorded potential change from a H2/air cell to a H2/5%H2-Ar concentration cell; (B) The I–V and power curves of the H2/5%H2-Ar concentration cell.
Thermodynamic evaluation on electrochemical synthesis of NH3 from H2/H2O and N2
In order to consider the potential for electrochemical synthesis of ammonia, a thermodynamic evaluation on synthesis of ammonia from H2 and N2 was carried- out38. In theory, the reaction is spontaneous at a temperature below ~175°C when the partial pressure of H2 and N2 is 1 bar (Fig. 2A). It is expected that ammonia would be produced as long the applied voltage is higher than the electrode over-potential. When water is used for electrochemical synthesis of ammonia, the positive standard Gibbs free energy change indicates the reaction is non-spontaneous and applied potential is required (Fig. 2A). The corresponding required voltage for electrochemical synthesis of NH3 from H2 or H2O is shown in Fig. 2B. At 25°C, a minimum voltage of 1.17 V is required for electrochemical synthesis of ammonia from liquid water and N2 at partial pressure of 1 bar.

(A) The Gibbs free energy change for electrochemical synthesis of ammonia from N2 and H2, N2 and H2O (gaseous or liquid) at pressure of 1 bar; (B) The minimum applied voltage required for electrochemical synthesis of ammonia from N2 and H2 at pressure of 1 bar (the negative voltage at a temperature below 200°C means spontaneously generated voltage), N2 and H2O (gaseous or liquid).
Synthesis of ammonia from H2 and N2
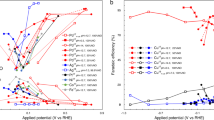
Ammonia was first synthesised from conventional precursors, H2 and N2 using the electrochemical cell. As shown in Fig. 3A, an initial current of 58 mA cm−2 was observed even at an applied voltage of 0.2 V. The current decreases after 3 minutes, possibly due to the reaction between produced ammonia and the membrane. Instead of decreasing, the current gradually increases when 0.4V is applied indicating the reaction has completed. A current density of 390 mA cm−2 has been achieved at room temperature when 1.2 V is applied (Fig. 3A). The ammonia formation rates at different applied voltage are shown in Fig. 3B. The highest ammonia formation rate of 3.1 × 10−5 mol m−2 s−1 was observed at 0.2 V which is about three orders of magnitude higher than the previously reported value reported by Kordali et al30 and is comparable to those reported by Liu et al31. As shown in Fig. 3C, the formed ammonia increased with time. The amount of generated ammonia was 1.13 × 10−5 mol after applying 0.2 V for 1 hour (Fig. 3C) which is higher than the estimated maximum amounts of ammonia that could be generated from the decomposition of the NH4-form of the membrane (9.14 × 10−6 mol) if all the current arose from NH4+ transport which is unlikely, or estimated dissolved ammonia (8.02 × 10−6 mol) (please see supplementary information). This result demonstrates that the generated ammonia is from the electrosynthesis process.

(A) Current density of a N2, Pt | Nafion 211 | Pt, H2 cell under different applied voltages. Cathode was supplied with N2, anode was supplied with H2. (B) The ammonia formation rate at N2 and H2 sides, total ammonia formation rate and Faraday efficiency. (C) The relationship between formed NH3 and time of a N2, Pt | Nafion 211 | Pt, H2 cell under different applied voltages. Cathode was supplied with N2, anode was supplied with H2.
The higher ammonia formation rate at lower voltage may be due to the lower hydrogen ion supply at the cathode which gives more time for formation of ammonia according to reaction (2). Between 0.6 and 1.2 V, the formed ammonia slightly increased at higher voltage (Fig. 3B). Although ammonia was mainly observed at the N2 side, a small amount of ammonia was also observed at the H2 side when the formation rate was relatively high. One of the possible reasons is that, ammonia is very soluble in water, at higher formation rate, some of the formed ammonia at N2 side may in situ dissolve in water, diffuse to the H2 side then brought out by the flowing H2. On the other hand, there could be some cross-over effects too which is common in electrochemical cells based on polymer electrolytes.
Synthesis of ammonia from H2 and air
Air contains 78% N2 therefore it would be better to synthesise ammonia directly from air without the separation process. When N2 at the cathode was replaced by air, a stable current density of 142 mA cm−2 was observed when the cell voltage was 0.2 V (Fig. 4A). At 1.0 V, the current density stabilised at 537 mA cm−2. This indicates that the membrane is fairly stable. Comparing to the H2/N2 cell, the current densities are higher for the H2/air cell, possibly due to the extra driving force from O2 in air. Interestingly, ammonia was also produced on the air side with a small amount at the H2 side when the formation rate is relatively high (Fig. 4B). At the same cell voltage, the ammonia formation rates are also higher than those for H2/N2 cell. The formed ammonia increased against time (Fig. 4C). This experiment indicates that ammonia can be synthesised directly from air without gas separation. This is consistent with thermodynamic evaluation concluding reaction (3) is spontaneous at a temperature below 175°C when the partial pressure of N2 is at 1 bar although it is slightly lower in air (Fig. 3A). Another parallel reaction at the cathode is formation of water between proton and O2 in air; however, if a selective catalyst for ammonia synthesis is used, the reaction can be kinetically in favour of ammonia formation. Comparing to conventional Haber-Bosch process, due to different catalysts used along with various synthesis conditions, the oxygen poisoning on ammonia synthesis catalysts is not an issue.

(A) Current density of an Air Pt | Nafion 211 | Pt, H2 cell under different applied voltages. Cathode was supplied with air, anode was supplied with H2. (B) The ammonia formation rate at air and H2 sides, total ammonia formation rate and Faraday efficiency. (C) The relationship between formed NH3 and time of an Air, Pt | Nafion 211 | Pt, H2 cell under different applied voltages. Cathode was supplied with air, anode was supplied with H2.
Synthesis of ammonia directly from H2O and air
Water is the most abundant source for hydrogen. It would be a better choice if we can directly synthesise ammonia from air and water bypassing the hydrogen production stage. Therefore H2 at the anode was replaced by water. At 1.2 V, the current density of the cell was 47 mA cm−2, lower than that for the H2/N2 cell, possibly due to the high electrode polarisation at the water side (Fig. 5A). The ammonia formation rates increase at higher cell voltage (Figs. 5B). The ammonia formation rate is slightly lower than that when H2 was fed at the anode. The highest ammonia formation rate was observed at low applied voltage when H2 was used at anode (Figs. 3B & 4B); however, when water was supplied at the anode, the ammonia formation rate increased against applied voltage (Fig. 5B). When a dc voltage was applied to the cell, hydrogen was pumped to the cathode through transfer of protons in the electrolyte membrane. At high applied voltage, the high hydrogen flow rate may limit the lifetime of hydrogen species at the electrode/electrolyte/gas interfaces therefore the ammonia formation rate is relatively lower (Fig. 5C). In conclusion, for the first time, this experiment clearly indicates that ammonia can be directly synthesised from air and water at room temperature and one atmosphere.

(A) Current density of an Air, Pt | Nafion 211 | Pt, H2O cell under different applied voltages. Cathode was supplied with air, anode was supplied with H2O. (B) The ammonia formation rate at air and H2O sides, total ammonia formation rate and Faraday efficiency. (C) The relationship between formed NH3 and time of an Air, Pt | Nafion 211 | Pt, H2O cell under different applied voltages. Cathode was supplied with air, anode was supplied with H2O.
Discussion
In most reports, H2 and N2 were commonly used as precursors for electrochemical synthesis of ammonia while H2 production and N2 separation are essential4. H2 production can be bypassed if H2O was used as a precursor; however, the reaction between H2O and N2 to form ammonia is thermodynamically non-spontaneous under normally pressure (Fig. 2B); however, this can be achieved through electrochemical process because the applied voltage provides extra driving force. Although there are a few reports on electrochemical synthesis of ammonia from N2 and H2O at 570°C15 or 300°C22,23, the formed ammonia tends to decompose to N2 and H2 because thermodynamically the decompositiontemperature of ammonia is around 175°C (Fig. 2A). Therefore, a synthesis temperature below 175°C is required in order to avoid decomposition of formed ammonia.
In order to demonstrate that the produced ammonia is from the electrochemical process, the maximum amount of dissolved ammonia has been estimated. It has been reported that the maximum H2O uptake of Nafion 117 membrane was 20.64 vol% at room temperature, investigated by small angle neutron scattering technology39. It is assumed that Nafion 211 membrane would exhibit similar behaviour. The maximum dissolved ammonia in the absorbed water in the used Nafion membrane was estimated to be 8.02 × 10−6 mol (please see supplement information) which is smaller than the generated ammonia in the first experiment, from H2 and N2 while applied at 0.2 V for 1 hour (1.13 × 10−5 mol, Fig. 3C). This value is also higher than the formed ammonia from possible decomposition of NH4+-form Nafion (9.14 × 10−6 mol). The total ammonia from decomposition of NH4+-form Nafion and dissolved NH3 in absorbed water is 1.72 × 10−5 mol which is significantly smaller than the total measured ammonia 7.15 × 10−5 mol (Figs. 3C, 4C and 5C). It should be noted that the NH4+-form Nafion membrane was washed by de-ionisedwater until no ammonia was detected in outlets of the cell. Considering the amounts of generated ammonia in experiments, it is clear that the collected ammonia cannot be from the dissolved ammonia from absorbed water in or the decomposition of ammonium Nafion membrane.
When H2 and N2 were used for electrochemical synthesis of ammonia, at the oxidation electrode, hydrogen loses electrons while protons are formed.

The formed protons will transfer though the H+/NH4+-form Nafion to the other side to react with N2 to electrochemically form NH3;

The overall reaction is:

However, the reaction between proton and N2 depends on both thermodynamics and kinetics. Considering the over-potential on both electrodes, if the ‘net’ potential difference between the two electrodes is above the value displayed in Fig. 2B, then thermodynamically reaction (3) should happen. However, a lot of reactions are under kinetic control particularly at low temperatures. The Faraday efficiency of reaction (3) is shown in Fig. 3B. It is about 2% when 0.2 V voltage was applied while decreased to less than 1% when higher voltage was applied. When air was used at the oxidation electrode or water at the reduction electrode, the Faraday efficiency for ammonia formation was both less than 1% (Figs. 4B and 5B) which means only a small portion of supplied electricity was converted into ammonia. The Faraday efficiency also increased when the current across the cell is relatively low indicating higher voltage may also facilitate reaction (2) (Fig. 5B) when water instead of hydrogen was flowed at the cathode. More efficient catalysts at the N2/air side are required. The protons at the reduction electrode not only react with N2 to form NH3, but can receive electrons to form H2 as well;

When 0.2 V is applied, only 2% applied electricity was converted to ammonia while the other 98% was converted to H2. The H2 flow rate at the reduction electrode is proportional to the current across the cell. When higher voltage was applied, the current across the cell also increased (Fig. 3A). Most of the protons were converted to H2 again because the dwelling time of protons on the Pt/C electrode will be shorter thus the overall Faraday efficiency for ammonia formation decreased (Fig. 3B).
When N2 at the reduction electrode was replaced by air, besides reaction (4), another important reaction is between protons and oxygen in the air;

This is also the cathode reaction for a H2/O2 fuel cell. This reaction indicates that, under certain conditions, a small amount of ammonia may be formed when air was used as oxidant in hydrogen fuel cells. It has been reported that ammonia can passivate the oxygen reduction reaction (5) at the cathode of a proton exchange membrane fuel cell40. On the other hand, the passivation of ammonia on Pt/C catalysts for reaction (5) may supress the formation of H2O, which may favour the competitive reaction between protons and N2 in air to form ammonia. From this point of view, if a suitable catalyst is identified to suppress the formation of H2O according to reaction (5) while in favour of reaction (3), air can be directly used as nitrogen sources for electrochemical synthesis of ammonia.
Production of hydrogen through electrolysis for electrochemical energy storage has been widely investigated. Water can be used for direct electrochemical synthesis of ammonia. Then the reaction at the oxidation electrode is:

The formed protons will transfer through the proton-conducting membrane, react with N2 in air to form ammonia while O2 is formed at the oxidation electrode.
The overall reaction is:

While ammonia is produced at the air side, O2 is also produced at the water side which can be used for other applications such as oxyfuel combustion.
It should be noted that this is just a starting point to directly synthesis ammonia from air and water at room temperature although theoretically Pt is not among the best catalysts for ammonia synthesis32. In the future, other low cost ammonia synthesis catalysts such as Co3Mo3N and Ni2Mo3N41 can be used to replace Pt for selective ammonia synthesis under mild conditions. The acidity of the H+/NH4+-form Nafion membrane would be much weaker than the H+-form Nafion allowing selection of a large range of catalysts for ammonia synthesis. This is a low temperature, low pressure process with flexibility in scale and location. This technology will break the link between ammonia industry and fossil fuels. Considering climate change and the depletion of fossil fuels used for synthesis of ammonia by conventional method, this is a renewable and sustainable chemical synthesis process for future.
Methods
Fabrication of electrochemical cell for ammonia synthesis
Nafion 211 membrane (DuPont®) was boiled in 3% H2O2 for 1 hour, rinsed by deionised water, boiled in de-ionised water for 2 hours then in 0.5 M H2SO4 for 1 hour. After rinsing with deionised water a few times, the membrane was stored in deionised water for cell fabrication.
Pt/C (E-Tek, 30 wt%) on SGL gas diffusion layer (GDL 10 BC) was used as both electrodes with a Pt loading of 1 mg cm−2. Some 5% Nafion suspension (Aldrich) and isopropanol were mixed with Pt/C catalysts for preparation of the catalytic layer. The membrane electrode assembly (MEA) with a working area of 1 cm2 was fabricated by hot pressing.
The MEA was put in electrochemical cell testing jig using graphite as bipolar plates. 35 wt% ammonia aqueous solution (Alfa Aesar) was pumped to both sides of the cell by a parasitic pump (Waterson Marlon 320) for one day to convert H+-form Nafion 211 membrane into NH4+-form. De-ionised water was then pumped to the cell for one week to clean up the residual ammonia and no ammonia can be detected from the outlets. A d.c. voltage of 40 mV was applied to the cell for 4 hours to activate the MEA and improve the electrode/electrolyte interfaces then air was flowed through both cathode and anode chambers overnight before ammonia synthesis experiments. All the presented experimental data were collected from the same MEA.
Ammonia synthesis and detection
H2 (or water) and N2 (or air) were passed through room temperature water first then filled into the chambers of the cell. The dc potential was applied by a Solartron 1470A electrochemical interface controlled by software Cell Test® for automatic data collection. The order for applied voltage was from low to high. The produced ammonia was collected by dilute H2SO4 (0.001 M). The concentration of  in the absorbed solution was analysed using Nessler's reagent (Aldrich). The produced ammonia was detected using an ammonia meter (Palintest 1000) and the rate of ammonia formation was calculated using the following equation.
in the absorbed solution was analysed using Nessler's reagent (Aldrich). The produced ammonia was detected using an ammonia meter (Palintest 1000) and the rate of ammonia formation was calculated using the following equation.

Where [NH4+] is the measured NH4+ ion concentration, V is the volume of the dilute H2SO4 for ammonia collection, t is the adsorption time and A is the effective area of the cell.
References
Lan, R., Irvine, J. T. S. & Tao, S. W. Ammonia and related chemicals as potential indirect hydrogen storage materials. Inter. J. Hydrogen Energy 37, 1482–1494 (2012).
Bastidas, D. M., Tao, S. W. & Irvine, J. T. S. A symmetrical solid oxide fuel cell demonstrating redox stable perovskite electrodes. J. Mater. Chem. 16, 1603–1605 (2006).
Pool, J., Lobkovsky, E. & Chirik, P. Hydrogenation and cleavage of dinitrogen to ammonia with a zirconium complex. Nature 427, 527–530 (2004).
Amar, I. A., Lan, R., Petit, C. T. G. & Tao, S. W. Solid-state electrochemical synthesis of ammonia: a review. J. Solid State Electrochem. 15, 1845–1860 (2011).
http://www.kbr.com/Newsroom/Publications/Articles/Carbon-Dioxide-Capture-and-Storage-in-the-Nitrogen-Syngas-Industries.pdf. (Access 28th August, 2012).
http://www.decc.gov.uk/assets/decc/11/stats/climate-change/4282-statistical-release-2010-uk-greenhouse-gas-emissi.pdf. (Access 28th August, 2012).
Waugh, K. C., Butler, D. & Hayden, B. E. The mechanism of the poisoning of ammonia-synthesis catalysts by oxygenates O2, CO and H2O - An in-situ method for active surface determination. Catal. Lett. 24, 197–210 (1994).
Jennings, J. Catalytic ammonia synthesis: fundamentals and practice. (Springer, 1991).
Chatt, J., Pearman, A. & Richards, R. The reduction of mono-coordinated molecular nitrogen to ammonia in a protic environment. Nature 253, 39–40 (1975).
Yandulov, D. & Schrock, R. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science 301, 76 (2003).
Avenier, P. et al. Dinitrogen dissociation on an isolated surface tantalum atom. Science 317, 1056 (2007).
Nishibayashi, Y. et al. Buckminsterfullerenes: A non-metal system for nitrogen fixation. Nature 428, 279–280 (2004).
Laplaza, C. & Cummins, C. Dinitrogen cleavage by a three-coordinate molybdenum (III) complex. Science 268, 861 (1995).
Himmel, H. & Reiher, M. Intrinsic dinitrogen activation at bare metal atoms. Angew. Chem. Inter. Edition 45, 6264–6288 (2006).
Schrock, R. Catalytic reduction of dinitrogen to ammonia by molybdenum: theory versus experiment. Angew. Chem. Inter. Edition 47, 5512–5522 (2008).
Pickett, C. & Talarmin, J. Electrosynthesis of ammonia. Nature 317, 652–653 (1985).
Furuya, N. & Yoshiba, H. Electroreduction of nitrogen to ammonia on gas-diffusion electrodes modified by Fe-phthalocyanine. J. Electroanalytical Chem. 263, 171–174 (1989).
Furuya, N. & Yoshiba, H. Electroreduction of nitrogen to ammonia on gas-diffusion electrodes modified by metal-phthalocyanines. J. Electroanalytical Chem. 272, 263–266 (1989).
Norby, T. Solid-state protonic conductors: principles, properties, progress and prospects. Solid State Ionics 125, 1–11 (1999).
Kreuer, K. D. Proton conductivity: Materials and applications. Chem. Mater. 8, 610–641 (1996).
Iwahara, H., Esaka, T., Uchida, H. & Maeda, N. Proton conduction in sintered oxides and its application to steam electrolysis for hydrogen production. Solid State Ionics 3–4, 359–363 (1981).
Tao, S. W. & Irvine, J. T. S. A stable, easily sintered proton-conducting oxide electrolyte for moderate-temperature fuel cells and electrolyzers. Adv. Mater. 18, 1581–1584 (2006).
Marnellos, G. & Stoukides, M. Ammonia synthesis at atmospheric pressure. Science 282, 98–100 (1998).
Skodra, A. & Stoukides, M. Electrocatalytic synthesis of ammonia from steam and nitrogen at atmospheric pressure. Solid State Ionics 180, 1332–1336 (2009).
Murakami, T., Nohira, T., Goto, T., Ogata, Y. H. & Ito, Y. Electrolytic ammonia synthesis from water and nitrogen gas in molten salt under atmospheric pressure. Electrochim. Acta 50, 5423–5426 (2005).
Murakami, T. et al. Electrolytic synthesis of ammonia from water and nitrogen under atmospheric pressure using a boron-doped diamond electrode as a nonconsumable anode. Electrochem. & Solid State Lett. 10, E4–E6 (2007).
Amar, I. A., Lan, R., Petit, C. T. G., Arrighi, V. & Tao, S. W. Electrochemical synthesis of ammonia based on a carbonate-oxide composite electrolyte. Solid State Ionics 182, 133–138 (2011).
Perman, E. & Atkinson, G. The Decomposition of Ammonia by Heat. Proceedings of the Royal Society of London 74, 110–117 (1904).
Kundu, P. P. & Pal, A. Cation exchange polymeric membranes for fuel cells. Reviews in Chemical Engineering 22, 125–153 (2006).
Kordali, V., Kyriacou, G. & Lambrou, C. Electrochemical synthesis of ammonia at atmospheric pressure and low temperature in a solid polymer electrolyte cell. Chem. Comm. 1673–1674 (2000).
Zhang, Z. F., Zhong, Z. P. & Liu, R. Q. Cathode catalysis performance of SmBaCuMO5+δ (M = Fe, Co, Ni) in ammonia synthesis. J. Rare Earths 28, 556–559 (2010).
Skulason, E. et al. A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction. Phys. Chem. Chem. Phys. 14, 1235–1245 (2012).
Burns, R. & Hardy, R. Nitrogen fixation in bacteria and higher plants. Molecular Biology, Biochemistry and Biophysics 21, 1–189 (1975).
Benemann, J. & Valentine, R. The pathways of nitrogen fixation. Advances in Microbial Physiology 8, 59–104 (1972).
Hongsirikarn, K., Goodwin, J. Jr., Greenway, S. & Creager, S. Influence of ammonia on the conductivity of Nafion membranes. J. Power Sources 195, 30–38 (2010).
Moskvich, Y. N., Polyakov, A. M. & Sukhovsky, A. A. The NMR-study of ionic motions and conductivity mechanisms in protonic conductors MHSeO4 and M3H(AO4)2 . Ferroelectrics 81, 1161–1164 (1988).
Pawowski, A., Pawlaczyk, C. & Hilczer, B. Electric conductivity in crystal group Me3H(SeO4)2 (Me: NH4+, Rb+, Cs+). Solid State Ionics 44, 17–19 (1990).
Binnewies, M. & Milke, E. Thermochemical data of elements and compounds. (Wiley Online Library, 1999).
Young, S. K., Trevino, S. & Beck Tan, N. C. Small-angle neutron scattering investigation of structural changes in nafion membranes induced by swelling with various solvents. Journal of Polymer Science Part B: Polymer Physics 40, 387–400 (2002).
Halseid, R., Vie, P. J. S. & Tunold, R. Effect of ammonia on the performance of polymer electrolyte membrane fuel cells. J. Power Sources 154, 343–350 (2006).
Hargreaves, J. S. J. & McKay, D. A comparison of the reactivity of lattice nitrogen in Co3Mo3N and Ni2Mo3N catalysts. J. Mol. Catal. A-Chem. 305, 125–129 (2009).
Acknowledgements
This work was funded by EPSRC SuperGen ‘Delivery of Sustainable Hydrogen’ project (EP/G01244X/1).
Author information
Authors and Affiliations
Contributions
S.W.T. and R.L. drafted and J.T.S.I. revised the manuscript. S.W.T. and J.T.S.I. conceptualized the study. R.L. and S.W.T. performed synthesis, characterization and analysis.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Lan, R., Irvine, J. & Tao, S. Synthesis of ammonia directly from air and water at ambient temperature and pressure. Sci Rep 3, 1145 (2013). https://doi.org/10.1038/srep01145
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01145
This article is cited by
-
Efficient catalyst-free N2 fixation by water radical cations under ambient conditions
Nature Communications (2024)
-
V2O4 Nanowires/rGO Hybrid for Electrocatalytic Nitrogen Fixation
Catalysis Letters (2024)
-
Converting N2 molecules into NH3 with TiO2/Fe3O4 composite covered with a thin water layer under ambient condition
Scientific Reports (2023)
-
Recent advances and challenges of nitrogen/nitrate electro catalytic reduction to ammonia synthesis
Frontiers in Energy (2023)
-
Promotion of biological nitrogen fixation activity of an anaerobic consortium using humin as an extracellular electron mediator
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
