
Abstract
We investigated the properties of clonally-expanded mouse hair follicle stem cells (HF-SCs) in culture. The expansion method, suppression of asymmetric cell kinetics (SACK), is non-toxic and reversible, allowing evaluation of the cells' asymmetric production of differentiating progeny cells. A tight association was discovered between non-random sister chromatid segregation, a unique property of distributed stem cells (DSCs), like HF-SCs and a recently described biomarker, Lgr5. We found that nuclear Lgr5 expression was limited to the HF-SC sister of asymmetric self-renewal divisions that retained non-randomly co-segregated chromosomes, which contain the oldest cellular DNA strands, called immortal DNA strands. This pattern-specific Lgr5 association poses a potential highly specific new biomarker for delineation of DSCs. The expanded HF-SCs also maintained the ability to make differentiated hair follicle cells spontaneously, as well as under conditions that induced cell differentiation. In future human cell studies, this capability would improve skin grafts and hair replacement therapies.
Similar content being viewed by others


Introduction
The hair follicle is among the best-studied stem cell compartments in mammals1,2. Hair follicle stem cells (HF-SCs) renew several major components of the skin, including sebaceous glands, the epidermis, the hair follicle and hair3,4,5, making them ideal for skin restoration applications. HF-SCs might yield cosmetic and functional advances in skin grafting for burn victims; and their transplantation might also treat male-patterned baldness. Pursuit of such advances has been thwarted by one vexing problem. HF-SCs are highly refractory to ex vivo expansion, even when isolated after molecular marking6,7,8. Recently, we reported that stem cells from the whisker follicles of mice could be readily expanded by the method of suppression of asymmetric cell kinetics (SACK)9. Based on the SACK principle that a significant barrier to the ex vivo expansion of distributed stem cells (DSCs)10,11 is their asymmetric self-renewal12,13, we used guanine ribonucleotide precursors to shift HF-SCs from their default state of asymmetric self-renewal, which in culture leads to their dilution and loss, to symmetric self-renewal, which promotes their exponential expansion9. Here, we report newly defined properties of SACK-expanded cell strains from hair follicles that further illustrate their DSC character. These include asymmetric expression of the recently described HF-SC biomarker Lgr5 and tightly associated non-random sister chromatid segregation. Non-random segregation is a highly specific property of DSCs that previously was not detected in analyses of mouse hair follicles performed in situ14,15. Our findings raise the possibility that Lgr5 marks stem cells that undergo asymmetric self-renewal with non-random segregation in the hair follicle. Under conditions that promote asymmetric self-renewal, arrested progeny cells produced by HF-SCs spontaneously undergo complex differentiation, which is an essential property for future applications in medicine and cosmetics.
Results
Nuclear Lgr5 expression is limited to the cycling HF-SC after asymmetric self-renewal divisions
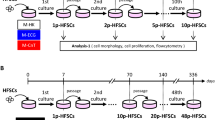
The detailed procedure for SACK expansion of HF-SCs has been reported9. Briefly, we excised intact mouse whisker follicles, removed adherent external cells to prevent dermal fibroblast growth, transected them and cultured them in medium supplemented to 400 μM with one of three SACK agents, xanthosine (Xs), xanthine (Xn), or hypoxanthine (Hx). Epitheliod cells readily grew out of transected follicles. Cultures supplemented with Xn reached confluency first. During a next 7-day culture period after whole-culture passage, Hx and Xn supplementation produced 5- and 6-fold more cells, respectively, than control conditions without SACK agents. Xs supplementation did not significantly increase cell outgrowth. Limiting dilution cloning12 was employed to evaluate the SACK-dependency of expansion cultures and to derive clonal cell strains. Only the Xn-supplemented cultures showed a significant increase in the number of cell clones under the SACK condition (2.5-fold; p < 0.05, one-tailed Fisher's exact test). Of 17 propagated Xn-derived cell clones (out of 50 total), 15 survived expansion to full-scale cultures for cryo-preservation. Clonal strains have been cultured for more than 40 population doublings in Xn-supplemented medium without crises or senescence, whereas control cultures without SACK agents underwent a typical crisis16 at 15 population doublings (data not shown).
Two Xn-derived clonal cell strains, 3C5 and 5B8, were evaluated more extensively for HF-SC characteristics. In Xn-supplemented medium, 90% and 83% of 3C5 cells and 5B8 cells, respectively, were Lgr5-positive in indirect in situ immunofluorescence (ISIF) studies (e.g., see Figs. 2, 5A, 5B; also Supplementary Information Figs. S1 and S2). Lgr5, a proposed G-protein coupled receptor, is a recently reported biomarker for murine HF-SCs8. Lgr5 was detected in both the cytoplasm and nucleus of cells. In an immunohistochemical analysis of human gastrointestinal tract tissues, Lgr5 sub-cellular localization was reported to be cytoplasmic17. However, its localization has also been reported as nuclear in other tissues (e.g., thyroid gland and cultured A-431 cells18). Nuclear localization of other G-protein coupled receptors has been described as well19. Both nuclear and cytoplasmic fluorescence required the anti-Lgr5 antibody; and both were prevented when the antibody was blocked with peptide antigen (Fig S3).

Nuclear Lgr5 expression is limited to the cycling stem cell sister of hair follicle stem cell asymmetric self-renewal divisions.
Shown are epifluorescence micrographs from parallel sister pair (SPr) and cytochalasin D (CD) analyses for hair follicle stem cell strain 3C5 cultured under conditions that either suppress asymmetric self-renewal divisions (left) or promote them (right). Examples of respective Co-SYM and Co-ASYM patterns of nuclear Lgr5 and cyclin A detection are given. DAPI, nuclear DNA fluorescence. CyA, ISIF with specific antibodies for cyclin A. Lgr5, ISIF with antibodies for Lgr5. ISIF was performed simultaneously for cyclin A and Lgr5. Merge, overlay of DAPI, cyclin A and Lgr5 epifluorescence images. Phase, corresponding phase micrographs. Scale bar, 50 microns.

Induced differentiation properties of SACK-expanded, clonal hair follicle stem cell strains.
Strain 3C5 (A) and 5B8 (B) cells were evaluated for expression of Lgr5 and the hair follicle-epidermis biomarkers keratin 5 (K5), keratin 10 (K10) and Filaggrin, either under conditions of active cell proliferation (UNDIFF) or after differentiation by serum reduction (DIFF) as detailed in Methods. Phase, phase micrographs of cells under the two conditions. Shown are immunofluorescence micrographs developed with either single or dual antibodies directed against the indicated specific biomarkers. The colors of a micrograph's biomarker labels match the corresponding fluorescence colors that indicate detection of the respective biomarker in the micrograph. DAPI, all cells were counterstained to detect nuclear DNA by fluorescence for overlay with respective fluorescence micrographs. In LGR5/DAPI micrographs, arrows denote examples of cells with nuclear Lgr5 fluorescence. In dual biomarker micrographs, arrows denote examples of cells that display fluorescence corresponding to both biomarkers. Scale bar, 200 microns.
We used cytochalasin D (CD) assays11,20,21 and sister pair (SPr)11,12,21 to estimate changes in the frequencies of asymmetric self-renewal versus symmetric self-renewal by the expanded cell strains. Asymmetric self-renewal, division which continuously produces non-stem committed progenitors while preserving stem cell phenotype, is a gnomonic for DSCs22. The SPr assay evaluates the cycling status of sister cells produced from divisions at low cell densities; and the complementary CD assay evaluates the cycling capability of sister nuclei when cytokinesis is prevented. As shown in Figs. 1 and 2, SPr and CD assays can detect asymmetric self-renewal divisions by individual cells, if the non-stem cell sister undergoes a cell cycle arrest as a part of its differentiation program. Therefore, in culture, asymmetric self-renewal can be defined by cell divisions in which continued cycling by the stem cell sister is denoted by nuclear cyclin A expression and the arrest of non-stem cell sisters is noted by a lack of nuclear cyclin A expression (e.g., Fig. 1f, CyA)11,21. Symmetric self-renewal divisions are defined by sister cells that both show nuclear cyclin A expression11,21 (e.g., Fig. 1j, CyA).

Examples of complete data profiles for a cytochalasin D (CD) assay of 3C5 and 5B8 clonal hair follicle stem cell strains cultured under conditions that either promote or suppress asymmetric self-renewal.
Co-ISIF results with anti-cyclin A (CyA, red) and anti-LGR5 (green) antibodies are indicated by a square arrangement of four circles. Horizontally, the paired circles represent paired sister nuclei in a CD-arrested binucleated cell. The top pair indicates the fluorescence pattern observed for cyclin A; and the bottom pair indicates the corresponding staining pattern for LGR5. Open circles indicate that no significant fluorescence was detected. Closed circles indicate that fluorescence was detected for the respective biomarker. All 10 theoretically possible staining patterns are depicted (a–j). (A) conditions that promote asymmetric self-renewal (-Xn); (B) conditions that suppress asymmetric self-renewal (+Xn). n, total number of binucleated cells evaluated and the respective numbers for each possible pattern type; %, percent that each pattern type represents out of the total number of sister nuclear pairs evaluated. The solid rectangle highlights the “Co-SYM” designation for Lgr5 and cyclin A; and the dashed rectangle highlights to the “Co-ASYM” designation.
Although the SPr and CD assays can be used to quantify changes in the relative frequencies of asymmetric and symmetric self-renewal, they may underestimate absolute frequencies. We have noted that some newly-defined biomarkers indicate higher rates of asymmetric self-renewal than cyclin A11; and cyclin A-based asymmetric self-renewal frequencies are less than those determined by time-lapse microscopy11,23. These differences could be explained by transient initial expression of cyclin A expression in arresting sisters.
Fig. 1A shows an example of a data profile from a single CD analysis under Xn-free conditions, in which nuclear cyclin A expression and nuclear Lgr5 expression were evaluated together. There are 10 possible expression patterns for these two biomarkers in CD-arrested binucleated cells. These patterns emerge from the cell cycle status of the paired sister nuclei (which have been shown to behave independently for cyclin A and Lgr5 expression properties11) and cell-cycle dependency of the expression of the evaluated proteins. Most of the possible combinatorial patterns were observed infrequently (Fig. 1). The asymmetric co-expression (Fig. 1f, Co-ASYM) and symmetric co-expression (Fig. 1j, Co-SYM) patterns predominated. Hence, they were the primary basis for evaluating the self-renewal properties of HF-SC cultures11,21. Their analysis indicated that nuclear Lgr5 expression occurred primarily in cycling cells; and in the Xn-free condition evaluated, a higher fraction of these cells cycled asymmetrically, producing a non-cycling sister cell with reduced nuclear Lgr5.
In CD assays in Xn-free medium, 53 ± 12% of 3C5 cell divisions (n = 3; examined binucleated cells range = 59–66) showed asymmetric co-expression of nuclear cyclin A and nuclear Lgr5. Nuclear Lgr5 was restricted to the cycling sister. The non-dividing cells of asymmetric self-renewal divisions showed primarily cytoplasmic Lgr5 localization (See Fig. 2, Co-ASYM, SPr). For sister cell pairs and paired CD nuclei that displayed symmetric cyclin A expression, nuclear Lgr5 expression was also symmetric (Fig. 2, Co-SYM). In CD analyses with Xn supplementation to suppress asymmetric self-renewal, the frequency of divisions with asymmetric co-expression decreased to 25 ± 8% (n = 3; examined binucleated cells range = 55–64). This frequency corresponds to 75% symmetric self-renewal, which manifests the SACK effect. This highly significant difference in self-renewal pattern (p < 0.0004; two-tailed Fisher's exact test) is consistent with nuclear Lgr5 marking HF-SCs that shift between symmetric self-renewal and asymmetric self-renewal in response to Xn. In fact, under Xn-free conditions, 3C5 and 5B8 cells showed, respectively, 90% and 96% concordance between asymmetric nuclear Lgr5 expression and directly corresponding asymmetric cyclin A expression (Fig. 1A, f versus d, g and h). The sister cells produced by asymmetric self-renewal divisions that are negative for both cyclin A and nuclear Lgr5 are consistent with differentiating non-stem cells (See later).
Asymmetrically self-renewing HF-SCs employ non-random sister chromatid segregation
An even more specific gnomonic for DSCs, that is also highly associated with asymmetric self-renewal, is non-random sister chromatid segregation10,11,24,25,26. Non-in vivo analyses suggested that non-random segregation did not occur in examined mouse pelage hair follicles14,15. These past studies may have overlooked cells with this property because of the low sensitivity of in vivo assays. We used the sensitive method of label retention followed by CD binucleate segregation analysis (“label retention-CD”)20,27 to investigate non-random segregation by SACK-expanded HF-SCs. Cells were labeled for approximately one cell generation (i.e., average cell cycle time, 24 hours) with the thymidine analogue bromodeoxyuridine (BrdU). Thereafter, BrdU was removed and culturing was continued for 4 cell generations before arresting cells as binucleates with CD to evaluate retention and segregation of BrdU-labeled DNA detected by ISIF.
Three labeling schemes were compared (Figure 3A). In scheme-I, cells were maintained throughout in Xn-supplemented medium. In scheme-II, cells were grown for approximately 6 generations in Xn-free medium before labeling with BrdU and thereafter maintained in Xn-free medium. For scheme-III, which was a “pulse-chase” procedure, cells were labeled in Xn-supplemented medium; but for the rest of the analysis they were maintained in Xn-free medium with a 10-fold molar excess of thymidine. Scheme-III limits continued BrdU incorporation from internal pools.

Label retention-CD analysis for detection of non-random segregation of immortal DNA strands in hair follicle stem cells.
(A) Diagrammed are the three BrdU-labeling schemes (I–III) employed, as described in the text, to evaluate the Xn-dependency of non-random sister chromatid segregation by 3C5 hair follicle stem cells. The number of generation times (GT; 1GT∼24 hours) for each stage of the analyses (horizontal solid arrows and lines) is indicated (dotted arrows). The times of replacement of cultures with BrdU-, thymidine chase (Thd)-, or CD-supplemented culture medium are noted by vertical lines. At the indicated stages, cells were cultured in medium that either promoted asymmetric self-renewal (-Xn [↑ASR]) or suppressed it (+Xn [↓ASR]). (B) Examples of epifluorescence micrographs of cells evaluated in scheme III for the segregation pattern of BrdU-labeled DNA by ISIF (BrdU). Shown are three examples of binucleated cells showing retention and non-random segregation of BrdU-labeled immortal DNA (NRandom) and one example showing symmetric inheritance indicative of random segregation (Random). DAPI, nuclear DNA fluorescence. Merge, overlaid DAPI and anti-BrdU images. Binucleation was confirmed by phase microscopy (Phase). Scale bars, 25 microns. (C) Quantitative summary of the percentage of CD-induced binucleated cells showing non-random segregation of retained BrdU-labeled DNA (%NR). The mean of three independent experiments is plotted for schemes I and II. Error bars denote the standard deviation of the data.
Figure 3B shows examples of label retention-CD images indicative of non-random segregation (Nrandom). BrdU-substituted DNA strands, detected by ISIF, are retained and segregated primarily to one sister nucleus over 4 cell generations. Based on both logical inference20,27 and direct experimental analyses26, the BrdU-retaining nucleus is the HF-SC nucleus. In the former case, we establish in this report, as previously20,27, that immortal DNA strands are quantitatively detected in cycling cells after several generations of asymmetric self-renewal divisions that produce one cycling sister and one arrested sister. It can be inferred that, in order to achieve such an inheritance outcome, the labeled immortal DNA strands must be inherited by the cycling sisters of asymmetric self-renewal divisions. Otherwise, later cycling cells would not contain them. In the latter experimental case, recently, we showed directly that a biomarker that is also asymmetrically detected between the sisters of asymmetric self-renewal divisions is co-asymmetric with both cyclin A, which marks the cycling stem cell sister and co-segregated immortal DNA strands with high concordance26. This result demonstrates directly that the cycling stem cell inherits the immortal DNA strands.
Random segregation is denoted by sister nuclei with similar BrdU content (See Fig. 3B, Random)20,26,27. In scheme-I, the continuous SACK condition, only 13 ± 1.5% (n = 3; Fig. 3C, I) of BrdU-positive binucleated cells showed a non-random segregation pattern. In contrast, in scheme-II, the continuous SACK-free condition, non-random segregation was 2.6-fold more frequent (34 ± 5%; n = 3; Fig. 3C, II; p < 0.003, Student's unpaired t test).
Non-random segregation detection by label retention-CD analysis requires that stem cells label while symmetrically self-renewing and then shift to asymmetric self-renewal stably for sufficient cell generations to release label that may continue to incorporate from internal pools after the change to BrdU-free medium. Therefore, continued incorporation of BrdU from internal pools in schemes I and II results in lower sensitivity for detection of non-random segregation. With this qualification, the scheme-I result suggested that, under SACK conditions, only ∼17% (13% non-random segregation/75% symmetric self-renewal in CD assay) of HF-SCs spontaneously shifted from symmetric self-renewal to asymmetric self-renewal during ≤ 96 hours of culture; whereas under SACK-free conditions, 72% (34% non-random segregation/47% symmetric self-renewal in CD assay) made the symmetric to asymmetric shift.
The scheme-III procedure is optimal for detection of non-random segregation, as it greatly reduces continued incorporation of BrdU during the period of asymmetric self-renewal and maximizes the frequency of symmetric self-renewal during the labeling period and asymmetric self-renewal during the chase period. With Xn-free conditions during the chase period, 44% of binucleated cells showed the non-random segregation pattern (Fig. 3C, III, -Xn); whereas Xn supplementation in parallel reduced the frequency to 19% (Fig. 3C, III, +Xn; p = 0.0025; two-tailed Fisher's exact test). Since binucleated cells derived from cells that cycled asymmetrically with non-random segregation during the labeling period would be unlabeled at the end of scheme-III, these data indicate that approximately 44% of randomly segregating cells can be shifted to non-random segregation by removal of Xn. Conversely, 57% ([44−19]/44) of cells undergoing non-random segregation can be converted to random segregation by Xn. These shift fractions match very well the respective 37% and 52% shifting fractions between asymmetric versus symmetric self-renewal in response to Xn determined by CD assays (p = 0.88; two-tailed Fisher's exact test). This link between self-renewal pattern and chromosomal segregation pattern observed for natural HF-SCs was predicted by earlier studies with genetically engineered cultured cell models20,27.
Quantitative digital imaging of anti-BrdU fluorescence in individual sister nuclei of BrdU-positive binucleated cells supported the above interpretation of the frequency data. Fig. 4A shows the distribution of mean fluorescence intensity (MFI) of scheme-III cells after the 24 hour BrdU-labeling period. At the completion of the experiment, nuclei in cells chased with Xn supplementation show a significant decrease in MFI (Fig. 4B). Approximately half the nuclei in binucleated cells from the Xn-free chase condition show a similar reduction in MFI. However, the MFI of the rest of the Xn-free chase distribution is consistent with nuclei with a BrdU content that approximates 1/2 of the starting level (Fig. 4C). This BrdU content is consistent with nuclei from HF-SCs that initiated non-random segregation on average one division after BrdU-labeling. Consistent with this interpretation, whereas the distribution of percent difference between the MFI of paired nuclei ( = 100% x [A–B]/A, where A ≥ B) for chase with Xn was unimodal with a median of 19% (Fig. 4D), for chase without Xn, the distribution was bimodal (Fig. 4E). Approximately half of the binucleates had percent differences that matched the distribution of differences for the Xn chase (Compare Fig. 4D and 4E, left peak)). These binucleates correspond to products of random segregation. The other half of binucleates had percent differences that ranged from approximately 68 to 98 percent (Fig. 4E, right peak) and quantitatively accounted for the 44% frequency of binucleates with the non-random segregation pattern for BrdU, as shown in Fig. 3.

Quantitative evaluation of the suppression of hair follicle stem cells' non-random segregation by the SACK agent Xn.
The pulse-chase label retention-CD experiment described in the text and Fig. 3 was quantified by digital imaging of fluorescence due to anti-BrdU antibodies. (A) Distribution of the mean fluorescence (Fl) intensity of nuclei in mononucleated cells after 24 hours of BrdU-labeling. (B) Distribution of the mean fluorescence intensities of nuclei in binucleated cells chased in Xn-supplemented medium. (C) Distribution of the mean fluorescence intensities of nuclei in binucleated cells chased in Xn-free medium. p, for statistical confidence by the K-S test that the B and C distributions are distinct. (D) Distribution of the % differences between sister nuclei in binucleated cells chased in Xn-supplemented medium. (E) Distribution of the % differences between sister nuclei in binucleated cells chased in Xn-free medium. p, for statistical confidence by the K-S test that the D and E distributions are distinct. M, median. The mean fluorescence intensity for nuclei in binucleated cells that were unlabeled, but otherwise went through the entire ISIF procedure, was 0.7 ± 1.0.
Since suppression of asymmetric cell kinetics is never 100%, at all times some cells in cultures of SACK-derived DSC strains will be committed to differentiation. Of course, the critical factor for success in the SACK approach is achieving sufficient suppression, so that >50% of all stem cell divisions are symmetric. In some cases, non-stem progeny cells may also undergo symmetric divisions (e.g., transient amplifying cells). If this occurred at a high rate, SPr and CD assays based on cycling cell markers like cyclin A would be inadequate for detecting asymmetrically self-renewing stem cells. In this setting, biomarkers that distinguished stem cells from differentiated cells with a different basis than cycling status would be required. Whether Lgr5 has such capability is not yet clear, because essentially all asymmetric self-renewal divisions for HF-SC strains yield a non-cycling, non-stem sister under the present culture conditions. Because non-random segregation does not suffer from these uncertainties and is strictly associated with asymmetric self-renewal, when determined by pulse-chase label retention-CD, it provides a more specific biomarker for DSCs. By this conservative measure, we estimate that at least 44% of cycling cells in SACK-derived hair follicle cell cultures are HF-SCs.
Confirmation of the epidermal lineage of HF-SCs
We evaluated the expression of three independent markers of differentiated epidermal lineage cells, keratin 5, keratin 10 and filaggrin, as well as Lgr5, in cultures of HF-SCs. Conditions of active proliferation at higher cell density (compared to SPr and CD assays) were compared to conditions of serum reduction, which is well-known to induce epithelial cells to withdraw from the cell cycle and initiate differentiation programs. Xn-free medium was used for both conditions to favor production of non-stem progeny cells (see Methods). Keratin 5 is a basal layer marker, keratin 10 is a spinous layer marker and filaggrin marks the granular layer28.
As noted earlier, during active growth, essentially all cells expressed Lgr5 (Fig. 5, UNDIFF, LGR5/DAPI; Supplementary Information, Figs. S1 and S2). In respective 3C5 and 5B8 cultures, 34 ± 5.6% (n = 3) and 40 ± 9.2% (n = 3), of the cells had nuclear Lgr5 expression. Serum reduction caused a marked reduction in Lgr5 expression, but a subpopulation of positive cells with reduced expression was still detectable (Fig. 5, DIFF, LGR5/DAPI). The character of these cells was not been evaluated further at this time. They could either indicate inefficient differentiation or persistent undifferentiated HF-SCs, which are predicted for asymmetric self-renewal29.
Filaggrin fluorescence was detectable at low levels in cells of actively cycling cultures of both HF-SC strains (Figs. 5A and 5B, UNDIFF, Filaggrin/DAPI), but keratin 5 and keratin 10 were only detectable at very low levels in rare cells in actively cycling 5B8 cultures (Fig. 5B, UNDIFF, K5/DAPI, K10/DAPI; Also compare Supplementary Information Figs. S1 and S2, UNDIFF). After serum reduction (Fig. 5, DIFF), both HF-SC strains displayed a large number of cells with high expression of each epidermal marker. Cells expressing markers for two different epidermal layers were also detectable (Fig. 5, DIFF, K10/Filaggrin/DAPI; K10/K5/DAPI; arrows).
HF-SCs spontaneously produce differentiating progeny cells
For success in transplantation applications, DSCs must retain their ability to resume asymmetric renewal of functional, tissue-specific, differentiated tissue cells. Since the cell kinetics and chromosome segregation effects of SACK agents are readily reversed upon removal of the agents, the efficiency of differentiation of non-stem cell progeny can be evaluated after in vivo transplantation or, in vitro, by applying culture conditions that promote differentiation along specific cellular lineages. Under SACK-free conditions, HF-SC cultures spontaneously produce differentiating progeny in a cell density-related manner (Fig. 6). This property was most evident in cell colonies that showed heterogeneity in cell morphology and function. At the periphery of colonies, where cell division is maintained at a higher rate, cells retained an undifferentiated appearance (Figs. 6A and 6C). However, further inward from the edge, where cell division rates decrease, cells with a differentiated morphology appeared that stained more intensely with the vital dye crystal violet (Fig. 6B). Cells in highly dense regions produced light-reflective vesicles (Fig. 6D) that stained with oil-red (Fig. 6E), indicative of sebocyte differentiation with production of sebum30. In addition, transmission electron microscopy analysis of cells maintained under these conditions revealed a high frequency of fully formed primary cilia, a differentiated cell feature that is unusual for many types of cultured cells (Fig. 6F)31.

Spontaneous differentiation properties of SACK-expanded hair follicle stem cells.
(A–C) Phase micrographs of crystal violet-stained colonies grown under SACK agent-free conditions. (A) At colony edges, where there is active cell division, few differentiated cells are present (40X magnification). (B) In the center of colonies, where cells are growth-arrested, a high degree of differentiation is evident by dark purple cells, which contain light-reflective vesicles (40X). (C) Higher magnification micrograph of cells at the periphery of a differentiating colony (100X). (D) Phase micrograph of unstained light reflective vesicles (arrows; 100X). (E) Oil red-positive vesicles (arrows; 100X). (F) Transmission electron micrograph showing example of a primary cilium (arrow).
Discussion
Here, we add another DSC type to the list of those with non-random sister chromatid segregation. Knowledge of how widespread this unique function is among diverse DSC types will better inform its role in angiosperm (flowering plants) and mammalian evolution. In addition to the many accounts of immortal DNA strand co-segregation in both cultured cells20,27,32,33,34,35,36 and stem cell compartments in tissues37,38,39, there are reports of failure to detect it in mouse epidermis40, the mouse hematopoietic system41, mouse neocortical precursor cells42 and mouse hair follicles14,15. However, only one of these studies42 considered the essential co-dependency of immortal DNA stand co-segregation on asymmetric self-renewal as a possible explanation for their observations. If the examined cells were not undergoing asymmetric self-renewal, both the original immortal DNA strand concept24 and our studies (herein and refs. 20, 26 and 27) predict that non-random segregation would not be observed.
Of course, there might be other causes for the apparent contradiction as well, including possible disruption of co-segregation by artificial molecular-genetic assays14,15; the use of toxins40; failure to evaluate toxicity in dual-labeling experiments that combine chloro-deoxyuridine and iodo-deoxyuridine14,41; misinterpretation of cell kinetics data in dual-label experiments14,41; and dismissing rare outlier cells that did not conform to random chromosome segregation15,41. For example, in populations enriched for mouse hematopoietic stem cells, Kiel and others41, in fact, detected a significant number of cells with labeling kinetics consistent with non-random chromosome segregation. However, because of an erroneous statistical analysis, they dismissed these cells. Their statistical analysis improperly treated dependent cellular events as independent ones, leading to their misinterpretation of their data.
Lgr5-positive, bone fide HF-SCs have been identified in the mouse hair follicle8. Finding that cells clonally expanded from mouse hair follicles by regulation of asymmetric self-renewal display both Lgr5 expression and non-random segregation connects Lgr5 to the tight association between DSC asymmetric self-renewal and non-random segregation. However, the connection involves more than simple co-expression. Specifically, this connection is asymmetric localization of nuclear Lgr5 expression to the stem cell sister, which maintains the immortal DNA strands after asymmetric self-renewal divisions. In HF-SC cultures, nuclear Lgr5 is expressed in both sisters during symmetric self-renewal divisions, which are associated with random chromosome segregation.
The observed co-asymmetric localization of nuclear Lgr5 with cyclin A is not a chance occurrence. Fig. 1 shows all the possible observable dual nuclear staining patterns for cyclin A and Lgr5. The respective relative frequencies of each diagrammed pattern from a-j expected based on chance occurrence are 1∶2∶1∶2∶1∶2∶2∶2∶2∶1. For example, there is only one way that both sisters can be negative for both markers (a); but there are two ways by which they could be symmetrically negative for cyclin A and asymmetric for nuclear Lgr5 detection (b). Based on this analysis, if the Co-ASYM (f) and Co-SYM (j) patterns occurred by chance, their expected frequencies would be 12.5% (2/16 x 100%) and 6.25% (1/16 x 100%). The observed frequencies in the experiment described in Fig. 1A were 51%, 39% and 24%, 30%, respectively. In the worst case (i.e., 24%), the probability of this occurrence by chance is p = 0.013 (Fisher's exact two-tailed test). The fact that the frequencies are altered by Xn supplementation is another indication that their occurrence is not due simply to chance.
If Lgr5 is also expressed in committed progenitor cells, as has now been considered43, then its expression per se may over-estimate the number of DSCs in tissues in vivo. As such, future investigations of DSC location and number in vivo might benefit from the use of cytochalasin D to identify binucleated cells with co-asymmetric localization of nuclear Lgr5 and cyclin A expression. Because it will specifically identify cells undergoing asymmetric self-renewal and non-random segregation, this approach might also identify tissue DSCs with a high degree of specificity in many different tissues. This issue may be particularly relevant to a recent report of the failure to detect non-random chromosome segregation in mouse small intestinal crypt cells designated to be stem cells based on their Lgr5 expression44. Although other technical problems might also be at fault (e.g., labeling toxicity, inadequate chase period), to the extent that the identified cells fail to undergo asymmetric self-renewal (because they are in large part committed progenitor cells as has been suggested44), the report's conclusions will accordingly be erroneous.
The presented serum reduction studies clearly establish the epidermal lineage of the HF-SCs. These studies also show that these DSCs are multipotent, possessing the ability to produce progeny cells that reside in three morphologically and functionally distinct layers of the epidermis. The production of cells co-expressing markers of different epidermal layers is a further demonstration of the ability of the cells to project a diverse epidermal development program inclusive of committed progenitors and terminal cells. Finally, the persistence of Lgr5-positive cells under conditions that generally induce differentiation fits with the concept that an essential aspect of DSCs is their ability to remain undifferentiated when their progeny are committed29.
The ability of the SACK-expanded mouse HF-SCs to spontaneously produce differentiating progeny cells is an important predictor for efficacy in transplantation applications. Without specific in vitro instruction, the cells are able to produce progeny cells engendered with complex morphology and function (e.g., sebum production). In a more complex in vivo environment, these attributes could result in production of de novo hair follicles and/or other skin appendages that are lacking in manufactured human skin grafts. Of course, human HF-SCs will be required. We have shown that SACK-based methods for expansion of rat hepatic stem cells12 were readily adapted for derivation of human hepatic stem cells45. Therefore, we are optimistic that the same can be achieved for human HF-SCs.
An important consideration for future clinical development of SACK-expanded human HF-SCs would be their genetic stability. Thus far, we have not systematically investigated this property of SACK-expanded mouse HF-SCs. However, the observed proliferation and differentiation properties of the cells do not indicate transformed cell properties (e.g., anchorage independent growth; growth factor independence). By design, the SACK method selects against rare variant cells that have acquired mutations that promote transformed cell proliferation (e.g., p53 mutations). This discrimination is accomplished by the method's prime effect of keeping cultured DSCs in a exponential growth state (i.e., symmetric self-renewal) that safeguards against mutated cells achieving the growth advantage required to overtake normal DSCs12,13.
Methods
Hair follicle stem cell strains
Mouse whisker follicle stem cell strains were clonally derived by the SACK method and maintained at sub-confluent cell densities in Xn-supplemented medium as described9. Specifically, the routine culture medium included: 3 parts Dulbecco's Modified Eagle Medium and 1 part Ham's F12 (Invitrogen [Gibco], Inc., Carlsbad, CA); and was supplemented with 10% dialyzed fetal bovine serum (dFBS), penicillin and streptomycin per the supplier's recommendation (Invitrogen [Gibco], Inc.), 10 ng/ml human epidermal growth factor (Invitrogen, Inc.) and 400 μM xanthine (Xn; Sigma Chemical Co., St. Louis, MO). All surgical procedures were performed according to humane protocols approved by the Animal Care and Use Committee at the Massachusetts Institute of Technology, where the HF-SC expansion studies were conducted.
Self-renewal pattern assays
SPr and CD ISIF assays with specific anti-cyclin A and anti-Lgr5 antibodies were performed as previously described11,21,26. Mouse anti-mouse cyclin A monoclonal antibody (Abcam, Inc., Cambridge, UK; cat# ab38) was diluted 1∶200 in PBS containing 2% goat serum. Alexa Fluor® 568-conjugated goat anti-mouse IgG (Invitrogen, Inc., Carlsbad, CA) diluted 1∶300 in blocking solution was used as the secondary antibody. Cells stained for cyclin A detection were subsequently stained in the same manner with goat anti-Lgr5 polyclonal antibody (Santa Cruz, Biotechnology, Inc., Santa Cruz, CA; catalogue# sc-68580) diluted 1∶50, followed by Alexa Fluor® 488-conjugated donkey anti-goat IgG (Invitrogen, Inc., Carlsbad, CA) diluted 1∶300. Control analyses that omitted anti-cyclin A and/or anti-Lgr5 antibodies were evaluated to ensure that all detected fluorescence required specific antibodies. Confirmation of the specificity of the Lgr5 antibodies has been described previously11 (See also Fig. S3).
Non-random segregation analyses
Label retention-CD analyses were performed as described previously20,26,27 with minor modifications. For analyses without a chase, cells were labeled with 20 μM BrdU. For analyses with a chase, cells were labeled with 5 μM BrdU and chased with 50 μM thymidine. For CD treatment, cells were cultured for 14 hours in medium supplemented to 2 μM CD. Thereafter, they were fixed with 70% ice-cold ethanol for 30 minutes. All subsequent incubations and washes were performed at room temperature. After washing with PBS, the cells were treated with 2N HCl for 10 minutes. After washing again with PBS, they were incubated in blocking solution (PBS, 0.5% BSA, 0.05% Tween-20) for 10 minutes. The blocked cells were incubated for 3 hours with anti-BrdU antibodies (Chemicon 3424) diluted 1∶100 in blocking solution. After washing with blocking solution, incubation with goat anti-mouse IgG-488 nm antibodies, diluted 1∶300 in blocking solution, was maintained for 45 minutes. The cells were then washed sequentially with blocking solution and PBS before mounting with 4′-6-diamido-2-phenylindole (DAPI)-containing VectaShield® mounting media (Vector Laboratories, Inc., Burlingame, CA). Epifluorescence images were captured with a Leica DMR microscope and Leica DC300F digital camera system. The mean pixel intensity of BrdU-labeled nuclei was quantified using NIH Image J software.
Induced cell differentiation analyses
For the active proliferation state, 10,000 routinely maintained cells were plated in each well of a standard 2-well chamber culture slide in routine culture medium. Sixteen to 24 hours later, the medium was replaced with Xn-free medium and culture was continued for an additional 48 hours before fixation and ISIF analyses.
For cell differentiation, cultures slides as prepared above were cultured for an additional 5 days until slide cell monolayers were 1-day post confluency. The monolayers were then rinsed and replaced with Xn-free medium supplemented with only 0.5% dFBS. After 48 hours, the cell cultures were replaced with the same medium. 48 hours later (corresponding to 96 hours of serum reduction), the differentiated cells were fixed and examined by ISIF. ISIF was performed with specific anti-keratin 5 (AbCam, Inc., Cambridge, UK; rabbit monoclonal, cat# ab52635; 1∶200 dilution); anti-keratin 10 (AbCam, Inc.; mouse monoclonal, cat# ab9025; 1∶50 dilution) and anti-filaggrin (Covance; rabbit polyclonal, cat# FRB-417P; 1∶1000 dilution) antibodies as described earlier for self-renewal pattern assays. All secondary antibodies (Invitrogen, Inc., Carlsbad, CA) were diluted 1∶300.
Transmission electron microscopy
TEM analyses were performed as described46 with the modifications of embedding in epon-812 resin and examining 70 nm sections.
Statistical analyses
Fisher's exact tests were performed at http://www.danielsoper.com/statcalc/calc29.aspx. Statistical parameters for anti-BrdU fluorescence intensity histograms were determined using the NIH Image J program. The Kolmogorov-Smirnov (K-S) test for distinct distributions was performed at http://www.physics.csbsju.edu/stats/KS-test.n.plot_form.html.
References
Paus, R. & Cotsarelis, G. The biology of hair follicles. New Eng. J. Med. 341, 491–497 (1999).
Fuchs, E. Skin stem cells: rising to the surface. J. Cell Biol. 180, 273–284 (2008).
Taylor, G., Lehrer, M. S., Jensen, P. J., Sun, T.-T. and Lavker, R. M. Involvement of follicular stem cells in forming not only the follicle but also the epidermis. Cell 102, 451–461 (2000).
Oshima, H., Rochat, A., Kedzia, C., Kobayashi, K., & Barrandon, Y. Morphogenesis and renewal of hair follicles from adult multipotent stem cells. Cell 104, 233–245 (2001).
Celso. et al. Characterization of bipotential epidermal progenitors derived from human sebaceous gland: contrasting roles of c-myc and β-catenin. .Stem Cells 26, 1241–1252 (2008).
Tumbar, T., et al. Defining the epithelial stem cell niche in skin. Science 303, 359–363 (2004).
Morris. et al. Capturing and profiling adult hair follicle stem cells. .Nature Biotech. 22, 411–417 (2004).
Jaks. et al. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. .Nature Gen. 40, 1291–1299 (2008).
Sherley, J. L. & King, J. Methods for ex vivo propagation of somatic hair follicle stem cells. .U.S. Patent No. 7,655,465 B2 (2010).
Sherley, J. L. A new mechanism for aging: chemical “age spots” in immortal DNA strands in distributed stem cells. Breast Disease 29, 37–46 (2008).
Noh, M., Smith, J. L., Huh, Y. H. & Sherley, J. L. A resource for discovering specific and universal biomarkers for distributed stem cells. PLoS ONE 6(7): e22077. 10.1371/journal.pone.0022077 (2011).
Lee, H.-S., et al. Clonal expansion of adult rat liver epithelial stem cells by suppression of asymmetric cell kinetics (SACK). Biotech. & Bioeng. 83, 760–771 (2003).
Paré, J.-F. & Sherley, J. L. in Current Topics in Developmental Biology vol. 73 (ed. Schatten, G) 141–171 (Elsevier, 2006).
Sotiropoulou, P. A., Candi, A. & Blanpain, C. The majority of multipotent epidermal stem cells do not protect their genome by asymmetrical chromosome segregation. Stem Cells 11, 2964–2973 (2008).
Waghmare, S. K. et al. Quantitative proliferation dynamics and random chromosome segregation of hair follicle stem cells. EMBO J. 27, 1309–1320 (2008).
Todaro, G. J. & Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17, 299–313 (1963).
Becker, L., Huang, Q. & Mashimo, H. Immunostaining of Lgr5, an intestinal stem cell marker, in normal and premalignant human gastrointestinal tissue. TheScientificWorld J. 8, 1168–1176 (2008).
Berglund, L. et al. A gene-centric human protein atlas for expression profiles based on antibodies. Mol. Cell Proteomics 10, 2019–2027 (2008).
Boivin, B., Chevalier, D., Villeneuve, L. R., Rousseau, E. & Allen, B. G. Functional endothelin receptors are present on nuclei in cardiac ventricular myocytes. J. Biol. Chem. 278, 29153–29163 (2003).
Merok, J. R., Lansita, J. A., Tunstead, J. R. & Sherley, J. L. Co-segregation of chromosomes containing immortal DNA strands in cells that cycle with asymmetric stem cell kinetics. Cancer Res. 62, 6791–6795 (2002).
Taghizadeh, R. et al. CXCR6, a newly defined biomarker of tissue-specific stem cell asymmetric self-renewal, identifies more aggressive human melanoma cancer stem cells. PLoS ONE 10.1371/journal.pone.0015183 (2010).
Sherley, J. L. in Stem Cell Repair and Regeneration. (eds Habib N. A., Gordon M. Y., Levicar N., Jiao L., Thomas-Black G.) 21–28 (Imperial College Press 2005).
Rambhatla, L. et al. Cellular senescence: ex vivo p53-dependent asymmetric cell kinetics. J. Biomed. Biotech. 1, 27–36 (2001).
Cairns, J. Mutation selection and the natural history of cancer. Nature 255, 197–200 (1975).
Charville, G. W. & Rando, T. A. Stem cell aging and non-random chromosome segregation. Phil. Trans. R. Soc. B 366, 85–93 (2011).
Huh, Y. H. & Sherley, J. L. Molecular cloaking of H2A.Z on mortal DNA chromosomes during non-random segregation. Stem Cells 29, 1620–1627. 10.1002/stem.707 (2011).
Rambhatla, L., Ram-Mohan, S., Cheng, J. J., & Sherley, J. L. Immortal DNA strand co-segregation requires p53/IMPDH-dependent asymmetric self-renewal associated with adult stem cells. Cancer Research 65, 3155–3161 (2005).
Blanpain, C. & Fuchs, E. Epidermal stem cells of the skin. Annu. Rev. Cell Dev. Biol. 22, 339–373 (2006).
Sherley, J. L. Stem cell differentiation: What does it mean? Proc. Second Joint EMBS-BMES Conf. 1, 741–742 (2002).
Xia . et al. Isolation of human sebaceous glands and cultivation of sebaceous gland-derived cells in an in vitro model. .J. Invest. Dermatol. 93, 315–321 (1989).
Alieva, I. B. & Vorojev, I. A. Vertebrate primary cilia: a sensory part of centrosomal complex in tissue cells, but a “sleepy beauty,” in cultured cells. Cell Biol. Internat. 28, 139–150.
Karpowicz, P., et al. Support for the immortal strand hypothesis: neural stem cells partition DNA asymmetrically in vitro. J. Cell Biol. 170, 721–732 (2005).
Armakolas, A. & Klar, A. J. S. Cell type regulates selective segregation of mouse chromosome 7 DNA strands in mitosis. Science 311, 1146–1149 (2006).
Conboy, M. J., Karasov, A. O., & Rando, T. A. High incidence of non-random template strand segregation and asymmetric fate determination in dividing stem cells and their progeny. PLOS 5, 1120–1126 (2007).
Pine, S. R., Ryan, B. M., Varticovski, L., Robles, A. I., & Harris, C. C. Microenvironmental modulation of asymmetric cell division in human lung cancer cells. Proc. Natl. Acad. Sci. USA 107, 2195–2200 (2010).
Hari, D. et al. Isolation of live label-retaining cells and cells undergoing asymmetric cell division via nonrandom chromosomal cosegregation from human cancers. Stem Cells Devel. 10.1089/scd.2010.0455 (2011).
Potten, C. S., Owen, G., & Booth, D. Intestinal stem cells protect their genomes by selective segregation of template DNA strands. J. Cell Sci. 115, 2381–2388 (2002).
Smith, G. Label-retaining epithelial cells in mouse mammary gland divide asymmetrically and retain their template DNA strands. Development 132, 681–687 (2005).
Capuco, A. V. Identification of putative bovine mammary epithelial stem cells by their retention of labeled DNA strands. Exp. Biol. Med. 232, 1381–1390 (2007).
Kuroki, T. & Murakami, Y. Random segregation of DNA strands in epidermal basal cells. Jpn. J. Cancer Res. 80, 637–642 (1989).
Kiel, M. J. et al. Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature 449, 238–242 (2007).
Fei, J.-F. & Huttner, W. B. Nonselective sister chromatid segregation in mouse embryonic neocortical precursor cells. Cerebral Cortex 19, 149–154 (2009).
Snippert, H. J. et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143, 134–144 (2010).
Escobar, M. et al. Intestinal stem cells do not protect their genome by asymmetric chromosome segregation. Nature Comm. 10.1038/ncomms1260 (2011).
Sherley, J. L. & Panchalingam, K. Methods for ex vivo propagation of adult hepatic stem cells. U.S. Patent No. 7,824,912 (2010).
Park, H. et al. Effects of electrical stimulation in C2C12 muscle constructs. J. Tissue Eng. Regen. Med. 2, 279–287.10.1002/term.93 (2008).
Acknowledgements
We thank N. Watson (W. M. Keck Foundation Biological Imaging Facility at the Whitehead Institute, Cambridge, MA) for technical assistance with transmission electron microscopy analyses. This work was supported by National Science Foundation grant #984332 and NIH-NIGMS Director's Pioneer Award #5DP1OD000805. J.C. was supported by NIH-NIGMS Director's Pioneer Award Supplement #3DP1OD000805-03S1 issued under the American Recovery and Reinvestment Act of 2009.
Author information
Authors and Affiliations
Contributions
J.L.S. led the scientific development of the research and worked with Y.H.H. to design, analyze and interpret cell kinetics and chromosome segregation experiments. Y.H.H. performed and quantified all SPr and CD assays and developed statistical analyses for them. Y.H.H. and J.C performed and quantified scheme-I and scheme-II label retention-CD analyses and performed statistical analyses. Y.H.H. performed and quantified scheme-III label retention-CD analyses and performed statistical analyses. J.K. derived the HF-SC strains and conducted analyses of their differentiation properties. J.L.S. and Y.H.H. assembled and wrote the manuscript. All authors reviewed the final manuscript and participated in its editing led by J.L.S.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Huh, Y., King, J., Cohen, J. et al. SACK-Expanded Hair Follicle Stem Cells Display Asymmetric Nuclear Lgr5 Expression With Non-Random Sister Chromatid Segregation. Sci Rep 1, 176 (2011). https://doi.org/10.1038/srep00176
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00176
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
