
Abstract
The Fukushima Daiichi nuclear power plant (DNPP) accident caused massive releases of radioactivity into the environment. The released highly volatile fission products, such as 129mTe, 131I, 134Cs, 136Cs and 137Cs were found to be widely distributed in Fukushima and its adjacent prefectures in eastern Japan. However, the release of non-volatile actinides, in particular, Pu isotopes remains uncertain almost one year after the accident. Here we report the isotopic evidence for the release of Pu into the atmosphere and deposition on the ground in northwest and south of the Fukushima DNPP in the 20–30 km zones. The high activity ratio of 241Pu/239+240Pu (> 100) from the Fukushima DNPP accident highlights the need for long-term 241Pu dose assessment and the ingrowth of 241Am. The results are important for the estimation of reactor damage and have significant implication in the strategy of decontamination.
Similar content being viewed by others

Introduction
On March 11, 2011, a catastrophic earthquake (M 9.0) occurred in the northwest Pacific about 130 km off northeastern Japan, followed by a gigantic tsunami, which caused serious damage of the electric system of the Fukushima Daiichi nuclear power plants (DNPP). As a consequence, the cooling systems of nuclear reactors failed, resulting in hydrogen explosions on March 12 and 14 in the Unit 1 and 3 reactors, respectively. On March 15, other explosions happened in the Unit 4 reactor building and the Unit 2 reactor. These explosions of the Fukushima DNPP caused serious releases of radionuclides into the atmosphere1,2,3. The released high volatility fission products including 129mTe, 131I, 134Cs, 136Cs and 137Cs were carried together with the air parcel and subsequent wet and dry depositions, caused accumulation of them on the ground4,5. Consequently, their released amounts and distributions on the ground, as well as the impact on the environment have been studied to provide scientific basis for radiation dose estimation and prediction of their behavior and fate in the environment. For the non-volatile radionuclides, Pu isotopes attracted great public attention in the Fukushima DNPP accident because Pu isotopes present a large risk for internal radiation exposure via ingestions of contaminated agricultural crops, in particular for 241Pu (a beta-emitter, T1/2 = 14.4 years), with its decay, the ingrowth of 241Am (alpha and gamma-emitter, T1/2 = 432.7 years) will present a new radiation risk. In addition, the accurate determination of Pu isotopic composition may provide important information for the estimation of reactor damage, considering the fact that high radiation levels make it impossible to directly measure damage to the melted reactor cores6. As discussed by Powers et al.7 after the Chernobyl accident, isotope compositions and activity ratios of different radionuclides could be useful to obtain information on the situation of the nuclear fuel, such as the fuel burn-up and the inventory of radionuclides in the reactor and thus on the accident mechanisms. For the Fukushima DNPP accident, a preliminary investigation found no significant increase of activities of 239+240Pu in the soil samples and 241Pu, the principle isotope contributing to the dose due to external exposure from radioactivity deposition after the accident, was not considered, although the 238Pu/239+240Pu activity ratio suggested possible Pu contamination in the northwest of the Fukushima DNPP8. Therefore, the release of Pu isotopes into the environment from the Fukushima DNPP accident needs to be clarified.
According to the air dose monitoring data from the MEXT (Ministry of Education, Culture, Sports, Science and Technology) and the atmospheric dispersion simulation by SPEEDI (System for Prediction of Environmental Emergency Dose Information), a high concentration plume of released radionuclides moved to the northwest from the power plant during the daytime on March 15, 2011 and a large amount of radionuclides were deposited on the ground by precipitation5. In addition, on March 21, 2011, high deposition rates were observed in the Kanto Plain, i.e., Ibaraki, Tochigi, Saitama and Chiba Prefectures and Tokyo9. To understand any deposition of Pu isotopes on the ground and to elucidate its isotopic composition, we collected surface soil samples for the determination of activities of 137Cs and Pu and Pu atom ratios (240Pu/239Pu, 241Pu/239Pu) in: Chiba, Kamagaya and Mito Cities in the Kanto Plain; the Evacuation-Prepared Area (J-Village, 20 km south of Fukushima DNPP); and the Deliberate Evacuation Area (S1, in Katsurao Village, 25 km WNW of Fukushima DNPP; S2, in Namie Town, 26 km NW of Fukushima DNPP; and S3, in Iitate Village, 32 km NW of Fukushima DNPP) (Fig. 1). The determination of Pu isotopes was done using a sector-field ICP-MS10. Details of the separation and purification of Pu are described elsewhere (see Methods Section).

Map showing the locations of soil sampling sites.
Results
The results of activities of 137Cs, 239+240Pu and 241Pu, the atom ratios of 240Pu/239Pu and 241Pu/239Pu and the activity ratio of 137Cs/239+240Pu in the soil and litter samples were summarized in Table 1. Data of 137Cs activity for the J-Village samples were cited from Tagami et al.11 and data for global fallout and soil in Tokyo and Sapporo were cited from Kelley et al.12. Pu isotopic composition data for atmospheric fallout in Japan were cited from Zhang et al.13 and for the Chernobyl accident from Muramatsu et al.14 and Ketterer et al.15, respectively. For the samples collected in Fukushima Prefecture, activities of 239+240Pu ranged from 0.019 to 1.400 mBq/g, within the typical global fallout 239+240Pu activity range of 0.15 to 4.31 mBq/g observed in Japanese soils before the Fukushima DNPP accident16. However, high activities of 241Pu ranging from 4.52 to 34.8 mBq/g were detected in samples of the J-Village surface soil (0–2 cm) and of litter at sites S2 and S3 (Table 1). 241Pu was released into the environment through atmospheric nuclear weapons tests in the last century. Due to its short half-life of 14.4 years, the activity of 241Pu in Japanese soils is quite low (ca. 1.2 for 241Pu/239+240Pu activity ratio, 241Pu decay corrected to March 15, 2011). Therefore, the finding of high 241Pu activities in these samples suggested an additional Pu input. The 240Pu/239Pu and 241Pu/239Pu atom ratios found in these samples ranged from 0.303 to 0.330 and from 0.103 to 0.135, respectively. They were significantly higher than those of global fallout (0.180±0.007, 1σfor 240Pu/239Pu atom ratio and 0.00194±0.00014, 1σ for 241Pu/239Pu atom ratio)12 and the atmospheric fallout deposition in Japan from 1963 to1979 (0.1922±0.0044, 1σ for 240Pu/239Pu atom ratio and 0.00287±0.00056, 1σ for 241Pu/239Pu atom ratio)13, indicating new Pu input from the Fukushima DNPP accident. We noted that in the surface soil (0–1 cm) under the litter layer at sites S3 and S2, no 241Pu was determined and 240Pu/239Pu atom ratios were 0.144 and 0.177, respectively, close to the global fallout value of 0.180. This phenomenon indicated that the released Pu deposited in the litter layer, had not reached the underlying surface soil by May 2011 when the samples were collected. We considered that the atom ratios of 240Pu/239Pu and 241Pu/239Pu found in the litter layer reflected the isotopic composition of the released Pu from the Fukushima DNPP accident.
Discussion
Compared to the Pu isotopic composition seen after the Chernobyl accident14,15, the Fukushima accident Pu had a slightly higher 241Pu/239Pu atom ratio, but lower ratio of 240Pu/239Pu (Fig. 2). However, due to the large amount of 239+240Pu (about 8.7×1013 Bq) released from the Chernobyl accident17, the activity ratio of 241Pu/239+240Pu of the Chernobyl accident (83±5)15,18is much lower than that of the Fukushima DNPP accident (107.8, average of S2 and S3 litter, 241Pu decay corrected to March 15, 2011). The Pu atom ratios increase with the increase of the fuel burn-up time in the reactor. The relatively higher 241Pu/239+240Pu activity ratio of the Fukushima DNPP accident might be because of the damage to the Unit 3 reactor, which had a mixed core, containing both uranium fuel and mixed uranium and plutonium oxide (MOX) fuel; the latter was about 6% of the core fuel. The additional production of 241Pu from the 239Pu fuel may have enhanced the 241Pu/239+240Pu activity ratio and 241Pu/239Pu atom ratio inside the reactor during normal operation before the accident.

Mixing plot of 241Pu/239Pu atom ratio vs. 240Pu/239Pu atom ratio for litter and surface soil samples collected in the 20-30 km zones of Fukushima prefecture, Japan and a comparison of isotopic composition with those of the Chernobyl accident and the global fallout sources.
Error bars are ± 1 standard deviation. Data on the Chernobyl accident are cited from Muramatsu et al.14 and Ketterer et al.15. Data on the global fallout are cited from Kelley et al.12. Data on atmospheric fallout in Japan are cited from Zhang et al.13; these data were obtained from atmospheric fallout reference material prepared from samples collected at 14 stations through Japan in 1963-1979 by the Meteorological Research Institute (MRI), Japan.
The atom ratios of 240Pu/239Pu and 241Pu/239Pu found in the surface soil of J-Village were slightly lower than those in litter samples in Namie Town (S2) and Iitate Village (S3) in the NW direction of the Fukushima DNPP. The plot of 241Pu/239Pu vs. 240Pu/239Pu for the data of Table 1 for the global fallout, the soil in J-Village and the litter at sites S2 and S3 could be described by a linear function (r2 = 0.9901): 241Pu/239Pu = 0.9024×(240Pu/239Pu)−0.1656 (Fig. 2). It indicated that the Pu in J-Village surface soil (0–2 cm) contained a small proportion of global fallout Pu.
Using a two end-member mixing model (see Methods Section) based upon the work of Krey19, we found the percentage of Fukushima-derived 239+240Pu in the J-Village soil was 87 %; and the other 13 % 239+240Pu was of global fallout origin. We noted that Pu activities in the J-Village surface soil were ca. one order of magnitude lower than those in northwest of Fukushima DNPP.
In the samples that showed Pu contamination from the Fukushima accident, we detected extremely high 137Cs activities. They ranged from 1.15×104 to 4.65×106 mBq/g (Table 1). The activity ratios of 137Cs/239+240Pu for these samples ranged from 1.95×105 to 2.53×107 and they were 1–3 orders of magnitude higher than that of the Chernobyl accident (770, 137Cs corrected for decay to June, 1997)14, indicating that the release of 239+240Pu from the Fukushima DNPP accident was very small. This was supported by the 239+240Pu activity data in Table 1; even in the samples with high 137Cs contamination, the detected 239+240Pu activities were still in the typical activity range of the global fallout. To understand the differences of Pu emissions between the Fukushima DNPP accident and the Chernobyl accident, we made a rough estimation on the amount of atmospheric release of Pu and the percentage of core inventory released. The estimation was made based on the average of 137Cs/239+240Pu activity ratios (1.48×107) observed in litter samples at site S2 and S3, relative to the total amount of 137Cs releases, 1.5×1016 Bq and 3.58×1016 Bq, estimated by METI (Ministry of Economy, Trade and Industry, Japan)20 and Stohl et al.3, respectively, assuming 137Cs and Pu isotopes followed same deposition mechanism and no significant variation of 137Cs/239+240Pu activity ratio during the release and deposition. It should be noted that there is no attempt to make an accurate estimation on the release of Pu from the Fukushima DNPP accident due to the limited data on the deposition of Pu, but rather a rough estimation to obtain the information on the order of magnitude of Pu release from the accident. As shown in Table 2, the amounts of released 239+240Pu and 241Pu were 1.0×109 − 2.4×109 Bq and 1.1×1011 − 2.6×1011 Bq, respectively. These values are very close to those estimated by METI20 and about 4 orders of magnitude lower than those of the Chernobyl accident.17,18,21 Kirchner et al.22 recently calculated the mean fuel inventory of Pu isotopes in the Fukushima DNPP reactors using ORIGEN-ARP module of the SCALE-5.1 code system, this made it possible to estimate the percentages of the amounts of released Pu isotopes to core inventory with the information of fuel load in Unit 1, Unit 2 and Unit 3 reactors (in total 250 t). It was found that although the inventories of Pu isotopes in the reactors in the Fukushima DNPP were ca. 3.5 times those in the Chernobyl No. 4 reactor23, the percentages of core inventory released for both 239+240Pu and 241Pu were about 5 orders of magnitude lower than those of the Chernobyl accident. These results suggested that for the Fukushima DNPP accident, the plutonium emitted into the environment was mainly due to the release of Pu associated with fuel fragments as a consequence of the hydrogen explosions, as suggested by Kirchner et al.22.
MEXT has estimated the 239+240Pu dose of external exposure and inhalation from resuspension as 0.12 mSv for a person living for 50 years in the contaminated area8. On the other hand, the 241Pu/239+240Pu activity ratio of the Fukushima-derived Pu was found to be higher than 100. The additional dose contribution from 241Pu has to be estimated. As an example, assuming a similar contamination of 241Pu in the surface soil as that in the litter layer and using the method of IAEA-TECDOC-95524, we estimated the 241Pu dose for a person living for 50 years in the vicinity of S2 site to be 0.44 mSv, about 4 times the 239+240Pu dose.
241Pu is a beta-emitting isotope, as a result of241Pu decay the increase of 241Am may significantly enhance the alpha-activity level in the contaminated area for a certain period of time. Using the 241Pu/239+240Pu activity ratio of the Fukushima-derived Pu (107.8), we made a prognostic prediction on the ingrowth of 241Am (Fig. 3). Details of the theoretical calculation on the decay of 241Pu and ingrowth of 241Am are described in Methods Section. The result showed that the 241Am/239+240Pu activity ratio would increase quickly reaching a value of 1 in the year 2018 and it would reach a maximum value of 3.12 in the year 2081, followed by a gradual decrease. This calculated maximum value of 3.12 is almost one order of magnitude higher than that of the expected global fallout 241Am/239+240Pu in the year 204225. Furthermore, the increased amount of 241Am may remain in the surface soil for decades together with Pu isotopes. In our previous study on the migration of 241Am and Pu released from the atomic bomb detonation in Nagasaki26, we found that the 241Am/239+240Pu activity ratio (0.036±0.006) detected in a soil core (0–15 cm) in Nishiyama area, Nagasaki, Japan in 2008 approached the expected maximum value27, indicating that 241Am and Pu were still together in the soils after six decades and showing no significant difference regarding their downward migration behavior (Fig. 3). In addition, a more efficient transfer of 241Am into plants may be expected. A recent study showed that the coefficients of 241Am transfer from soil to wild plants28, particularly to legumes, are 3–5 times higher than those of 239,240Pu. Therefore, it is highly necessary to investigate the distribution and surface activity of 241Pu inside the 20 km zone, where much higher 241Pu could be expected. This is important for the long-term dose assessment of actinides and will have important implications in the strategy for decontamination procedures.

Curves of the calculated activity ratios of 241Pu/239+240Pu and 241Am/239+240Pu from the Nagasaki atomic bomb Pu, the global fallout Pu and the Fukushima DNPP accident Pu with elapsed time.
The 241Am ingrowth from the Nagasaki atomic bomb detonation was based on the initial 241Pu/239+240Pu activity ratio estimated by Yamamoto et al.27. The 241Am/239+240Pu activity ratio (0.036 ±0.006) detected in a soil core collected in Nishiyama area, Nagasaki, Japan in 2008 approached the calculated maximum value, indicating that 241Am and Pu were still together in the soils after 6 decades. 241Am from the global fallout source was expected to reach the maximum 241Am/239+240Pu activity ratio of 0.36 in the year 204225. The theoretic calculation indicated that 241Am/239+240Pu activity ratio would quickly approach the value of 1 by 7 years after the Fukushima DNPP accident and it would reach a maximum value of 3.18 in the year 2081.
For soil samples collected in Mito, Chiba and Kamagaya Cities, although 137Cs activities were significantly higher than the activity level before the accident (Table 1), for example, the Kamagaya soil sample 2 (0–2 cm) collected on the grounds of a Japanese shrine, near the drain pipes from a building roof, had a 137Cs activity of 11429±88 mBq/g, the 239+240Pu activities and 240Pu/239Pu atom ratios were the typical values of the global fallout and no 241Pu could be detected. We concluded this Pu was consistent with global fallout origin. If any, the Fukushima source contribution to the total Pu activity was negligible.
In addition to the atmospheric releases, the cooling of the reactors with fresh water and seawater and the release of highly contaminated water from the damaged reactor buildings resulted in the direct discharges of radionuclides into the Pacific Ocean29,30. The water-soluble 137Cs released from the atmospheric fallout and the directly discharged radioactive waste water caused serious contamination in the marine environment31. However, information on the distribution of plutonium in the marine environment is very limited. It remains unknown if there was a Pu contamination derived from the release of radioactive waste water. Starting from May, 2011, MEXT monthly reported the monitoring results of 239+240Pu and 238Pu activities in seawater and sediments in the 15 km zone in the Pacific off Fukushima32. As the monitoring was conducted using the analytical methods for emergency monitoring, the reported activities of 239+240Pu and 238Pu in seawater were always lower than the detection limits (<0.55 mBq/L for 239+240Pu and <0.61 mBq/L for 238Pu). Although the 239+240Pu activities in sediments ranged from 0.015 to 0.97 mBq/g, were within the range of 239+240Pu activities observed in sediments in the Japanese coastal area before the Fukushima DNPP accident33, no information on the Pu isotopic composition is available for source identification. Obviously, the possible release of Pu isotopes and their impact on the marine environment need further studies.
Methods
Soil and litter sampling
Soil samples (0–2, 5–7 and 10–12 cm) were collected in the Evacuation-Prepared Area (J-Village, 20 km south of Fukushima DNPP) in April, 2011. The soil samples from J-Village were collected in a flower garden. The collection of the organic layer and soil samples in a forest floor was carried out in a deciduous broad-leaved forest at 3 sampling locations differing in distance and direction from the nuclear power plant in the Deliberate Evacuation Area (S1, in Katsurao Village, 25 km WNW of Fukushima DNPP, N37°29′02.8″ E140°45′46.4″; S2, in Namie Town, 26 km NW of Fukushima DNPP, N37°34′17.1″ E140°47′39.9″; and S3, in Iitate Village, 32 km NW of Fukushima DNPP, N37°36′20.0″ E140°45′17.1″) in May, 2011. For each location, 3 samples were collected several meters apart for organic layer and soil samples. The organic layer samples were divided into 2 parts, the upper litter layer (AOL) and the lower fermentation + humus layer (AOF, AOH). From under the organic layer, a 5-cm soil core was taken at 0–5 cm depth. The soil cores were sliced into 1–5 cm thick slices using a spatula. The upper litter layer and surface soil sample (0–1 cm) were analyzed for Pu isotopes.
Soil samples, including a core sample down to 13 cm (0–1, 1–3, 3–5 and 5–13 cm were collected on the campus of the National Institute of Radiological Sciences (NIRS) in Chiba City in April, 2011. The surface soil samples from Mito City (0–1 cm) were collected in the playground of a public park and the those from Kamagaya City (0–2, 2–5 cm) were collected in the garden of a private residence and the grounds of a Japanese shrine in August, 2011.
All samples were oven dried at 80°C overnight and passed through a 2 mm mesh sieve. The soil from the flower garden had no large stones and almost all the fraction could pass through the sieve. All samples were measured for 137Cs activity after passing the 2 mm mesh sieve. For Pu isotope analysis, organic matter in the samples was decomposed by heating in a Muffle oven at 450°C for 6 hours.
Analytical procedures for 137Cs and Pu isotopes
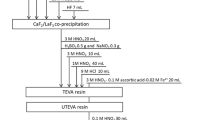
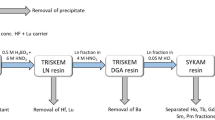
137Cs activity was determined using a Ge detection system (Seiko EG&G) for 3600 s for most cases. The 137Cs activity was determined using its peak at 661.6 KeV. A mixed gamma standard solution (Amersham, QCY-46) was used for an efficiency correction and reference standard materials IAEA-156, 373 and 375 were used for an accuracy check11. Pu isotope separation and purification were done using a modified method based on the MEXT method for Pu analysis in environmental samples34. Briefly, 3.0–7.0 g samples (soil and litter) were mixed with 1 pg 242Pu tracer and digested by heating on a hot-plate using 10 MHNO3-1 M HF. After the digestion, the solution was heated to dryness and the residues were dissolved in 30 ml 8 M HNO3. NaNO2 was added to adjust the Pu oxidation state and then 0.3 g boric acid was added to convert unreacted HF to BF4−. Dowex 1×8 anion resins were used in the separation and purification processes. After loading the sample solution on the first Dowex 1×8 column (2 ml), sequential elution of U, Th and Pu was conducted using 35 ml 8 M HNO3, 35 ml 10 M HCl and 35 ml NH4I-HCl solution, respectively. The obtained Pu fraction was heated to dryness after adding 5 ml HNO3. The residue was dissolved in 10 ml 4 M acetic acid and this solution was loaded onto the second Dowex 1×8 column (2 ml). Then 20 ml 4 M acetic acid was used to wash the column and all the eluted solution was collected for Pu analysis. The collected acetic acid solution (30 ml) was heated to dryness and the residue was dissolved in 0.7 ml 4% HNO3 for Pu isotope analysis using a highly sensitive APEX/SF-ICP-MS analytical system10. Sediment standard reference materials IAEA-368 (marine sediment standards, International Atomic Energy Agency) and SRM-4354 (freshwater lake sediment standards, American National Standards Institute of Technology) were used for analytical method validation13,35.
Two end-member mixing model for source apportionment of the Fukushima DNPP accident source Pu
Since two isotopically distinctive Pu sources, global fallout and the Fukushima DNPP accident fallout, were identified in this study, it is possible to evaluate the individual relative contribution from global and the Fukushima DNPP accident Pu based on 240Pu/239Pu ratios. We used a simple two end-member mixing model, which is similar to the one described by Krey19, to calculate the relative contribution of the Fukushima DNPP accident source Pu:

where (Pu) = activity of 239+240Pu; R = 240Pu/239Pu atom ratio; and subscripts F, G and S refer to Fukushima DNPP accident, global fallout and the soil sample in J-Village, respectively. This equation converts 240Pu/239Pu atom ratio data to activity ratios of 239+240Pu from two sources. The constant 3.674 is the ratio of the specific activities of 240Pu to 239Pu obtained from IAEA-recommended half-lives of 24110±30 and 6563±7 years for 239Pu and 240Pu, respectively. We defined

where (Pu)T is the total activity of 239+240Pu in the sample and (Pu)G consists of the global fallout. Hence, the percentage of the Fukushima DNPP accident source Pu could be obtained from the following equation:

Kelley et al.12 reported the mass ratio of 240Pu/239Pu in global fallout to be 0.180 ± 0.007, based on a world-wide program of sampling conducted at 21 sites in 1970 and 1971 between 30°N and 70°N. The 240Pu/239Pu ratio of the Fukushima DNPP accident obtained in this study was 0.327. Therefore, the percentage of the Fukushima DNPP accident source Pu could be calculated using the 240Pu/239Pu atom ratio of 0.303 in the surface soil in the J-Village to be 87%.
Theoretical calculation of the decrease of 241Pu/239+240Pu activity ratio and the increase of 241Am/239+240Pu activity ratio with time
We calculated the decrease of 241Pu/239+240Pu activity ratio with time using the equation given below:

where A means the activity and the subscripts t and 0 mean the values at time t and t0, respectively. It is assumed that A239+240Pu(t)≈A239+240Pu(0). λ241Pu = 0.0483 y−1.
For the calculation of the increase of 241Am/239+240Pu activity ratio with time, we used the following equation:

where A means the activity and the subscripts t and 0 mean the values at time t and t0, respectively. It is assumed that A239+240Pu(t)≈A239+240Pu(0). λ241Pu = 0.0483 y−1 and λ241Am = 0.0016 y−1. The activity ratios of241Pu/239+240Pu at t = 0 for Nagasaki atomic bomb, the global fallout and the Fukushima DNPP accident were 1.2127, 12.825 and 107.8 (this study), respectively.
References
NSC, Trial estimation of emission of radioactive materials (I-131, Cs-1137) into atmosphere from Fukushima Dia-ichi nuclear power station. http://www.nsc.go.jp/NSCenglish/geje/2011%200412%20press.pdf (accessed 2011.6.27)
Chino, M. et al. Preliminary estimation of release amounts of 131I and 137Cs accidentally discharged from the Fukushima Daiichi nuclear power plant into the atmosphere. J. Nucl. Sci. Tech. 48, 1129–1134 (2011).
Stohl, A. et al. Xenon-133 and caesium-137 releases into the atmosphere from the Fukushima Dai-ichi nuclear power plant: determination of source term, atmospheric dispersion and deposition. Atmos. Chem. Phys. Discuss. 11, 28319–28394 (2011).
Japanese Ministry of Education, Culture, Space, Science and Technology (MEXT), Monitoring information of environmental radioactivity level. Distribution map of radioactive Caesium (2011) http://radioactivity.mext.go.jp/ja/distribution_map_around_FukushimaNPP/ (accessed 2011.8.30)
Kinoshita, N. et al. Assessment of individual radionuclide distributions from the Fukushima nuclear accident covering central-east Japan. Proc. Natl. Acad. Sci. 108, 19526–19529 (2011).
Cyranoski, D., Brumfiel, G. Fukushima impact is still hazy. Nature 477, 139–140 (2011).
Powers, D. A, Kress, T. S. & Jankowski, M. W. The Chernobyl source term. Nucl. Saf. 28, 10–28 (1987).
Japanese Ministry of Education, Culture, Space, Science and Technology (MEXT), Monitoring information of environmental radioactivity level. Distribution map of Plutonium and 90Sr (2011) http://radioactivity.mext.go.jp/ja/distribution_map_around_FukushimaNPP/ (accessed 2011.9.30)
Hirose, K. Fukushima Daiichi nuclear power plant accident: summary of regional radioactive deposition monitoring results. J. Environ. Radioact. in press (2012). doi:10.1016/j.jenvrad.2011.09.003
Zheng, J. & Yamada, M. Inductively coupled plasma-sector field mass spectrometry with a high-efficiency sample introduction system for the determination of Pu isotopes in settling particles at femtogram levels. Talanta 69, 1246–1253 (2006).
Tagami, K. et al. Specific activity and activity ratios of radionuclides in soil collected about 20 km from the Fukushima Daiichi Nuclear Power Plant: Radionuclide release to the south and southwest. Sci. Total Environ. 409, 4885–4888 (2011).
Kelley, J. M., Bond, L. A. & Beasley, T. M. Global distribution of Pu isotope and Np. Sci. Total Environ. 237/238, 483–500 (1999).
Zhang, Y. S. et al. Characterization of Pu concentration and its isotopic composition in a reference fallout material. Sci. Total Environ. 408, 1139–1144 (2010).
Muramatsu, Y. et al. Concentrations of 239Pu and 131Pu and their isotopic 240 determined by ICP-MS in soils collected from the Chernobyl 30-km zone. Environ. Sci. Technol. 34, 2913–2917 (2000).
Ketterer, M. E., Hafer, K. M. & Mietelski, J. W. Resolving Chernobyl vs. global fallout contributions in soils from Poland using plutonium atom ratios measured by inductively coupled plasma mass spectrometry. J. Environ. Radioact. 73, 183–201 (2004).
Muramatsu, Y., Yoshida, S. & Tanaka, A. Determination of Pu concentration and its isotope ratio in Japanese soils by HR-ICP-MS. J. Radioanal. Nucl. Chem. 255, 477–480 (2003).
Kruger, F. W., Albrecht, L., Spoden, E. & Weiss, W. Der Ablauf des Reaktorunfalls Tschernobyl 4 und die weitraumige Verfrachtund des freigesetzten Materials: Neuere Erkenntnizze und ihre Bewertund, in Zehn Jahre nach Tshernobyl, eine Bilanz. (eds. A. Bayer, A. Kaul, & Chr. Reiners) 3–22 (Gustav Fisher, Stuttgart;1996).
IAEA, International nuclear safety advisory group. Summary Report on the Post-Accident Review Meeting on the Chernobyl Accident. IAEA Safety Series No. 75-INSAG-1, IAEA Press, Vienna, P. 34 (1986).
Krey, P. W. Remote Pu contamination and total inventories from Rocky Flats. Health Phys. 30, 209–214 (1976).
METI (Ministry of Economy, Trade and Industry), Data on the amount of released radioactive materials, 2011. http://www.meti.go.jp/press/2011/10/20111020001/20111020001.pdf (accessed 2012.1.26)
Harrison, R. M. et al. Atmospheric Pathways. in SCOPE 50, Radioecology after Chernobyl, Biogeochemical Pathways of Artificial Radionuclides. (eds. S. F. Warner & R. M. Harrison) 55–100 (John Wiley & Sons, Chichester; 1993).
Kirchner, G. K., Bossew, P. & De Cort, M. Radioactivity from Fukushima Dai-ichi in air over Europe; part 2: what can it tell us about the accident? J. Environ. Radioact. in press (2012). doi:10.1016/j.jenvrad.2011.12.016
Devell, L., Guntay, S. & Powers, D. A. The Chernobyl reactor accident source term: development of a consensus view, CSNI Report, Organisation for Economic Co-Operation and Development (OECD), OCDE/GD (96) 12. (1995).
IAEA, Generic Assessment Procedures for Determining Protective Actions during a Reactor Accident. IAEA, Vienna, IAEA-TECDOC-955. ISSN 1011–4289 (1997).
Livingston, H. D., Schneider, D. L. & Bowen, V. T. 241Pu in the marine environment by a radiochemical procedure. Earth Planet. Sci. Lett. 25, 361–367 (1975).
Zheng, J., Yoshida, S., Yamada, M. & Aono, T. Current status of 241Am in Nishiyama soil: six decades after Nagasaki A-bomb explosion. Proceedings of the Tenth Workshop on Environmental Radioactivity. 2009–8, 39–44 (2009).
Yamamoto, M., Komura, K. & Sakanoue, M. Discrimination of the plutonium due to atomic explosion in 1945 from global fallout plutonium in Nagasaki soil. J. Radiat. Res. 24, 250–258 (1983).
Skvernyuk, I. I., Orehova, M. G., Mastko, V. P., Kudryashov, V. P. & Gaponenko, V. I. Plutonium in plants of natural coenosis of south-east Byelorussian Polesye. Proceedings of Polish-Ukrainian-Byelorussian International Conference on Natural Environment of Polesye: Present Situation and Changes. Vol. 2. 335–337 (Brest, Belarus;2002).
Tsumune, D., Tsubono, T., Aoyama, M. & Hirose, K. Distribution of oceanic 137Cs from the Fukushima Daiichi nuclear power plant simulated numerically by a regional ocean model. J. Environ. Radioact. in press (2012). doi:10.1016/j.jenvrad.2011.10.007
Buesseler, K., Aoyama, M. & Fukasawa, M. Impacts of the Fukushima nuclear power plants on marine radioactivity. Environ. Sci. Technol. 45, 9931–9935 (2011).
Honda, M. C. et al. Dispersion of artificial caesium-134 and -137 in the western North Pacific one month after the Fukushima accident. Geochem. J. 46, e1–e9 (2012).
Japanese Ministry of Education, Culture, Space, Science and Technology (MEXT), Monitoring information of environmental radioactivity level. Distribution of radionuclides in marine environment (2011) http://radioactivity.mext.go.jp/ja/around_TEPCO_FukushimaNPP_seawater/ (accessed 2012.1. 11)
Zheng, J. & Yamada, M. Sediment core record of global fallout and Bikini close-in fallout Pu in Sagami Bay, western northwest Pacific margin. Environ. Sci. Technol. 38, 3498–3504 (2004).
Japanese Ministry of Education, Culture, Space, Science and Technology (MEXT), Rapid analysis of Pu in environmental samples. 2002. http://www.kankyo-hoshano.go.jp/series/main_pdf_series_28.html (accessed 2011.4.25)
Wu, F. C., Zheng, J., Liao, H. Q., Yamada, M. & Wan, G. J. Anomalous plutonium isotopic ratios in sediments of Lake Qinghai from the Qinghai-Tibetan Plateau, China. Environ. Sci. Technol. 45, 9188–9194 (2011).
Acknowledgements
This study was partially supported by the Agency for Natural Resources and Energy, the Ministry of Economy, Trade and Industry (METI), Japan.
Author information
Authors and Affiliations
Contributions
J. Z., K. T., Y. W. and S. U. designed the study. J. Z. conducted Pu isotope analyses and wrote the manuscript. K. T., T. A., Y. W. and N. I. analysed 137Cs activity. Y. K., S. F., S. Y. and S. I. participated in the soil sampling, discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Zheng, J., Tagami, K., Watanabe, Y. et al. Isotopic evidence of plutonium release into the environment from the Fukushima DNPP accident. Sci Rep 2, 304 (2012). https://doi.org/10.1038/srep00304
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00304
This article is cited by
-
Effect of land use and vegetation coverage on level and distribution of plutonium isotopes in the northern Loess Plateau, China
Journal of Radioanalytical and Nuclear Chemistry (2023)
-
First application of plutonium in soil erosion research on terraces
Nuclear Science and Techniques (2023)
-
Analysis of particles containing alpha-emitters in stagnant water at torus room of Fukushima Dai-ichi Nuclear Power Station’s Unit 2 reactor
Scientific Reports (2022)
-
Quantitative assessment of the spatial distribution of 239+240Pu inventory derived from global fallout in soils from Asia and Europe
Journal of Geographical Sciences (2022)
-
Rapid method for sequential determination of Pu and Am in soil and sediment samples by sector-field inductively coupled plasma mass spectrometry
Journal of Radioanalytical and Nuclear Chemistry (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
