
Abstract
In mammals, the livers regenerate after chemical injury or resection of hepatic lobe by hepatectomy. How liver regeneration is initiated after mass loss remains to be defined. Here we report that following liver injury, activated platelets deploy SDF-1 and VEGF-A to stimulate CXCR7+ liver sinusoidal endothelial cell (LSEC) and VEGFR1+ myeloid cell, orchestrating hepatic regeneration. After carbon tetrachloride injection or hepatectomy, platelets and CD11b+VEGFR1+ myeloid cells were recruited to LSECs, and liver regeneration in both models was impaired in thrombopoietin-deficient (Thpo−/−) mice repressing production of circulating platelets. This impeded regeneration phenotype was recapitulated in mice with either conditional ablation of Cxcr7 in LSEC (Cxcr7iΔ/iΔ) or Vegfr1 in myeloid cell (Vegfr1lysM/lysM). Both Vegfr1lysM/lysM and Cxcr7iΔ/iΔ mice exhibited suppressed expression of hepatocyte growth factor and Wnt2, two crucial trophogenic angiocrine factors instigating hepatocyte propagation. Of note, administration of recombinant thrombopoietin restored the prohibited liver regeneration in the tested genetic models. As such, our data suggest that platelets and myeloid cells jointly activate the vascular niche to produce pro-regenerative endothelial paracrine/angiocrine factors. Modulating this ‘hematopoietic–vascular niche’ might help to develop regenerative therapy strategy for hepatic disorders.
Similar content being viewed by others


Introduction
In mammals, the liver can undergo regeneration after either chemical injury or surgical resection of liver mass, a partial hepatectomy (PH) procedure.1–10 This regeneration process is governed by dynamic interplay between parenchymal hepatocytes and non-parenchymal cells (NPCs),1,2,4,11–14 including stellate cells,5,10 liver sinusoidal endothelial cells (LSECs),15–23 biliary epithelial cells24 and hematopoietic cells.25–29 As such, defining the multicellular interaction orchestrating liver regeneration might help to design therapeutic interventions for hepatic diseases.
LSECs lining hepatic sinusoidal vasculature are essential in choreographing liver organogenesis.3,15,30–35 During liver development and regeneration, LSECs produce endothelial-expressed paracrine (angiocrine)36–41 cues to regulate synchronized propagation of hepatocyte12,40 and resolve fibrosis.35–39 However, how angiocrine factor production from LSECs is triggered by liver injury remains to be defined.
Following injury, platelets are recruited to the damaged vasculature. In addition to their hemostatic function, platelets are circulating reservoirs of growth factors.26,42–47 We have previously shown that tissue injury triggers secretion of vascular endothelial growth factor-A (VEGF-A) and stromal-derived factor-1 (SDF-1) from platelets.45 VEGF-A binds to VEGF receptors 1 and 2 on vascular endothelial cells (ECs)48,49 and hematopoietic cells50–54 to stimulate production of growth factors. SDF-1 also activates its EC-specific cognate receptor CXCR7 to modulate vascular patterning and angiocrine factor production.55–57 The unique function of platelets as major reservoir for VEGF-A and SDF-1 led us to hypothesize that after liver injury, platelets release bioavailable VEGF-A and SDF-1 to prime LSECs and hematopoietic cells, enabling a hematopoietic–vascular niche orchestrating hepatic regeneration.
Materials and methods
Animals
C57BL/6J and LysM-Cre mice were obtained from Jackson laboratory. The Chd5(PAC)CreERT2 mice expressing tamoxifen-responsive CreERT2 driven by EC-specific VE-cadherin promoter58,59 were provided by Dr Ralf Adams. Thrombopoietin (TPO)-deficient (Thpo−/−) mice60 were kindly offered by Dr Frederic J de Sauvage (Genentech, Inc., San Francisco, CA, USA). Mice harboring loxP site-flanked exon 3 of Cxcr7 (Cxcr7LoxP/LoxP) were kindly provided by ChemoCentryx, Inc. (Mountain View, CA, USA). Floxed Vegfr1 mice were kindly provided by Dr Guo-Hua Fong. Rosa-CreERT2 animals expressing tamoxifen-responsive inducible Cre were described previously.15,39 The Chd5(PAC)CreERT2 mouse line was crossed with floxed Cxcr7 mice to generate Cxcr7iΔEC/iΔEC mice and control Cxcr7iΔEC/+ mice after treatment of tamoxifen at a dose of 250 mg kg−1 for 6 days, and interrupted for 3 days after the third dose. Mice were rested for at least 20 days after the last injection. Cxcr7LoxP/LoxP mice were also crossed with Rosa-CreERT2 mice to generate Cre+Cxcr7loxP/loxP mice, resulting Cxcr7 deletion in adult mice (Cxcr7iΔ/iΔ). Floxed Vegfr1 mice were bred with LysM-driven Cre (Jackson, Bar Harbor, ME, USA) to generate mice lacking Vegfr1 in myeloid cells (Vegfr1lyzM/lyzM). Deletion of target genes was corroborated by quantitative PCR. Investigators who performed mouse experiments and who analyzed the pattern of cell distribution were randomly assigned with samples, and they were blinded to the genotype of the animals or samples from various groups. All animal experiments were carried out following the guidelines of Institutional Animal Care and Use Committee at Weill Cornell Medicine.
Mouse liver regeneration and repair models
In all, 70% PH model was used as previously described.15 In brief, three most anterior lobes were resected without injuring the blood supply to the caudate and the right lobes after mice were anesthetized by 100 mg kg−1 intraperitoneal (i.p.) ketamine and 10 mg kg−1 xylazine followed by midline laparotomy. To induce liver injury, single injection of carbon tetrachloride (Sigma-Aldrich, St Louis, MO, USA) in oil at a concentration of 40% (0.64 mg ml−1) was injected to mice at a dose of 1.6 mg kg−1 to induce acute liver injury.35,61 Hepatic regeneration was assessed based on the following criteria: liver lobe weight, hepatocyte proliferation, alanine aminotransferase level and histology using hematoxylin and eosin staining. Six- to ten-week-old mice were utilized and compared.
Stimulation of thrombopoiesis
To stimulate thrombopoiesis, recombinant TPO, VEGF-A and/or SDF-1 (PeproTech, Rocky Hill, NJ, USA) was injected into Thpo−/− or WT mice i.p. at a dose of 25 μg kg−1 on a daily basis 10 days before PH or CCL4 injury and afterwards. Vehicle for individual cytokines was also injected as a control group. The degree of hepatic regeneration was evaluated with control group, including alteration in circulating platelets and parameters of hepatogenesis.
Immunostaining and morphometric analysis
Tissues were collected and cryopreserved as described15,35 for morphometric analysis.15,36 Mouse liver was fixed with 4% paraformaldehyde and cryopreserved in optimal cutting temperature compound. For immunofluorescent microscopy, the liver sections (10 μm) were blocked (5% donkey serum/0.3% Triton X-100) and incubated with anti-VE-cadherin (2 μg ml−1, R&D Systems, NE Minneapolis, MN, USA) and VEGFR3 (Eli Lilly and Co., New York, NY, USA) antibodies to identify LSECs.15,45 To reveal platelet activation, antibodies against CD41 and P-selectin (BD Biosciences, San Jose, CA, USA) were used, respectively. CD11b (BD Biosciences) and VEGFR1 antibodies (Eli Lilly and Co.) were used to detect myeloid cells.15,45,62 Id1 staining was performed with rabbit anti-human Id1 antibody (Biocheck, Foster City, CA, USA). After incubation in fluorophore-conjugated secondary antibodies (2.5 μg ml−1, Jackson ImmunoResearch, West Grove, PA, USA), sections were counterstained with 4,6-diamidino-2-phenylindole (Invitrogen, Carlsbad, CA, USA). No appreciable staining was observed in isotype IgG controls.
Cell proliferation in vivo was measured by 5-Bromo-2′-deoxyuridine (BrdU) uptake. Single dose of BrdU (Sigma, St Louis, MO, USA) at 50 mg kg−1 was i.p. injected to mice 1 h before killing. Liver lobes were removed, weighed and slice of tissues were incubated with 1 m HCl at room temperature for 1 h, neutralized with 10 mm Tris (pH 8.5) for 15 min. After incubation with secondary antibody (Jackson ImmunoResearch), cells incorporated with BrdU were identified as proliferating hepatocytes.
Image acquisition and analysis
Histology analysis of liver sections was captured with Olympus BX51 microscope (Olympus America, Center Valley, PA, USA), and fluorescent images were recorded on AxioVert LSM710 (Carl-Zeiss, Thornwood, NY, USA) confocal microscope. Fluorescent signals in slide were independently evaluated by two investigators from randomly selected fields of view. Parameters from each individual animal were measured and averaged.
Isolation and cultivation of LSECs
Mouse LSECs were isolated by previously described two-step collagenase perfusion technique with modifications.15 In brief, the liver was perfused with Liver Perfusion Medium (Invitrogen), and dissociated by Liver Digest Medium (Invitrogen). The NPCs were fractionated with Percoll (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) gradient centrifugation with 75% stock Percoll solution and 35% stock Percoll solution. LSEC faction was isolated by mouse LSEC-binding magnetic beads (Miltenyi, Auburn, CA, USA) and Dynabeads Magnetic Beads conjugated with anti-mouse CD31 antibody (MEC13.3, BD Biosciences). Expression of Id1, CXCR7, HGF and Wnt2 messenger RNA was determined. Primary human LSECs were procured from ScienCell Research Laboratories (catalog no. 5000, Carlsbad, CA, USA). Expression of factor VIII was validated by immunostaining. Akt-LSECs were derived from isolated LSECs that were transfected with the pCCL. PGK lentiviral vector with mouse constitutively active Akt1 (myristoylated Akt: myrAkt).63 After starving in serum-free medium, 500 000 LSECs were seeded and stimulated with 10 ng ml−1 SDF-1. LSECs were also treated with 30 μm Wortmannin (Sigma-Aldrich).
Flow cytometry analysis
Flow cytometry analysis of platelets and LSECs on isolated liver NPCs as previously described.15,45,62 In brief, retrieved livers from killed animals were minced, digested in liver digestion medium (Invitrogen), and filtered through a 30-μm strainer. Single-cell suspensions were preincubated with Fc block (CD16/CD32; BD Biosciences) and then incubated with primary antibodies recognizing mouse LSECs and hematopoietic cells, as well as rat IgG2aκ and IgG2aβ isotype control. Primary antibodies were directly conjugated to different Alexa Fluor dyes or Quantum Dots (BD Biosciences) using antibody labeling kits (Invitrogen). Labeled cell populations were measured by a LSRII flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Compensation for multivariate experiments was carried out with FACS Diva software (Becton Dickinson Immunocytometry Systems, Franklin Lakes, NJ, USA).
Gene expression analysis by real-time PCR
Total RNA was extracted using RNeasy kit (Qiagen, Germantown, MD, USA). After isolation, 500 ng of total RNA was transcribed into complementary DNA by using the superscript reverse transcriptase kit (Invitrogen). The detection of complementary DNA expression for the specific genes was performed by using the SYBR Green quantitative PCR (Applied Biosystems, Foster City, CA, USA). To selectively knockdown Cxcr7 in LSECs, shRNA lentiviruses were generated by cotransfecting 15 μg of shuttle lentiviral vector, 3 μg of pENV/VSV-G, 5 μg of pRRE and 2.5 μg of pRSV-REV in 293T cells.35 Viral supernatants were concentrated by ultracentrifugation and used to transduce LSECs.
Statistical analysis
All data were presented as the mean±s.e.m. Comparisons between different groups were made using one-way analysis of variance. Statistical significance was set at P<0.05. Each experiment was at least three times.
Results
We first examined the localization of platelet-derived VEGF-A and SDF-1 after carbon tetrachloride (CCl4)-induced liver injury. VEGF-A and SDF-1 were co-stained with platelet surface marker CD41 in the liver 24 h after CCl4 i.p. injection (Figures 1a and b). Compared to sham mice, CCl4 injection caused significant deposition of CD41+ platelets on VEGFR3+ LSECs, with the majority of them stained for VEGF-A and SDF-1 (Figure 1c). Flow cytometric analysis of hepatic NPCs showed that CD41+ platelets constituted 24% of NPCs in CCl4-injured but not sham mice (Figure 1d), and platelet activation marker, P-selectin, was presented on the surface of 73% of CD41+ platelets in the damaged liver. Thus, CCl4 injury caused recruitment and local activation of platelets secreting VEGF-A and SDF-1, which might activate hematopoietic and LSECs via VEGF-A and SDF-1 receptors.

Hepatotoxic injury recruits CD41+ platelets carrying VEGF-A and SDF-1 to liver vasculature. (a, b) After intraperitoneal (i.p.) injection of hepatotoxic agent carbon tetrachloride (CCl4), CD41+ platelets expressing VEGF-A and SDF-1 were recruited to VEGFR3+ liver sinusoidal endothelial cells (LSECs). Liver sections were stained with antibodies against platelet marker CD41, VEGF-A (a), SDF-1 (b) and LSEC-specific marker VEGFR3. After CCl4 injury, but not administration of PBS (control), CD41+SDF-1+VEGF-A+ platelets were associated with VEGFR3+ LSECs; scale bar, 50 μm. (c) Quantification of CD41+SDF-1+VEGF-A+ platelets associated with VEGFR3+ LSECs. N=5–7 mice per group. (d) Flow cytometry analysis of platelet accumulation in the CCl4-injured and control livers. Activation of CD41+ platelets was evidenced by surface expression of activation marker P-selectin.
To test the contribution of platelets in protecting against liver injury, we examined mice deficient of TPO (Thpo−/−) after CCl4 injection. Platelet number is decreased in Thpo−/− mice by 95% as compared to wild-type (WT) mice.60,64 Hepatocyte proliferation after CCl4 injection was significantly reduced in Thpo−/− mice than that of WT control group (Figures 2a–c). Meanwhile, hepatic injury was markedly increased, as indicated by elevation of plasma alanine aminotransferase activity (Figure 2d). Of note, the impaired hepatocyte proliferation and enhanced hepatic injury in Thpo−/− mice were rescued by injection of VEGF-A and/or SDF-1 (Figures 2a–d). Therefore, activated platelets recruited to the injured vascular bed supply SDF-1 and VEGF-A to stimulate hepatic repair.

Platelet-deficient mice exhibit impaired liver regeneration after CCl4 injury. (a, b) After CCl4 injury, proliferation (BrdU+) of hepatocytes was prohibited in thrombopoietin knockout mice (Thpo−/−) lacking platelets. Hepatocyte proliferation was enhanced in Thpo−/− mice by injection of VEGF-A and SDF-1. N=6–8 mice per group. (c) Severe centrilobular damage in the liver of Thpo−/− mice after CCl4 injury, as indicated by scattered cell debris in Thpo−/− mice relative to mild centrilobular necrosis in WT mice. VEGF-A and SDF-1 injection ameliorated the injury in Thpo−/− mice. Scale bar, 50 μm. (d) Increased hepatic injury (plasma alanine aminotransferase, ALT activity) in Thpo−/− mice following CCl4 injection, as compared to WT mice. These data imply that VEGF-A and SDF-1 produced by activated platelets protect against acute hepatotoxic injury. N=6–8 mice per group.
We then further examined the contribution of platelets to liver regeneration after 70% PH. Two days after PH, CD41+ platelets were similarly recruited onto VEGFR3+ LSECs (Figure 3a). There was a co-localization of SDF-1 and VEGF-A with CD41+ platelets on the surface of VEGFR3+ LSECs (Figures 3a and b). Proliferation of hepatocytes and liver mass restoration after PH were diminished in Thpo−/− mice relative to WT group (Figures 3c and d). These data implicate that platelets produce VEGF-A and SDF-1 to prime LSECs, eliciting liver regeneration.

After 70% partial hepatectomy (PH), platelets harboring VEGF-A and SDF-1 are associated with VEGFR3+ LSECs. (a, b) Liver sections were stained with antibodies to CD41 (green), VEGF-A and SDF-1 (red), and VEGFR3 (gray). After PH, but not sham operation (Sham), CD41+SDF-1+VEGF-A+ platelets are associated with VEGFR3+ LSECs. Scale bar, 50 μm. (c, d) BrdU incorporation in hepatocyte (c) and liver mass restoration (d) in Thpo−/− and WT mice at days 2 and 8 after PH, respectively. #P<0.05, compared to control group; N=5–8 mice per group.
SDF-1 confers its pro-angiogenic activity65 through the activation of two receptors, CXCR4 and CXCR7. CXCR7 expression is mainly enriched in ECs and subsets of lymphocytes,55,57,66,67 and CXCR7 activation in EC is essential for the production of pro-regenerative angiocrine factor in organ repair.35,39 Therefore, we examined the expression pattern and functional attributes of EC-specific SDF-1 receptor, CXCR7, after PH (Figure 4a). Immunostaining shows that the expression of CXCR7 was upregulated in LSEC 2 days post PH, compared to sham-operated mice (Figures 4a and b). Therefore, CXCR7 might serve as an inducible LSEC-specific SDF-1 receptor after acute liver injury.

After PH, SDF-1 receptor CXCR7 is upregulated in LSEC and contributes to hepatocyte proliferation. (a) Two days after PH, the liver sections were stained for endothelial-specific VE-cadherin (green fluorescence). VE-cadherin+ LSECs are co-localized with CXCR7+ LSECs (red fluorescence). Scale bar, 50 μm. (b) Cxcr7 mRNA level in isolated LSECs was examined at indicated time after PH. The Cxcr7 expression level of sham LSEC was arbitrarily defined as 1. N=5–7 mice per group. (c, d) Mice harboring loxP-flanked Cxcr7 were crossed with endothelial cell-specific Cdh5-PAC-CreERT2 mice.58 Generated offsprings were treated six times with tamoxifen injection (250 mg kg−1) to induce Cxcr7 deletion (Cxcr7iΔEC/iΔEC).15 Mice carrying endothelial haplodeficiency of Cxcr7 (Cxcr7iΔEC/+) were used as control group. N=4 mice per group. (e–g) Inducible knockout of Cxcr7 in LSEC abrogated hepatocyte proliferation (e, f) and restoration of liver mass at indicated time after PH (g). Prohibition of liver mass recovery in Cxcr7iΔEC/iΔEC mice persisted for up to 90 days after PH; N=6–8 mice per group, P<0.01 between control and Cxcr7iΔEC/iΔEC group.
To test functional contribution of CXCR7 in liver regeneration, we selectively deleted Cxcr7 in ECs of adult mice using an inducible tamoxifen-responsive CreERT2 that is specifically expressed in ECs58 (Figure 4c). Mice expressing floxed Cxcr7 were bred with mouse line carrying EC-specific VE-Cadherin-CreERT2/Cdh5(PAC)CreERT2.58 I.p. injection of tamoxifen to resulting offsprings induced 96% of Cxcr7 deletion in ECs of adult mice (Figure 4d). These mice lacking Cxcr7 in ECs (Cxcr7iΔEC/iΔEC) were subjected to PH, and liver regeneration was compared with control mice harboring endothelial haplodeficiency of Cxcr7 (Cxcr7iΔEC/+). Hepatocyte proliferation and liver mass regeneration were markedly reduced in Cxcr7iΔEC/iΔEC mice, as compared to those of control mice (Figures 4e–g). Thus, endothelial CXCR7 might be essential for promoting liver regeneration after PH.
After PH, LSECs produce hepatic-active paracrine/angiocrine growth factors such as HGF and Wnt2.15 This angiocrine function of LSECs in liver regeneration depends on the activation of transcription factor inhibitor of DNA binding 1 (Id1) in LSEC.15,37 We then assessed the effect of SDF-1 on cultured human LSECs. SDF-1 induced both upregulation and nuclear enrichment of Id1 protein in LSEC, which was abrogated by genetic silencing of Cxcr7 (Figures 5a and b). In addition, SDF-1-dependent Id1 upregulation in LSEC was recapitulated by Akt overexpression and suppressed by Wortmannin, an inhibitor of PI3 kinase–Akt pathway (Figure 5b). These data imply that SDF-1 stimulates CXCR7 in LSEC to trigger Akt-dependent activation of Id1 angiocrine pathway.

SDF-1 activates CXCR7 on LSEC, inducing Akt-dependent activation of Id1 angiocrine pathway. (a) CXCR7 expression is essential for SDF-1-mediated Id1 nuclear induction. Early passages of primary human factor VIII+ LSECs (hLSECs) were stimulated with 10 ng ml−1 of SDF-1. shRNA silencing of Cxcr7 in human LSEC abolished nuclear accumulation of Id1 after SDF-1 treatment. Note SDF-1-induced translocation of Id1 from the cytoplasm to nuclei (arrow head). Scale bar, 10 μm. (b) PI3 kinase inhibitor Wortmannin blocked SDF-1-mediated upregulation of Id1, implicating that SDF-1 induction of Id1 is Akt-dependent. N=3–5 independent experiments.
We then assessed whether CXCR7 is responsible for inducing Id1-dependent production of angiocrine factors for liver repair. Given that CXCR7 is specifically upregulated in LSECs after liver injury,36 we utilized a tamoxifen-inducible Rosa-CreERT2 system to ablate Cxcr7 in adult mice (Figure 6a). Cxcr7LoxP/LoxP mice were crossed with Rosa-CreERT2 mice to generate CreERT2+Cxcr7loxP/loxP mice. Injection of tamoxifen induced deletion of Cxcr7 in adult mice (Cxcr7iΔ/iΔ; Figure 6b), and liver regeneration was compared between WT and Cxcr7iΔ/iΔ mice after CCl4 injection. There were significantly lower extent of hepatocyte proliferation (BrdU incorporation) and higher degree of hepatic injury in Cxcr7iΔ/iΔ mice than those of WT mice (Figures 6c and d). Activation of Id1-HGF/Wnt2 angiocrine pathway was markedly prohibited in Cxcr7iΔ/iΔ mice, as compared to WT mice (Figure 6e). These results suggest that after liver injury, platelets supply SDF-1 to activate CXCR7+ LSECs, inducing the production of pro-regenerative angiocrine Wnt2 and HGF for hepatic repair (Figure 6f).

After CCl4-induced liver injury, CXCR7 stimulates angiocrine-mediated hepatic regeneration. (a, b) Mice harboring loxP site-flanked Cxcr7 were crossed with Rosa-CreERT2 and treated six times with tamoxifen injection (250 mg kg−1) to induce the deletion of Cxcr7 in adult mice (Cxcr7iΔ/iΔ).15 Transcriptional level of Cxcr7 in the liver is shown in b; N=6–9 mice per group. (c, d) Inhibition of SDF-1 signaling in LSEC abolished liver regeneration after CCl4 injury. In Cxcr7iΔ/iΔ mice, the decrease in cell proliferation was determined by staining for BrdU incorporation; N=6 mice per group. Scale bar, 50 μm. (e) Increased hepatic damage in Cxcr7iΔ/iΔ mice after CCl4 injury was associated with abrogated induction of Id1-Wnt2/hepatocyte growth factor (HGF) angiocrine pathway; N=5 mice per group. (f) Platelet-dependent activation of LSECs leads to the generation of hepatogenic angiocrine factors. Upon hepatotoxic injury, activated platelets generate SDF-1 to turn on Id1 pathway in LSECs, resulting in angiocrine production of HGF and Wnt2 that initiates hepatocyte propagation and liver repair.
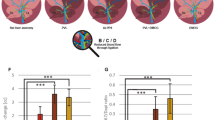
In the CCl4-injured liver, platelets carry both SDF-1 and VEGF-A. Hence, we investigated the contribution of VEGF-A receptors in mediating liver repair. VEGFR1 activation mediates early protection after CCl4 liver injury.16,68 Thus, we tested the contribution of VEGFR1-expressing cells in the injured liver.54 CCl4 injection caused significant recruitment of VEGFR1+CD11b+ myeloid cells to the liver, adhering to VEGFR3+ LSECs (Figure 7a). To determine the contribution of VEGFR1+ myeloid cells to liver repair, Vegfr1loxP/loxP mice were crossed with LysM-driven Cre to generate mice lacking Vegfr1 specifically in myeloid cells (Vegfr1lysM/lysM) (Figures 7b and c). Liver damage was substantially increased in Vegfr1lysM/lysM, as evidenced by elevated plasma alanine aminotransferase activity (Figure 7d), and repeated injection of recombinant TPO in Vegfr1lysM/lysM mice mitigated the injury. Of note, hepatogenic angiocrine Id1 pathway was suppressed in Vegfr1lysM/lysM mice compared to control group, which was restored by TPO injection (Figure 7e). Thus, liver injury recruits platelets and VEGFR1+ myeloid cells to jointly activate LSEC niche, driving liver repair (Figure 7f).

Activated platelets recruit VEGFR1+ myeloid cells to reinforce angiocrine-mediated liver repair. (a) Recruited VEGFR1+ cells in the liver co-expressed CD11b and were associated with VEGFR3+ LSECs, as revealed by immunostaining. Scale bar, 50 μm. (b, c) Conditional knockout of Vegfr1 in myeloid cells abrogated hepatocyte proliferation after CCl4 injection. Floxed Vegfr1 mice were bred with LysM-driven Cre to generate mice lacking Vegfr1 in myeloid cells (Vegfr1lyzM/lyzM). Quantification of Vegfr1 transcriptional level in myeloid cells is shown in c; N=5 mice per group. (d) Exacerbated liver injury in Vegfr1lyzM/lyzM mice than WT control, as evidenced by elevated plasma level of alanine aminotransferase, ALT. Injection of thrombopoietin (+TPO) prevented liver parenchymal injury in Vegfr1lyzM/lyzM mice; N=5–7 mice per group. (e) Pro-regenerative Id1 angiocrine pathway was suppressed in Vegfr1lyzM/lyzM mice, which was elevated by thrombopoietin injection (+TPO). Compared to WT control mice, transcriptional level of Id1 was lower in Vegfr1lyzM/lyzM mice after CCl4 injury. *P<0.05, compared to Vegfr1lyzM/lyzM group. N=5–7 mice per group. (f) Schema depicting the contribution of hematopoietic–vascular niche for liver regeneration. Upon liver injury, activated platelets are recruited to the liver and produce SDF-1 to activate CXCR7+ LSECs, initiating endothelial paracrine/angiocrine-mediated liver repair. Activation of VEGFR1+ myeloid cells by platelet VEGF-A further stimulate Id1-Wnt2/HGF angiocrine pathway in LSEC, reinforcing liver regeneration.
Discussion
LSECs lining hepatic sinusoids actively participate in liver repair and regeneration. Activation of transcription factor Id1 in LSECs leads to the elaboration of hepatic-active angiocrine factors.15 In the mouse liver, we have identified a preferential distribution of SDF-1 receptor CXCR7 on LSECs, which was upregulated by PH. We have also revealed the functional role of endothelial CXCR7 in generating hepatic-active factors in both PH and CCl4 models. As such, both hepatotoxic injury and loss of liver mass stimulate CXCR7 activation in LSECs, eliciting hepatic regeneration, and repair.
Recruited platelets in the injured liver initiates LSEC angiocrine signaling to trigger hepatic reconstitution. Post injury, platelets serve as circulating sentinel cells to promote tissue repair.26,42–46 Here our study has implied a paradigm in which platelet-supplied SDF-1 activates CXCR7 on LSECs and initiates subsequent angiocrine signaling. The beneficial effect of platelets in liver repair is in agreement with both preclinical and clinical findings that favorable prognosis of hepatic function correlates with higher circulating platelet count.29 Whether recombinant TPO has the similar protective effect in infectious liver injury remains to be investigated.47
The cytoprotective effect of VEGFR1+ myeloid cells after acute injury was evidenced by increased injury in mice with myeloid-specific Vegfr1 knockout (Vegfr1lysM/lysM). The rescue effect of TPO injection might depend on both platelets and myeloid cells. The effect of VEGFR1+ myeloid cells on angiocrine function was evidenced by the diminished Id1 pathway in LSECs of Vegfr1lysM/lysM mice. Conceivably, increasing platelet number by TPO injection enhances platelet-dependent activation of myeloid cells, reinforcing endothelial activation and vascular niche-mediated liver regeneration.
The finding that, after liver injury, administration of TPO, SDF-1 and VEGF-A enhanced liver repair has clinical relevance. First, it is conceivable that in thrombocytopenic patients, liver repair is impaired. Thus, cautiously increasing the number of circulating platelet might offer tissue protection. Alternatively, after acute injury, administration of SDF-1 and VEGF-A might augment hepatocyte proliferation by stimulating angiocrine factor generation in LSECs. Furthermore, transfusion/transplantation of properly primed platelets or myeloid cells can possibly offer optimal cell therapy approaches.
Taken together, we demonstrate a pro-regenerative interplay between platelets, myeloid cells and LSECs in liver repair. Platelets play an instrumental role in priming both angiocrine function of LSECs and myeloid cells post injury. Thus, platelet activation enables a hematopoietic–vascular33 niche that orchestrates liver regeneration. Identifying critical pathways establishing this hepatogenic hematopoietic–vascular niche might aid in devising regenerative therapy for hepatic diseases.
References
Fausto N, Campbell JS, Riehle KJ . Liver regeneration. Hepatology 2006; 43: S45–S53.
Michalopoulos GK, DeFrances MC . Liver regeneration. Science 1997; 276: 60–66.
Hu J, Srivastava K, Wieland M, Runge A, Mogler C, Besemfelder E et al. Endothelial cell-derived angiopoietin-2 controls liver regeneration as a spatiotemporal rheostat. Science 2014; 343: 416–419.
Zaret KS, Grompe M . Generation and regeneration of cells of the liver and pancreas. Science 2008; 322: 1490–1494.
Friedman SL . Mechanisms of hepatic fibrogenesis. Gastroenterology 2008; 134: 1655–1669.
Diehl AM . Neighborhood watch orchestrates liver regeneration. Nat Med 2012; 18: 497–499.
Iredale JP . Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest 2007; 117: 539–548.
Bataller R, Brenner DA . Liver fibrosis. J Clin Invest 2005; 115: 209–218.
Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 2005; 115: 56–65.
Yang C, Zeisberg M, Mosterman B, Sudhakar A, Yerramalla U, Holthaus K et al. Liver fibrosis: insights into migration of hepatic stellate cells in response to extracellular matrix and growth factors. Gastroenterology 2003; 124: 147–159.
Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010; 467: 707–710.
Woo DH, Kim SK, Lim HJ, Heo J, Park HS, Kang GY et al. Direct and indirect contribution of human embryonic stem cell-derived hepatocyte-like cells to liver repair in mice. Gastroenterology 2012; 142: 602–611.
Liu L, Yannam GR, Nishikawa T, Yamamoto T, Basma H, Ito R et al. The microenvironment in hepatocyte regeneration and function in rats with advanced cirrhosis. Hepatology 2012; 55: 1529–1539.
Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 2009; 136: 1136–1147.
Ding BS, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 2010; 468: 310–315.
LeCouter J, Moritz DR, Li B, Phillips GL, Liang XH, Gerber HP et al. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science 2003; 299: 890–893.
Straub AC, Clark KA, Ross MA, Chandra AG, Li S, Gao X et al. Arsenic-stimulated liver sinusoidal capillarization in mice requires NADPH oxidase-generated superoxide. J Clin Invest 2008; 118: 3980–3989.
Wang L, Wang X, Xie G, Wang L, Hill CK, Deleve LD . Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J Clin Invest 2012; 122: 1567–1573.
Iwakiri Y, Groszmann RJ . Vascular endothelial dysfunction in cirrhosis. J Hepatol 2007; 46: 927–934.
Gracia-Sancho J, Lavina B, Rodriguez-Vilarrupla A, Brandes RP, Fernandez M, Bosch J et al. Evidence against a role for NADPH oxidase modulating hepatic vascular tone in cirrhosis. Gastroenterology 2007; 133: 959–966.
Huebert RC, Vasdev MM, Shergill U, Das A, Huang BQ, Charlton MR et al. Aquaporin-1 facilitates angiogenic invasion in the pathological neovasculature that accompanies cirrhosis. Hepatology 2010; 52: 238–248.
Apte U, Zeng G, Muller P, Tan X, Micsenyi A, Cieply B et al. Activation of Wnt/beta-catenin pathway during hepatocyte growth factor-induced hepatomegaly in mice. Hepatology 2006; 44: 992–1002.
Ding BS, Liu CH, Sun Y, Chen Y, Swendeman SL, Jung B et al. HDL activation of endothelial sphingosine-1-phosphate receptor-1 (S1P1) promotes regeneration and suppresses fibrosis in the liver. JCI Insight 2016; 1: e87058.
Malato Y, Naqvi S, Schurmann N, Ng R, Wang B, Zape J et al. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J Clin Invest 2011; 121: 4850–4860.
Lee WY, Moriarty TJ, Wong CH, Zhou H, Strieter RM, van Rooijen N et al. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat Immunol 2010; 11: 295–302.
Lesurtel M, Graf R, Aleil B, Walther DJ, Tian Y, Jochum W et al. Platelet-derived serotonin mediates liver regeneration. Science 2006; 312: 104–107.
Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med 2012; 18: 572–579.
Aspinall AI, Curbishley SM, Lalor PF, Weston CJ, Liaskou E, Adams RM et al. CX(3)CR1 and vascular adhesion protein-1-dependent recruitment of CD16(+) monocytes across human liver sinusoidal endothelium. Hepatology 2010; 51: 2030–2039.
Kodama T, Takehara T, Hikita H, Shimizu S, Li W, Miyagi T et al. Thrombocytopenia exacerbates cholestasis-induced liver fibrosis in mice. Gastroenterology 2010; 138: 2487–2498, 2498. e1–7.
Sakaguchi TF, Sadler KC, Crosnier C, Stainier DY . Endothelial signals modulate hepatocyte apicobasal polarization in zebrafish. Curr Biol 2008; 18: 1565–1571.
Matsumoto K, Yoshitomi H, Rossant J, Zaret KS . Liver organogenesis promoted by endothelial cells prior to vascular function. Science 2001; 294: 559–563.
Red-Horse K, Crawford Y, Shojaei F, Ferrara N . Endothelium-microenvironment interactions in the developing embryo and in the adult. Dev Cell 2007; 12: 181–194.
Rafii S, Cao Z, Lis R, Siempos II, Chavez D, Shido K et al. Platelet-derived SDF-1 primes the pulmonary capillary vascular niche to drive lung alveolar regeneration. Nat Cell Biol 2015; 17: 123–136.
Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell 2011; 147: 539–553.
Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature 2014; 505: 97–102.
Rafii S, Butler JM, Ding BS . Angiocrine functions of organ-specific endothelial cells. Nature 2016; 529: 316–325.
Cao Z, Ding BS, Guo P, Lee SB, Butler JM, Casey SC et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell 2014; 25: 350–365.
Cao Z, Scandura JM, Inghirami GG, Shido K, Ding BS, Rafii S . Molecular checkpoint decisions made by subverted vascular niche transform indolent tumor cells into chemoresistant cancer stem cells. Cancer Cell 2017; 31: 110–126.
Cao Z, Lis R, Ginsberg M, Chavez D, Shido K, Rabbany SY et al. Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat Med 2016; 22: 154–162.
Hoehme S, Brulport M, Bauer A, Bedawy E, Schormann W, Hermes M et al. Prediction and validation of cell alignment along microvessels as order principle to restore tissue architecture in liver regeneration. Proc Natl Acad Sci USA 2010; 107: 10371–10376.
Cao Z, Fan-Minogue H, Bellovin DI, Yevtodiyenko A, Arzeno J, Yang Q et al. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res 2011; 71: 2286–2297.
Zimmerman GA, Weyrich AS . Signal-dependent protein synthesis by activated platelets: new pathways to altered phenotype and function. Arterioscler Thromb Vasc Biol 2008; 28: s17–s24.
Pang L, Weiss MJ, Poncz M . Megakaryocyte biology and related disorders. J Clin Invest 2005; 115: 3332–3338.
Bertozzi CC, Hess PR, Kahn ML . Platelets: covert regulators of lymphatic development. Arterioscler Thromb Vasc Biol 2010; 30: 2368–2371.
Jin DK, Shido K, Kopp HG, Petit I, Shmelkov SV, Young LM et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med 2006; 12: 557–567.
Furrer K, Rickenbacher A, Tian Y, Jochum W, Bittermann AG, Kach A et al. Serotonin reverts age-related capillarization and failure of regeneration in the liver through a VEGF-dependent pathway. Proc Natl Acad Sci USA 2011; 108: 2945–2950.
Lang PA, Contaldo C, Georgiev P, El-Badry AM, Recher M, Kurrer M et al. Aggravation of viral hepatitis by platelet-derived serotonin. Nat Med 2008; 14: 756–761.
Carmeliet P . Angiogenesis in life, disease and medicine. Nature 2005; 438: 932–936.
Ferrara N, Gerber HP, LeCouter J . The biology of VEGF and its receptors. Nat Med 2003; 9: 669–676.
Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Jung S et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell 2006; 124: 175–189.
Fischer C, Mazzone M, Jonckx B, Carmeliet P . FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer 2008; 8: 942–956.
Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005; 438: 820–827.
Stefater JA 3rd, Lewkowich I, Rao S, Mariggi G, Carpenter AC, Burr AR et al. Regulation of angiogenesis by a non-canonical Wnt-Flt1 pathway in myeloid cells. Nature 2011; 474: 511–515.
Fong GH, Zhang L, Bryce DM, Peng J . Increased hemangioblast commitment, not vascular disorganization, is the primary defect in flt-1 knock-out mice. Development 1999; 126: 3015–3025.
Miao Z, Luker KE, Summers BC, Berahovich R, Bhojani MS, Rehemtulla A et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc Natl Acad Sci USA 2007; 104: 15735–15740.
Yu S, Crawford D, Tsuchihashi T, Behrens TW, Srivastava D . The chemokine receptor CXCR7 functions to regulate cardiac valve remodeling. Dev Dyn 2011; 240: 384–393.
Sierro F, Biben C, Martinez-Munoz L, Mellado M, Ransohoff RM, Li M et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci USA 2007; 104: 14759–14764.
Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 2010; 465: 483–486.
Ramasamy SK, Kusumbe AP, Wang L, Adams RH . Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 2014; 507: 376–380.
Gurney AL, de Sauvage FJ . Dissection of c-Mpl and thrombopoietin function: studies of knockout mice and receptor signal transduction. Stem Cells 1996; 14 (Suppl 1): 116–123.
Yu C, Wang F, Jin C, Huang X, Miller DL, Basilico C et al. Role of fibroblast growth factor type 1 and 2 in carbon tetrachloride-induced hepatic injury and fibrogenesis. Am J Pathol 2003; 163: 1653–1662.
Hattori K, Heissig B, Wu Y, Dias S, Tejada R, Ferris B et al. Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1(+) stem cells from bone-marrow microenvironment. Nat Med 2002; 8: 841–849.
Kobayashi H, Butler JM, O'Donnell R, Kobayashi M, Ding BS, Bonner B et al. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol 2010; 12: 1046–1056.
Carver-Moore K, Broxmeyer HE, Luoh SM, Cooper S, Peng J, Burstein SA et al. Low levels of erythroid and myeloid progenitors in thrombopoietin-and c-mpl-deficient mice. Blood 1996; 88: 803–808.
Petit I, Jin D, Rafii S . The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol 2007; 28: 299–307.
Gerrits H, van Ingen Schenau DS, Bakker NE, van Disseldorp AJ, Strik A, Hermens LS et al. Early postnatal lethality and cardiovascular defects in CXCR7-deficient mice. Genesis 2008; 46: 235–245.
Wang J, Shiozawa Y, Wang Y, Jung Y, Pienta KJ, Mehra R et al. The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate cancer. J Biol Chem 2008; 283: 4283–4294.
Kato T, Ito Y, Hosono K, Suzuki T, Tamaki H, Minamino T et al. Vascular endothelial growth factor receptor-1 signaling promotes liver repair through restoration of liver microvasculature after acetaminophen hepatotoxicity. Toxicol Sci 2011; 120: 218–229.
Acknowledgements
We are grateful to Dr Ralf Adams at Max Planck Institute for generously providing Chd5(PAC)-CreERT2 mice. KS is supported by the Ansary Stem Cell Institute, the starr foundation TRI-Institution stem cell core project, the Empire State Stem Cell Board and New York State Department of Health grants (C026878, C028117, C029156, C030160), and by grants from the National Institute of Health (NIH) R01 (HL119872, HL128158). ZC was supported by Druckenmiller Fellowship from the New York Stem Cell Foundation and U54 CA163167 from NIH. This work was also supported by a National Scientist Development Grant from the American Heart Association (12SDG1213004), and R01HL130826 from NIH. SR is supported by the Ansary Stem Cell Institute and grant from the NIH R01 (DK095039).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Shido, K., Chavez, D., Cao, Z. et al. Platelets prime hematopoietic–vascular niche to drive angiocrine-mediated liver regeneration. Sig Transduct Target Ther 2, 16044 (2017). https://doi.org/10.1038/sigtrans.2016.44
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/sigtrans.2016.44
This article is cited by
-
The thrombopoietin mimetic JNJ-26366821 reduces the late injury and accelerates the onset of liver recovery after acetaminophen-induced liver injury in mice
Archives of Toxicology (2024)
-
Angiodiversity and organotypic functions of sinusoidal endothelial cells
Angiogenesis (2021)
-
Relevance of VEGFA in rat livers subjected to partial hepatectomy under ischemia-reperfusion
Journal of Molecular Medicine (2019)
-
Confounding influence of tamoxifen in mouse models of Cre recombinase-induced gene activity or modulation
Archives of Toxicology (2018)
