
Abstract
We report a new development of orthogonal scanning automated microscopy (OSAM) incorporating time-gated detection to locate rare-event organisms regardless of autofluorescent background. The necessity of using long-lifetime (hundreds of microseconds) luminescent biolabels for time-gated detection implies long integration (dwell) time, resulting in slow scan speed. However, here we achieve high scan speed using a new 2-step orthogonal scanning strategy to realise on-the-fly time-gated detection and precise location of 1-μm lanthanide-doped microspheres with signal-to-background ratio of 8.9. This enables analysis of a 15 mm × 15 mm slide area in only 3.3 minutes. We demonstrate that detection of only a few hundred photoelectrons within 100 μs is sufficient to distinguish a target event in a prototype system using ultraviolet LED excitation. Cytometric analysis of lanthanide labelled Giardia cysts achieved a signal-to-background ratio of two orders of magnitude. Results suggest that time-gated OSAM represents a new opportunity for high-throughput background-free biosensing applications.
Similar content being viewed by others


Introduction
Thanks to the extensive developments in fluorescent bioprobes and optical approaches to improve sensitivity, resolution and speed of analysis1,2, fluorescence imaging and single cell counting has become a fundamental research tool in the analytical fields of biotechnology and life sciences, from molecular biology discovery to clinical diagnostics3. In broad areas of microbiology, including infectious-disease diagnosis and anti-bioterrorism, fluorescence detection for quantification of target cells at low cost and high speed is increasingly popular. However this poses significant challenges for simple spectral-discrimination of probe fluorescence in anything other than highly purified samples, since intrinsic autofluorescence from background organisms and detritus obscures the visibility of fluorescence labelling due to spectral overlap4,5. This is especially true for detection of rare-event microorganisms in a matrix of non-target microorganisms which occur at frequencies typically less than one in 105 background cells, or as low as 1 in 109 background cells. For example, the number of residual circulating tumour cells (CTCs) in peripheral blood is a valuable indicator for progressive diagnosis of metastatic cancer patients. This requires a detection level of one residual cancer cell per 107 of bone marrow or peripheral blood stem cells2,6. Foetal cells present in maternal blood during pregnancy are an ideal source of genetic material for non-invasive prenatal diagnosis; however, the target foetal nucleated red blood cells (NRBCs) must be detected against the maternal cells at extremely low frequencies of 1 in 107 to 109 7,8,9. In water safety inspection, due to the very small number of organisms capable of infection, the methods of analysis must be sufficiently sensitive to detect a single microorganism (e.g. Cryptosporidium parvum and Giardia lamblia) in as many as 10 litres of water containing billions of non-target microorganisms and particles10.
The time-gated luminescence (TGL) technique uses the temporal domain to discriminate long-lifetime luminescence-labelled targets against short-lifetime autofluorescence. Detection of the luminescence signal is delayed typically by a few microseconds following termination of pulsed excitation, so that only the long-lived luminescence-labelled targets remain visible to the detector. Such background-free detection offers significant advantages for high-speed inspection of rare-event microorganisms11. In recent times a broad spectrum of long-lifetime luminescence bioprobes has become available in the forms of metal chelate complexes and nanoparticles, e.g. lanthanide, phosphorescence and charge transfer materials12,13,14,15,16. Use of temporal discrimination has been widely reported for time-resolved molecular assays and time-gated luminescence (TGL) bioimaging17,18,19,20,21,22,23. In particular, we recently demonstrated direct visual inspection of target microorganisms in background-free condition using very low-cost instrumentation24,25 which is sensitive enough for single nanoparticle imaging26.
Despite these advances, practical application of TGL techniques in many situations suffers from the low signal strength that is typical of the long-lifetime fluorophores used: recently-engineered luminescence probes used for time-gated luminescence imaging have typically more than 4 orders of magnitudes longer lifetimes than traditional dyes (lifetimes > 100 µs compared with ~ 10 ns) and consequently have much weaker emission than conventional fluorescent probes21,24. Thus time-gated detection usually requires long signal integration times to achieve sufficiently high signal levels to produce quality TGL microscopy images. Indeed, reported signal integration times range from 333 milliseconds for a top-of-the-range intensified-CCD camera21 up to 30 seconds for a standard (cooled) CCD camera27. Since significant time is also needed to inspect the whole area of a standard sample slide and analyse the 2-D images, the question arises: can time-gated luminescence bioimaging process a large number of samples (for example containing rare-event target organisms) with necessary accuracy/certainty on acceptable time scales?
We have recently reported a step-scanning technique for automated rare-event TGL inspection of the full sample area of a microscope slide28. Rather than recording every adjacent wide-field image across the slide, a step scanning and time-gated detection system was employed to locate a few positive “elements of interest” (containing potential target organisms) in an initial rapid scan of the sample, before time-gated luminescence imaging was applied only to those small areas of interest. Though this step-by-step scanning approach can minimise data storage and image analysis requirements, it still requires 70 ms dwell time at every analytical element and 47 minutes to perform a complete inspection of the slide sample, since it still involves “stop and watch” to acquire sufficient integrated signal from lanthanide luminescence of individual target cells.
Here we report a major advance in scanning TGL microscopy which delivers precise location of luminescent targets with high signal-to-background ratio and significantly improved coefficient of variation (CV), at sample processing speeds an order of magnitude higher than previously, using a novel on-the-fly orthogonal TGL scanning technique. The key performance parameters and detection limits are examined in detail and the technique demonstrated for both 5-micron and 1-micron diameter luminescent microspheres and for BHHCT-Eu chelate labelled Giardia lamblia cysts.
Results
TGL detection during on-the-fly scanning
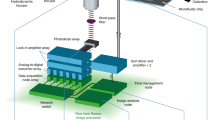
Figure 1 illustrates the spatial-temporal model of the TGL detection during on-the-fly scanning, demonstrated through an epi-fluorescent optical setup. The excitation source (UV LED) and single-element detector (gateable PMT) are switched on and off in a continuous TGL cycle at repetition rates (cycle rates) in the kilohertz (103 cycles/sec) range. Each TGL cycle, of duration TC, comprises an excitation pulse of duration TE (typically 10–100 μs) at which time the detector is turned off (gated). The detector is then turned on a short delay (TD ~ 10 µs) after cessation of the excitation pulse, to integrate the TGL signal for the remainder of the TGL cycle (duration TW) as an “area” value. Short-lifetime autofluorescence from the sample and any scattered light from the excitation source decay promptly during the delay period, thus their area values are always negligible. However, if a target particle or microorganism tagged with long-lifetime fluorophores moves into the detection field of view (FOV) of the microscope objective, its luminescence signal accumulates over the detection threshold so that the TGL decay pulse is recorded. The detector is again turned off just before the excitation is switched on for the next TGL cycle.

A schematic drawing illustrates the spatial-temporal relationship during on-the-fly TGL scanning.
A sample slide containing long-lifetime targets is scanned by a 2-D motorised stage moving over an epi-fluorescent optical setup (lower left). The transit of a target particle across the detection field of view (FOV) (upper left: overview under the FOV; upper right: displacement along scan axis x vs. time t) is correlated with the TGL detection sequences (lower right). The TGL cycles (duration TC) consist of gating periods (duration TG) and detection periods (duration TW). In each gating period, the FOV is excited for duration TE, with a delay time TD before the detector is switched on. P1 and P2 are the positions where a target particle enters and leaves the FOV and P is the position where the target gets closest to the centre of FOV during the scan. As the TGL cycle rate increased, x(P1) and x(P2) can be asymptotically determined from t1 and t2. Thus, the coordinate of the target along the scan axis is x(P) = (x(P1)+x(P2))/2.
t(P1) and t(P2) denote the times when the target enters or leaves the FOV. Thus, t(P2) − t(P1) represents the time interval (transit time) for which a target is potentially visible, this being set by the scan rate and size of the FOV. Note: t(P1) and t(P2) in general do not coincide with the beginning or end of a TGL cycle and therefore t(P2) − t(P1) usually does not coincide with whole TGL periods. However the signal contributed in the partial cycles at the very beginning or end of the transit is small and when TGL cycle rates are high, so that the cycle duration TC is very small compared to the whole transit time (TC ≪ t(P2) − t(P1)), the contributions from partial cycles are extremely small and can effectively be ignored. Therefore, t(P1) and t(P2) can be approximately represented by t1 and t2, which are the beginnings of the luminescence signal in the first and last TGL cycles captured from the target, respectively. Figure 2 depicts the real-time TGL signal recorded in our prototype demonstration at different TGL repetition rates (1 kHz for Figure 2a, 5 kHz for Figure 2b). As the cycle period is shortened, the recorded luminescence from a target particle takes the form of a pseudo-continuous signal, consisting of individual TGL decay pulses, which commences when the particle enters the FOV, rises to a peak when closest to the centre of the FOV, then diminishes to zero when the particle exits the FOV, as shown in Figure 2b.

Real-time TGL signals acquired during on-the-fly scanning.
(a) depicts the temporal waveform of time-gated luminescence (TGL) signal with a relatively-long detection time window, when a target passes across the FOV. One cycle is enlarged to illustrate the TGL detection scheme: An excitation pulse (blue) illuminates the interrogation field while a gating signal (grey) turns the detector off, leaving a delay period for residual excitation and autofluorescence to diminish. The long-lifetime TGL signal is recorded during the detection time window (experimental data plotted in red and fitted curve plotted in green). The profile (pink) shows here average intensity of luminescence decay varies as the target transits the FOV. (b) shows a real signal when the TGL cycle duration is 0.2 ms, along with its profile of average intensity.
In our prototype system, we chose a motorised stage (H101A, Prior Scientific, maximum translation velocity 24 mm/s) to move sample slides across the stationary objective. When a target profile is detected in the FOV, t1 and t2 relating to the first and last recorded TGL cycle can be used to identify the coordinates of entrance position P1 and exit position P2 on the continuous-scan axis and thus the position P of the target on the orthogonal axis where the particle is closest to the centre of the FOV. To take account of the effects of “accelerating-decelerating” of the motorised stage along the scan axis, we applied a kinematic calibration (see “Methods - Kinematic calibration of the motorised stage”) to precisely calculate the coordinate values of targets from detector pulse trains.
Orthogonal scanning strategy
Figure 3 illustrates the new 2-step orthogonal scanning strategy we have implemented for rapid detection and precise localisation of target particles or microorganisms. Note that while this approach is applicable to general sample scanning strategies (for example normal fluorescence detection on a solid-phase slide), its application in TGL mode is especially advantageous, since any signal from autofluorescence or scattered excitation light is eliminated, so that only rare-event targets (e.g. labelled microorganisms) in the wide field are visible to the detection system.

The schematics show 2-step orthogonal scanning strategy to discover and localise targets-of-interest.
(a) The sample is first examined in a serpentine pattern, of which the continuous movement is along X-axis, to obtain precise X coordinates as well as rough Y coordinates for every targets. (b) When a target particle moves across the FOV, on-the-fly TGL scanning delivers the X coordinates of P1 and P2, so that the precise X coordinate of the particle (at P) is known. (c) Then, the targets are scanned sequentially at precise X coordinates along Y-axis, to obtain precise Y coordinates. (d) The precise X coordinate obtained in the first step scanning ensures that each target translates in the exact middle of the FOV. Since on-the-fly TGL scanning provides the Y coordinates of P3 and P4, the precise Y coordinate of the particle (at P') is determined.
Referring to Figure 3a, the sample slide is first scanned across its whole area using a forth-and-back raster, or serpentine, pattern, with continuous motion along the X-axis and stepwise translation along the Y-axis. The interval between Y-axis scan lines is set by the size of the wide FOV of the microscope objective to ensure sufficient scan overlap. The target particle or organism is essentially identified when moving across the FOV along one of the scan lines, with its precise X-coordinate value calculated from the on-the-fly TGL signal. This 1st step also records the location coordinates along the Y-axis limited by the sequential index of the adjacent lines. Since target particles or organisms do not often appear on the exact centre of the wide field (see Figure 3b), a second orthogonal scan is then undertaken with continuous motion along Y-axis, but where the X-axis position is selected according to the X-coordinates determined for detection events identified in the initial X-axis scanning (see Figure 3c). Thus each Y-axis continuous scan line passes directly across each target (which then transits across the centre of the wide FOV, see Figure 3d), so that the precise Y-coordinate of the target can be determined from the on-the-fly TGL signal in that case. Since Y-axis scans are limited only to events detected in the X-axis scan, the total inspection time is substantially decreased.
It follows that the peak area (integration of the decay pulse in single TGL cycle) for the signal from each target is achieved when the target is exactly in the centre of the FOV during the Y-axis scan. This peak area, denoted as “pulse area” (µV·s), represents the maximum integrated luminescence (number of photons) emitted by the target during one TGL cycle. Note that although it would be possible to integrate the signal over all TGL cycles in the transit of the target across the FOV, this does not enhance the signal-to-background ratio, since the latter is also integrated over the same time.
Analytical speed
The processing time for each sample generally varies with the size of the scanning area, the size of the FOV of the microscope objective and the overall number of targets. The number of targets determines the number of Y-axis scan lines needed in the second stage of orthogonal scanning, though it does not affect the time taken for the first stage of serpentine-pattern scanning. The number of X-axis scans is determined by the interval between adjacent scan lines, this interval being determined by the diameter of the FOV of the objective and the overlap needed to avoid missing any targets. If the overlap is too small, the chance of overlooking a target is high, since some targets may pass by near the edge of the FOV, while excessive overlap incurs a substantial penalty in reduced scanning speed. Choice of X-axis line scan interval of 0.5 to 0.75 times the diameter of the FOV is a reasonable compromise. We selected 0.71 ( = 1/√2) representing the inscribed square of the circular FOV (the diameter of the FOV was 105 µm, while the interval was 75 µm). For this configuration, the processing time to scan each 15 mm x 15 mm sample slide (200 strips of 15 mm × 75 µm) was typically around 3.3 minutes (1 second per strip). We believe that sample analysis time can be further reduced to ~ 1 minute for the current system using new-generation motorised stages with faster translation velocity (e.g. MLS203-1, Thorlabs, maximal translation velocity 250 mm/s), without sacrifice to sensitivity or minimum detection levels.
Detection limit
The detection limit is defined as the minimum required pulse area to distinguish target particles or cells from background level. The TGL technique is highly effective in eliminating optical noise from stray light scattered from the microscope optics or the sample, as well as autofluorescence from the sample. Electronic noise is substantially removed from the detected luminescence signal since it varies randomly and rapidly in time and is averaged out in the integration of pulse areas. The major sources of background are expected to come from long-lived visible luminescence from the UV-LED29 and the long-lived visible luminescence from metal impurities in the glass microscope slide and cover slip21. The former can be substantially removed using spectral filtering on the source29.
The effect of luminescence from the microscope slide and cover slip was investigated in the present experiments with results illustrated in Figure 4a. Clean sets of glass (slides Part No. 7101, cover slips 18 mm × 18 mm, Sail Brand) as well as quartz substrates (slides Part No. FQM-7521, cover slips Part No. CFQ-2550, UQG Optics) were scanned as control samples with low signal-detection thresholds (8 and 2 μV·s for glass and quartz slides, respectively). The number of false-positive artefacts rose rapidly as the threshold was reduced. Here it is seen that quartz substrates generated a maximum pulse area only 1.97 μV·s (equivalent to 123 photoelectrons per TGL cycle, see Supplementary Discussion S1 online), 4 times lower than standard microscopic slide glass (8.03 μV·s, equivalent to 502 photoelectrons per TGL cycle). This effectively set the minimum detection level for our system. Since the only difference was in the substrate materials, the additional noise of 379 photoelectrons from the glass substrates was attributed to rare-earth impurities. The remaining 123 photoelectrons, which may still arise from the visible luminescence from UV LED, effectively limited the sensitivity of the prototype scanning system.

Determining the sensitivity of the prototype OSAM.
(a) draws the number of artefacts on a clean glass slide and clean quartz slide when varying the recording threshold of pulse area, indicating the detection limit on each substrate. (b) is a histogram of the luminescence intensity from a total number of 854 1-μm Eu-containing microspheres prepared on seven quartz slides, with the area threshold set to 3.0 μV·s. (c) shows the mapping result of a quartz slide carrying exact 36 1-μm Eu microspheres, which were selectively arranged by flow cytometer sorting to form a “MQ” pattern (CCD camera exposure time 150 ms).
Utilising quartz slides and cover slips and setting a threshold at 3.0 μV·s, we achieved 100% detection rate of 1-μm europium microspheres (FluoSpheres® F20882, Molecular Probes, Invitrogen) using a prototype orthogonal scanning automated microscopy (OSAM). Shown in Figure 4b, the mean pulse area and standard deviation were 17.7 μV·s and 5.4 μV·s, leading to 30.5% coefficient of variation (CV). The intensity histogram peak has a signal-to-background ratio (SBR) of 8.9, only requiring around 1,100 photoelectrons for unambiguous detection of the microspheres in our demonstration. The photoelectron number was used to estimate the number of photons emitted by europium ions from a 1-μm microsphere as 4.36×104 per TGL cycle. More details can be found in Supplementary Discussion S1 online.
The excitation power density used in the present experiments was measured as 2.7 W/cm2, while previously reported values using UV LEDs typically ranged from 0.6 ~ 2.5 W/cm2 21,24. However, because the on-the-fly TGL scanning requires a relatively high cycle rate for precise localisation of the targets, the duration of LED excitation pulse was set to only 80 μs, corresponding to a low excitation efficiency of about 3%. It follows that substantial scope remains to improve the SBR once higher power LEDs become available in the future.
To further demonstrate the capabilities of our prototype OSAM in eliminating both “false positive” and “false negative” errors, we prepared a unique sample for analysis using single-particle flow cytometer sorting. Figure 4c shows a recovered 36-dot “MQ” pattern with each dot representing an individual 1-μm microsphere. Some 10 slide samples were prepared with nominally 36 microspheres on each and examined using the prototype system (data shown in Supplementary Table S1 online). Between 34 and 38 microspheres were identified on each of the 10 predesigned “MQ” patterns. For samples with < 36 hits, each missing spot area was examined with great care, yet no undetected microspheres were found in the surrounding area. For samples with > 36 hits, the instrument returned to the positions of any extra hits, all of which were verified to be the target microspheres. Since all the detected microspheres were at least 3 times brighter than threshold (see Supplementary Table S1 online), these absences were attributed to inaccurate flow cytometer sorting.
Comparison between continuous scanning and step scanning
Since the continuous orthogonal scanning technique positions the target so that it transits through the centre of the FOV, the maximum integrated signal in a single TGL cycle (when the target is exactly in the centre of the FOV) is a true representation of the luminescence intensity emitted by the target. In contrast, in step-by-step scanning mode target locations in the FOV are random and integrated TGL signals are not a good representation of the true luminescence intensities of a target particle (see Supplementary Discussion S2 online). The continuous scanning mode therefore enhances the quantitative potential of TGL scanning microscopy, so that the mean and the CV of statistically measured luminescence intensities truly represent the average and the variation between individual target events.
Europium-doped 5-μm diameter FireRedTM microspheres (Newport Instruments) were used as calibration target particles to compare the performance between the prototype OSAM and the previous step-scanning setup. 14 sample slides were tested by step scanning first, which showed an average intensity of 129.5 μV·s and CV of 26.7% (Figure 5a), consistent with the previously reported result (CV = 27%)28. Step-scanning for each 15 mm × 15 mm sample slide took 47 minutes to process. The same 14 slides were then evaluated by the OSAM. In this case, recording maximum TGL signal (pulse area) by scanning 5-μm microspheres continuously across the centre of the FOV resulted in average intensity of 197.5 μV·s for the target population (Figure 5b), indicating a 1.5-fold enhancement in signal strength. The measured CV was 8.4% (compared to the intrinsic CV of 7.0%11,30), superior by a factor of at least 3 to the best obtained using step scanning. The processing time of the OSAM to scan each slide was about 3.3 minutes. This major acceleration (x 15) in analytical speed was attributed to more effective exploitation of the maximal translation velocity during continuous scans. The coordinates of each target determined by the successive orthogonal scans were used to retrieve the location of each microsphere for visual confirmation (Figure 5c).

Comparative TGL measurement of 5-μm europium microspheres and BHHCT-Eu chelate labelled Giardia cysts in two scanning modes.
(a) and (b) summarises the histogram results of the TGL intensity obtained from 5-μm europium microspheres by step scanning and continuous scanning, respectively. (d) and (e) sums up from seven slide samples in forms of histogram the distribution of luminescence intensity from a total number of 920 labelled Giardia cysts, analysed by step scanning and continuous scanning, respectively. (c) and (f) represent the imaging confirmation of the discovered microsphere and Giardia cyst after their locations were retrieved (CCD camera exposure time of 150 ms for luminescence imaging, 8 ms for bright-field imaging).The microspheres and Giardia cysts tested here were spiked on normal glass slides. The great contrast between the events and the threshold in all histograms indicates that the TGL scanning system is effectively free of false-negatives.
The capability of OSAM was also evaluated using a biological sample of immunoluminescence-labelled Giardia lamblia (a protozoan parasite infecting humans). Figure 5d and 5e depict TGL measurement of a total number of 920 Giardia cysts from the same 7 sample slides, by step scanning and continuous scanning, respectively. Although both approaches achieved 100% accurate detection, the step scanning measured an average pulse area of 123.5 μV·s, while that obtained by continuous scanning reached 192.0 μV·s (equivalent to 4.87×105 photons emitted per TGL cycle by europium complex per labelled cysts, see Supplementary Discussion S1) and weakest pulse area of 96.3 μV·s, 48 times higher than the minimal detection limit for quartz substrates. The distinctive signal-to-background ratio indicated unambiguous detection of Giardia cysts, free of false positive and negative errors. The CV result for continuous scanning (23.2%) was markedly superior to that for step scanning (38.4%), indicating greater accuracy of intensity quantification. Thanks to the precise localisation of target microorganisms, each Giardia event could be retrieved for bioimaging as shown in Figure 5f. The mapping and imaging results of a typical slide sample bearing 18 labelled Giardia cysts are given in Supplementary Fig. S1 and S2 online.
Discussion
We have successfully demonstrated on-the-fly TGL detection in a novel OSAM using UV LED excitation, enabling greatly reduced sample analysis time (3.3 minutes for a 15 mm × 15 mm sample area). Our approach removes a key constraint of TGL microscopy based on lanthanide probes whereby unacceptably long signal accumulation times have previously been required to produce the necessary detection levels and high signal-to-background ratio. We show here that the extremely low background levels achieved in OSAM allows TGL signals as weak as 123 photoelectrons per 100 μs to be distinguished from background with high contrast and precise target location. The intensity CV obtained for detected 5-μm microspheres was as low as 8.4%, close to the intrinsic CV of 7.0%. The reduced sample scan time coupled with enhanced sensitivity and locational accuracy indicates that OSAM represents a very practical approach to high speed analysis for biological cell samples. Significant scope remains to further lower the detection limit using UV laser excitation, improve the CV using linear encoders and autofocus optics and increase the speed through faster scanning mechanics.
These results also suggest that temporal domain multiplexing can be employed to expand spectral-domain multiplexing, which is currently limited to only a few (3–4) colours due to spectral overlapping of available (nanosecond-lifetime) fluorescent dyes. To achieve high-speed fluorescence analysis, it may now be possible to add at least four more practical colours in the time-gated domain, europium at 610–630 nm, terbium at 470–490 nm and 530–550 nm22, ruthenium at 550–700 nm31 and platinum at 660–740 nm32, to create up to 8 spectral channels in the visible. It is our belief that OSAM can take on a central role in a broad range of analytical and biosensing applications such as environmental sensing, pharmaceutical development and clinical diagnostics.
Methods
Optical configuration
The prototype OSAM has a similar optical and mechanical layout to that reported previously for step-scanning, but differs substantially in time-gating and detection strategy and technology, as well as scanning strategy. A motorised stage (H101A, Prior Scientific, maximal translation velocity vm = 24 mm/s) was implemented to mobilise the loaded slide samples across a stationary epi-fluorescent detection FOV. The excitation source was an ultraviolet light-emitting diode (UV-LED) with peak wavelength at 365 nm (NCCU033A, Nichia, 250 mW at 500 mA continuous injection current). The luminescence collected by the objective (NT38-340, Edmund Optics, 60×, NA = 0.75) was detected by a time-gated photomultiplier tube (PMT) (H10304-20-NF, Hamamatsu, cathode radiant responsivity R = 75 mA/W at λ = 612 nm, electron amplification gain GE = 106). Other optical elements included: a dichroic filter (400DCLP, Chroma) for the epi-fluorescent illumination; a fused silica condenser (f = 30 mm, d = 25 mm) to collimate the excitation beam; an UV-band-pass filter (UG5, SCHOTT) to purify the excitation; a visible-band-pass filter (9514-B, New Focus, 30-nm FWHM band at 624 nm) to purify the emission; and a convex lens (f = 40 mm, d = 25 mm) to focus the emission onto the PMT's photocathode window. In addition, a 45° mirror was inserted into the optical path to project the image onto a CCD camera (DS-Vi1, Nikon, 2 mega pixels) for image confirmation following the rapid sample scanning.
Electronic configuration
The purpose-built driving current supply of the UV-LED was synchronised with the bias voltage supply of the PMT via two analogue output channels of a PC-based multifunction data acquisition card (PCI-6251, National Instruments) to perform the TGL detection. The output current signal from the gated PMT was converted into a voltage signal via a low noise preamplifier (DLPCA-200, FEMTO, Germany; transimpedance gain GT = 105 Volts/Amp). The amplified signal and the gating TTL train were further digitised at sampling rate of 500 k samples/second/channel into two analogue input channels of the same data-acquisition card. The motorised stage controller (H129, Prior Scientific) was also interfaced with the same PC, to synchronise the scanning and the data collection.
TGL detection
For TGL configurations in all experiments, TC was set to 200 μs, yielding a maximum stage translation distance ( = vmTC) of 4.8 μm per TGL cycle, which was also the potential variation when locating a target. The duty ratio of TGL cycle was fixed at 50%, generating 100 μs for both the gating period (TG) and the signal collection window (TW). The excitation source was switched on for TE = 80 μs during the gating period with the delay time TD set to 5 μs. The TGL signals in each cycle were integrated after sampling to calculate “area” values. Only TGL cycles with area values exceeding a prearranged threshold were recorded, to thereby minimising background noise.
Preparation of microsphere samples
5-μm diameter FireRedTM microspheres (Newport Instruments) and 1-μm diameter FluoSpheres® microspheres (F20882, Molecular Probes) were used to prepare sample slides. The luminescence lifetimes of the spheres were 340 μs and 290 μs, respectively, when suspended in water and measured in bulk solution cuvette by a purpose-built spectrometer30. In each case, the original suspension was diluted with deionised water and subsequently mixed 1:1 with 2.5% polyvinyl alcohol (PVA). Every 10 μl sample from the mixture was spin-coated on a cover slip for 60 seconds at 800 rpm, which was then mounted on a standard microscopic slide (glass or quartz components). Each slide sample contained approximately 100 ~ 150 microspheres.
Preparation of patterned samples by flow sorting
Single 1-μm FluoSpheres microspheres sorted using a flow cytometer (FACSAriaTM, BD Biosciences, forward scattering and side scattering gated) were deposited on a sample slide in a specific pattern, notably 36 single microspheres arranged in individual positions so as to describe a predesigned “MQ” (Macquarie University) pattern. The positional sorting concept is illustrated in the Supplementary Video S1.
Preparation of labelled Giardia samples
Suspended Giardia cysts (6 ~ 9 μm in diameter, 105 in 18 μl, BTF-bioMérieux) were labelled with anti-Giardia antibodies (G203, cyst-wall specific, BTF-bioMérieux) conjugated to luminescent europium chelate BHHCT-Eu3+ according to the existing labelling protocol28. After centrifugal washing to remove excess reagent, the suspension containing labelled cysts was mixed 1:1 with 2.5% PVA solution followed by spin coating to prepare slides for analysis.
Kinematic calibration of the motorised stage
A sample slide of 5-μm microspheres was scanned with fixed-distance translation, to record the temporal sequences of TGL signal during each line scan. The spatial positions of the corresponding microspheres were precisely identified through fine adjustment based on the locations found by step scan. The data sets of displacement vs. time were fitted into 7th degree polynomial functions (R2 > 0.9999) for both directions along continuous scanning axis (see Supplementary Fig. S3 online), to calibrate the movement of the motorised stage at different speed from acceleration to deceleration.
References
Pepperkok, R. & Ellenberg, J. High-throughput fluorescence microscopy for systems biology. Nat. Rev. Mol. Cell Biol. 7, 690–696 (2006).
Hsieh, H. B. et al. High speed detection of circulating tumor cells. Biosens. Bioelectron. 21, 1893–1899 (2006).
Voura, E. B., Jaiswal, J. K., Mattoussi, H. & Simon, S. M. Tracking metastatic tumor cell extravasation with quantum dot nanocrystals and fluorescence emission-scanning microscopy. Nat. Med. 10, 993–998 (2004).
Aubin, J. E. Autofluorescence of viable cultured mammalian-cells. Journal of Histochemistry & Cytochemistry 27, 36–43 (1979).
Benson, R. C., Meyer, R. A., Zaruba, M. E. & McKhann, G. M. Cellular autofluorescence - is it due to flavins. Journal of Histochemistry & Cytochemistry 27, 44–48 (1979).
Gross, H. J., Verwer, B., Houck, D., Hoffman, R. A. & Recktenwald, D. Model study detecting breast cancer cells in peripheral blood mononuclear cells at frequencies as low as 10−7. Proc. Natl. Acad. Sci. U. S. A. 92, 537–541 (1995).
Bianchi, D. W., Zickwolf, G. K., Weil, G. J., Sylvester, S. & DeMaria, M. A. Male fetal progenitor cells persist in maternal blood for as long as 27 years postpartum. Proc. Natl. Acad. Sci. U. S. A. 93, 705–708 (1996).
Bajaj, S., Welsh, J. B., Leif, R. C. & Price, J. H. Ultra-rare-event detection performance of a custom scanning cytometer on a model preparation of fetal nRBCs. Cytometry 39, 285–294 (2000).
Johnson, K. L., Stroh, H., Khosrotehrani, K. & Bianchi, D. W. Spot counting to locate fetal cells in maternal blood and tissue: A comparison of manual and automated microscopy. Microsc. Res. Tech. 70, 585–588 (2007).
Veal, D. A., Deere, D., Ferrari, B., Piper, J. & Attfield, P. V. Fluorescence staining and flow cytometry for monitoring microbial cells. J. Immunol. Methods 243, 191–210 (2000).
Jin, D. Y. et al. Time-gated flow cytometry: an ultra-high selectivity method to recover ultra-rare-event mu-targets in high-background biosamples. J. Biomed. Opt. 14, 10 (2009).
Zhang, L., Wang, Y. J., Ye, Z. Q., Jin, D. Y. & Yuan, J. L. New class of tetradentate beta-diketonate-europium complexes that can be covalently bound to proteins for time-gated fluorometric application. Bioconjugate Chemistry 23, 1244–1251 (2012).
Selvin, P. R. Principles and biophysical applications of lanthanide-based probes. Annu. Rev. Biophys. Biomolec. Struct. 31, 275–302 (2002).
Song, B., Vandevyver, C. D. B., Chauvin, A.-S. & Buenzli, J.-C. G. Time-resolved luminescence microscopy of bimetallic lanthanide helicates in living cells. Organic & Biomolecular Chemistry 6, 4125–4133 (2008).
Buenzli, J.-C. G. Lanthanide Luminescence for Biomedical Analyses and Imaging. Chem. Rev. 110, 2729–2755 (2010).
Thibon, A. & Pierre, V. C. A highly selective luminescent sensor for the time-gated detection of potassium. J. Am. Chem. Soc. 131, 434–435 (2009).
Marriott, G., Clegg, R. M., Arndt-Jovin, D. J. & Jovin, T. M. Time resolved imaging microscopy phosphorescence and delayed fluorescence imaging. Biophys. J. 60, 1374–1387 (1991).
Beverloo, H. B. et al. Preparation and microscopic visualization of multicolor luminescent immunophosphors. Cytometry 13, 561–570 (1992).
Seveus, L. et al. Time-resolved fluorescence imaging of europium chelate label in immunohistochemistry and in situ hybridization. Cytometry 13, 329–338 (1992).
Hanaoka, K., Kikuchi, K., Kobayashi, S. & Nagano, T. Time-resolved long-lived luminescence imaging method employing luminescent lanthanide probes with a new microscopy system. J. Am. Chem. Soc. 129, 13502–13509 (2007).
Gahlaut, N. & Miller, L. W. Time-resolved microscopy for imaging lanthanide luminescence in living cells. Cytometry Part A 77A, 1113–1125 (2010).
Jin, D. Y. Demonstration of true-color high-contrast microorganism imaging for terbium bioprobes. Cytometry Part A 79A, 392–397 (2011).
Smolensky, E. D., Zhou, Y. & Pierre, V. C. Magnetoluminescent agents for dual MRI and time-gated fluorescence imaging. Eur. J. Inorg. Chem. 2141–2147 (2012).
Jin, D. Y. & Piper, J. A. Time-gated luminescence microscopy allowing direct visual inspection of lanthanide-stained microorganisms in background-free condition. Anal. Chem. 83, 2294–2300 (2011).
Jin, D. Y. in Methods in Cell Biology, Vol 102: Recent Advances in Cytometry, Part A: Instrumentation, Methods, Fifth Edition Vol. 102 Methods in Cell Biology (eds Darzynkiewicz Z., et al.) 479–513 (2011).
Deng, W. et al. Ultrabright Eu-doped plasmonic Ag@SiO2 nanostructures: time-gated bioprobes with single particle sensitivity and negligible background. Adv. Mater. 23, 4649–4654 (2011).
Jing, W. et al. Visible-light-sensitized highly luminescent europium nanoparticles: preparation and application for time-gated luminescence bioimaging. J. Mater. Chem. 19, 1258–1264 (2009).
Lu, Y. et al. Automated detection of rare-event pathogens through time-gated luminescence scanning microscopy. Cytometry Part A 79A, 349–355 (2011).
Jin, D., Connally, R. & Piper, J. Long-lived visible luminescence of UV LEDs and impact on LED excited time-resolved fluorescence applications. J. Phys. D-Appl. Phys. 39, 461–465 (2006).
Leif, R. C. et al. Calibration beads containing luminescent lanthanide ion complexes. J. Biomed. Opt. 14, 7 (2009).
Song, C. H. et al. Preparation and time-gated luminescence bioimaging application of ruthenium complex covalently bound silica nanoparticles. Talanta 79, 103–108 (2009).
Hennink, E. J., deHaas, R., Verwoerd, N. P. & Tanke, H. J. Evaluation of a time-resolved fluorescence microscope using a phosphorescent Pt-porphine model system. Cytometry 24, 312–320 (1996).
Acknowledgements
Authors thank to Olympus Australia for providing scanning stage and microscopy accessories, Newport Instruments (http://www.newportinstruments.com/) for providing FireRedTM microspheres and Prof. Jingli Yuan, Dalian University of Technology, for providing BHHCT-Eu3+ complexes. We gratefully acknowledge research funding and support from the Australian Research Council (Discovery Project DP 1095465) and Macquarie University. Dr. Dayong Jin acknowledges International Society for Advancement of Cytometry for support as an ISAC Scholar.
Author information
Authors and Affiliations
Contributions
Y.L., J.A.P., Y.H. and D.J. designed the research and wrote the main manuscript text. Y.L. conducted experiments. Y.L., P.X. and D.J. analysed the data. Y.L. and D.J. prepared figures and supplementary information. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Video S1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Lu, Y., Xi, P., Piper, J. et al. Time-Gated Orthogonal Scanning Automated Microscopy (OSAM) for High-speed Cell Detection and Analysis. Sci Rep 2, 837 (2012). https://doi.org/10.1038/srep00837
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00837
This article is cited by
-
Detection of rare prostate cancer cells in human urine offers prospect of non-invasive diagnosis
Scientific Reports (2022)
-
Practical Implementation, Characterization and Applications of a Multi-Colour Time-Gated Luminescence Microscope
Scientific Reports (2014)
-
On-the-fly decoding luminescence lifetimes in the microsecond region for lanthanide-encoded suspension arrays
Nature Communications (2014)
-
Single-nanocrystal sensitivity achieved by enhanced upconversion luminescence
Nature Nanotechnology (2013)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
