
Abstract
Chinese horseshoe crabs (Tachypleus tridentatus), ancient marine arthropods dating back to the mid-Palaeozoic Era, have provided valuable resources for the detection of bacterial or fungal contamination. However, excessive exploitation for the amoebocyte lysate of Tachypleus has dramatically decreased the population of the Chinese horseshoe crabs. Thus, we present sequencing, assembly and annotation of T. tridentatus, with the hope of understanding the genomic feature of the living fossil and assisting scientists with the protection of this endangered species. The final genome contained a total size of 1.943 Gb, covering 90.23% of the estimated genome size. The transcriptome of three larval stages was constructed to investigate the candidate gene involved in the larval development and validate annotation. The completeness of the genome and gene models was estimated by BUSCO, reaching 96.2% and 95.4%, respectively. The synonymous substitution distribution of paralogues revealed that T. tridentatus had undergone two rounds of whole-genome duplication. All genomic and transcriptome data have been deposited in public databases, ready to be used by researchers working on horseshoe crabs.
Design Type(s) | transcription profiling by high throughput sequencing design • sequence assembly objective |
Measurement Type(s) | whole genome sequencing • transcription profiling assay |
Technology Type(s) | DNA sequencing • RNA sequencing |
Factor Type(s) | developmental stage |
Sample Characteristic(s) | Tachypleus tridentatus • skeletal muscle tissue • larva • ocean biome |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
Horseshoe crabs in the world are represented by two extant populations following a particular geographical distribution1. Tachypleus tridentatus (2n = 26), Tachypleus gigas (2n = 28) and Carcinoscorpius rotundicauda (2n = 32) inhabit the Asian coastline of Southeast Asia; Limulus polyphemus (2n = 52) is distributed along the Atlantic coastline of North America2. Horseshoe crabs have the rare feature of providing blue blood, which has extensive infection-fighting properties that can be used to quantify gram-negative bacterial endotoxins or fungal contamination of medical products3. The horseshoe crabs show some features of crustaceans (crab shell and claws), but they are not crabs at all and are more closely related to spiders, scorpions and trilobites4,5. They belong to their own class called Merostomata, which shows three divisions of the body: the prosoma, opisthosoma and telson.
T. tridentatus, the tri-spine horseshoe crab, the largest of the living horseshoe crab species, was once widespread along the coast of Fujian, Hainan and Beibu Gulf Bay2, but now, the population has drastically decreased for various reasons, such as environmental pollution, decreasing coastline and excessive exploitation for blood extracts (amoebocyte lysate). T. tridentatus is now a ‘Grade II Protected Animal of China’ and is ‘protected aquatic wildlife’ in the Fujian and Guangxi Zhuang Autonomous Region. Although many horseshoe crab nature reserves have been established since 2001 in Guangdong Province around the northern South China Sea, more marine reserve networks for horseshoe crabs in the South and East China Seaboards are needed to establish with hope for protection for the habitat and recovery of this population of the T. tridentatus6.
The external morphology of horseshoe crabs has remained nearly unchanged since the Ordovician approximately 445 million years ago7. The morphological resemblance of a modern horseshoe crab to its ancient fossil suggests the question of whether the genome of the horseshoe crab has been as slow to evolve as the horseshoe crab’s outward appearance implies. Although several genome or transcriptome projects have been implemented on the horseshoe crabs with genotyping-by-sequencing or low-depth sequencing strategies, such as provided in the draft genome of the Atlantic horseshoe crab genome (L. polyphemus)8–10, the partial genome of three horseshoe crabs (C. rotundicauda, L. polyphemus and T. tridentatus)11, and the de novo transcriptome of T. tridentatus during embryonic development12, the lack of fully sequenced and finely annotated horseshoe crab genomic information has hindered the emergence of arthropod models for slow evolution, which has been widely discussed in the brachiopod Lingula anatina13 and the Sarcopterygii Latimeria chalumnae14.
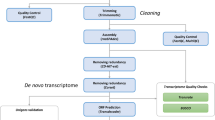
To understand the genomic feature of the living fossil and protect these endangered species, we have generated a draft genome and transcriptome of T. tridentatus. We extracted genomic DNA from leg muscle tissues, constructed 4 Paired-End (PE) and 5 Mate-Pair (MP) libraries, which were subsequently sequenced using the Illumina HiSeq 2500 or Hiseq 4000 platforms. Three stages (pre-trilobite, trilobite, and post-trilobite) of larva, containing two biological replicates for each stage, were chosen for further transcriptome sequencing. After filtering out the low quality, adaptor-contaminated or PCR duplication reads of genome sequencing, we applied the KMERFREQ_AR v2.0.415 and JELLYFISH v1.116 to evaluate the genome size, heterozygosis and repetition by calculating the frequency of 17-mers and then used the Platanus v1.2.417 to assemble the genome using all clean reads from PE and MP libraries. Another round of gap-filling steps was performed based on the assembly results, utilizing GapCloser v1.12-r615. The final assembled genome size was 1.94 Gb, containing 736,826 contigs (N50 = 52.2 kb) and 671,877 scaffolds (N50 = 2.76 Mb), which represented approximately 90.16% of the genome estimated via k-mer analysis. The BUSCO v2.118 and de novo assembled transcript evaluation demonstrated the genome was of considerable completeness and high gene region coverage. The transposable elements accounted for 39.96% of the T. tridentatus genome, whereas the DNA transposons accounted for the largest portion, 23.06%. The structural annotation of the genome yielded 29,134 genes, and approximately 83.37% of these genes were functionally annotated with at least one of four sources (InterPro, KEGG, SwissProt and TrEMBL). The synonymous substitution (Ks) distribution of the T. tridentatus paralogues indicated the Chinese horseshoe crabs had undergone two rounds of WGDs, as has its Atlantic counterpart. The release of the T. tridentatus genome and transcriptome in this study provides a crucial resource for future efforts to adopt better strategies to conserve the endangered Asian horseshoe crab and to take it as the model species to foster study on marine chelicerates.
Methods
Tissue sampling
For genomic sequencing, an adult T. tridentatus (Fig. 1) was acquired from the Guangxi Key Laboratory of Beibu Gulf Marine Biodiversity Conservation. Leg skeletal muscle tissue was collected and then stored in liquid nitrogen for immediate DNA extraction. For transcriptome sequencing, six fertilized eggs were collected by laboratory spawning and incubated with standard procedures under appropriate temperature and salinity19. Three larval stages were collected according to Sekiguchi’s definition20: ‘pre-trilobite’, ‘trilobite’ and ‘post-trilobite’. Two biological replicates for each larval stage were collected and stored in liquid nitrogen for immediate RNA extraction.

The morphology of T. tridentatus.
Library construction and sequence quality control
Genomic DNA was extracted from frozen muscle tissues of T. tridentatus using a genomic DNA isolation kit (Qiagen, Hilden, Germany) and the manufacturer’s protocol and then stored at −80 °C until library preparation. Subsequently, 4 pair-end libraries with insert sizes of 270 bp, 300 bp, 500 bp and 800 bp, and 5 mate-pair libraries with insert sizes of 2 kb, 5 kb, 10 kb, 20 kb and 40 kb were generated (Data Citation 1), sequenced by BGI (Shen Zhen) on Illumina Hiseq 2500 or Hiseq 4000, following the manufacturer’s instruction. Finally, we generated a total of 554.2 Gb raw bases from 19 lanes (Table 1). The raw reads of the PE and MP libraries were then filtered by SOAPnuke v1.521 according to the following criteria: 1) reads with adapter contamination or polymerase chain reaction duplicates were discarded, 2) reads with more than 30% low-quality bases (Q value ≤ 15) for the pair-end reads and more than 20% low-quality bases (Q value ≤ 10) for the mate-pair reads were removed, 3) reads with more than 1% N bases were removed, and 4) matching lengths of read 1 and read 2 were removed when 10 bp had at least a 10% mismatch. After sequences had been pre-processed with SOAPnuke v1.5 software, a total of 202.67 Gb clean pair-end and 71.54 Gb mate-pair reads were obtained (Table 1).
The total RNA of each larval development was extracted separately according to the TRIzol protocol (Invitrogen). Samples were sequenced by BGI (Shen Zhen) on an Illumina HiSeq 2500 platform (Data Citation 1). Paired-end reads were generated with a read length of 100 bp. A total of 436.63 Mb RNA-Seq reads were produced (Table 2). The raw reads of the transcriptome libraries were filtered by SOAPnuke v1.5 according to the following criteria: 1) reads with more than 1% N bases were removed; 2) reads with more than 20% low-quality bases (Q value ≤ 10) were removed; and 3) reads with adapter contamination were discarded. After pre-processing with SOAPnuke software, a total of 393.57 Mb RNA-seq reads were generated with an average Q20 ≥ 96% (Table 2).
Characteristics of the T. tridentatus genome
A total of 91.34 Gb clean reads from libraries for insert sizes of 300 bp and 500 bp were chosen for use in k-mer analysis using KMERFREQ_AR v2.0.415 and JELLYFISH v1.116 with a k length of 17. The genome size (G) of T. tridentatus was estimated by the following formula: G = k-mer number/k-mer depth, where the k-mer number is the total numbers of k-mers, and k-mer depth refers to the depths of the main peak in the k-mer frequency distribution. The k-mer frequency distribution indicated that T. tridentatus was a diploid species with low heterozygosity and repetition (Fig. 2). The frequency of 17-mers using KMERFREQ_AR v2.0.415 estimated a genome size of 2.15 Gb, whereas the JELLYFISH v1.116 approach yielded an estimated genome size of 2.14 Gb (Supplementary Table 1). The result of JELLYFISH was subsequently delivered to GenomeScope22, indicating the heterozygosity of the T. tridentatus genome was approximately 0.65%. The estimated genome size of T. tridentatus is much smaller than L. polyphemus, which is estimated to be 2.74 Gb (2.8 pg) based on the biochemical method23.

The distribution was determined with KMERFREQ_AR using a k-mer size of 17.
Genome assembly
For genome assembly, the contig construction, scaffold connection and gap-closer of the T. tridentatus genome was performed based on multiple k-mer values automatically optimized by the Platanus v1.2.417, with clean reads from pair-end and mate-pair libraries. All applications were used with the default parameters except for the initial k-mer 37. Furthermore, one more round of gap-closer was performed using GapCloser v1.12-r615 to fill the remaining gaps in the scaffolds. The final assembly contained 671,877 scaffolds and included a total length of 1.94 Gb, which represented approximate 90.16% of the genome estimated by the k-mer analysis. The sizes of the longest scaffold and contig were 18,230,544 and 1,165,240 bp, respectively, and 87.92% of the assembled sequences with lengths longer than 10 kb were among the 2,573 scaffolds. The contig and scaffold N50s were 52,179 and 2,761,313 bp, respectively, whereas the percentage of the gap in the genome was 1.55% (Table 3).
Transcriptome assembly
The de novo transcriptome assembly of each larval stage was performed using Trinity v2.4.024 with default parameters. The Trinity assemble resulted in 669,788 transcripts with an average N50 ~1 Kb (Supplementary Table 2). The transcriptome assembly of the same stage was then clustered to remove redundancies and form the Unigenes using the TGI Clustering Tool (TGICL) v2.125 with default parameters. The TGICL cluster generated 273,085 transcripts with an average N50 ~1.5 Kb (Supplementary Table 2). The genome-guided transcriptome assembly of each larval stage was performed with HiSat2 v2.2.1.026,27 and StringTie v1.3.427,28. The genome index was built using HISAT2-build, and then, the clean transcriptome reads were mapped to the genome using HiSat2, and the alignment result of each larval stage was merged to form one single BAM file using SAMtools v1.329. Finally, the genome-guided transcriptome assembly was performed using StringTie with the single BAM file.
Repeat annotation
Prior to gene prediction, the transposable elements were identified in the genome of T. tridentatus. The transposable elements were identified by a combination of homology and ab initio-based methods. For the homology method, RepeatMaskerv4.0.530 was adopted to identify the transposable elements against the RepBase library31 (release-20170127), while RepeatProteinMask v4.0530 was applied to identify the transposable elements against the TE protein databases attached with RepBase library. For the ab initio-based method, the de novo library of T. tridentatus genome was constructed using LTR_FINDER v1.0732 and REPEATMODELER v1.0.833. Subsequently, RepeatMasker v4.05 was used to identify and classify different categories of repetitive elements against the de novo library. Finally, the transposable elements of the same category identified by these two methods were integrated via sequence overlap. The percentage of the T. tridentatus genome covered by transposable elements was 39.96%, with a total length of 776,373,528 bp; meanwhile, the DNA transposons took up the largest portion— 23.06% (Supplementary Table 3).
Gene prediction
The MAKER34 application was used to predict the gene model by integrating the homology, transcriptome and ab initio gene predictions. MAKER was initially run in the est2genome = 1 and protein2genome = 1 model, which created a gene model directly from the transcript and protein evidence. The transcript evidence was based on de novo transcriptome assembly of the three larval stages, which included 273,085 transcripts (Supplementary Table 2), whereas the 96,006 protein sequences were collected from the genome of four arthropods deposited at NCBI: Limulus polyphemus10,35, Mesobuthus martensii36, Stegodyphus mimosarum37 and Ixodes scapularis38. The option ‘rm_gff’ was filled out with the transposable element file acquired from the repeat annotation step, and the expected max intron size for evidence alignments was set to 30,000 according to the max intron size of L. polyphemus. The initial run produced 25,252 gene models, and 2,000 high-confidence gene models were randomly selected according to the criterion determined by the maker2zff (an application in MAKER pipeline) default parameters, except for the maximum annotation edit distance (AED) of 0.1. These high confidence gene models were then used to train the parameters of SNAP39 (release-2013-11-29) and AUGUSTUS v3.3.140 software with each pipeline. A second round of MAKER was run with the training parameters, est2genome = 0 and protein2genome = 0 mode. All other parameters were the same as the first round except that we used ‘est_gff’ with the genome guiding transcriptome assembly to replace the ‘est’ option.
Gene function annotation
Protein sequences from the predicted gene models were searched against the KEGG41 (v84.0), SwissProt and TrEMBL databases42 (release2017-09) with E-value threshold of 1e-5. The domain annotations were applied using InterProScan43 (5.16-55.0) and by searching against the public databases Pfam44, PRINTS45, ProDom46, PIRSF47, PANTHER48, TIGRFAM49, SUPERFAMILY50, ProSitePatterns51, ProSiteProfiles52, Coils53 and SMART54. Approximately 83.37% of these genes were functionally annotated with at least one of these sources, with 21,414 InterProScan entries, 19,195 KEGG entries, 15,283 SwissProt entries and 23,687 TrEMBL entries (Supplementary Table 4).
Whole-genome duplications in L. polyphemus and T. tridentatus
The synonymous substitutions (Ks) distribution had been used to infer WGD in plant and vertebrates55,56. To inspect the evidence for the whole-gene duplication of the Chinese and Atlantic horseshoe crabs, we identified the paralogues of each species and calculated the substitutions per synonymous site (Ks) distribution of paralogous pairs with a Python script available online: https://github.com/EndymionCooper/KSPlotting. The main steps were listed as follows. 1) Sequence similarity was determined using an all-vs-all comparison of protein sequences, performed using BLASTP + v2.5057 with an E-value threshold of 1e-5. The paired genes were retained under the criterion that the shorter sequence was at least 50% of the longer sequence and that the alignment length was at least 50% of the shorter sequence. 2) Paralogue gene family construction was determined by the paralogue gene families that were built through single-linkage clustering. In brief, the genes with multiple alignment and associated matches were grouped into the same paralogue categories. After the paralogues had been identified, all possible pairs of protein sequences in each paralogue family were aligned using MUSCLE58 v3.8.31 with default parameters, and then the multiple alignments of the amino acid sequences were converted to the corresponding coding sequences (CDS). 3) Synonymous substitutions (Ks) values of each paired paralogue gene were calculated using CODEML59. Only gene pairs with a Ks estimate of <3 were considered for further analysis. A paralogous gene family of n members was derived from n-1 possible duplication events, but the number of probable pairwise Ks comparisons within a family was n × (n-1)/2, which could result in misconception of the ages of duplication events. The Ks values of each paralogous group were corrected to remove redundancy using a hierarchical clustering approach60,61, leaving the paralogous groups that contained the representative duplication events. We also retrieved all-vs-all alignment using the reciprocal best blast hit (RBH) criterion to identify the orthologous genes, and then, we applied KaKs Calculator62 with the method of Yang and Nielsen63 to estimate synonymous substitution rates (Ks) of the orthologs.
Code availability
The software versions, settings and parameters are described below.
1) KMERFREQ_AR: version 2.04, k-mer size of 17; 2) JELLYFISH: version 1.1, k-mer size 17; 3) GenomeScope: parameters used were k-mer length 17; read length 100; maximum k-mer coverage 1000; 4) Platanus: version 1.2.4, parameters used were contig Platanus contig -t 20 -k 37 -s 10 -u 0.1 -d 0.5 -m 400 -f <insert size 270 bp pair-end reads> <insert size 300 bp pair-end reads> <insert size 500 bp pair-end reads> <insert size 800 bp pair-end reads> ; scaffold -u 0.1 -c contig.fa -b contigBubble.fa -IP1 <insert size 270 bp pair-end reads> -IP2 <insert size 300 bp pair-end reads> -IP3 <insert size 500 bp pair-end reads> -IP4 <insert size 800 bp pair-end reads> -OP5 <insert size 2 k mate-pair reads> -OP6 <insert size 5 k mate-pair reads> -OP7 <insert size 10 k mate-pair reads> -OP8 <insert size 10 k mate-pair reads> -OP9 <insert size 20 k mate-pair reads> -OP10 <insert size 40 k mate-pair reads> -a1 270 -a2 300 -a3 500 -a4 800 -a5 2000 -a6 5000 -a7 10000 -a8 10000 -a9 20000 -a10 40000; gap_close -c scaffold.fa -IP1 <insert size 270 bp pair-end reads> -IP2 <insert size 300 bp pair-end reads> -IP3 <insert size 500 bp pair-end reads> -IP4 <insert size 800 bp pair-end reads>; 5) Gap Closer: version 1.12, parameters used were -l 150, in configFile: asm_flags = 4; PE125 lib: rd_len_cutoff = 100, pair_num_cutoff = 7, map_len = 45; PE150 libs: rd_len_cutoff = 135, pair_num_cutoff = 10, map_len = 50; 6) BUSCO: version 2.1, arthropod default parameters, arthropoda_odb9; 7) SOAPnuke: version 1.5, genome sequences pre-processing: pair-end library: -l 15 -q 0.3 -n 0.1 -d -i -S; mate-pair library: -l 10 -q 0.2 -n 0.1 -d -i -S; RNA-Seq sequences pre-processing: -l 10 -q 0.2 -n 0.1 -i; 8) Trinity: version 2.4.0, default parameters; 9) TGICL: version 2.1, -l 40 -c 10 -v 25 -O ‘-repeat_stringency 0.95 -minmatch 35 -minscore 35’; 10) HiSat2: version 2.2.1.0, --max-intronlen50000 --sensitive --dta --dta-cufflinks --phred64 --no-mixed --no-discordant; 11) StringTie: version 1.3.4, default parameters; 12) SAMtools: version 1.3, default parameters; 13) RepeatMasker: version 4.0.5 (with RepBase library release-20170127); 14) RepeatModeler: RepeatModeler-open-1.0.8; 15) LTR_FINDER: version 1.07; 16) MAKER: version 2.3.10, first run, parameters: est = merged transcripts from Trinity assemblies for 3 larval stages, protein = 4 arthropod proteins, rmlib = all transposon elements, est2genome = 1, protein2genome = 1; predicted 25,252 genes, randomly select 2,000 to train Augustus and SNAP. Second run: est_gff = merged transcripts gff from StringTie assemblies for three larval stages, protein = 4 arthropod proteins, rmlib = all transposon elements, est2genome = 0, protein2genome = 0, snaphmm = SNAP training parameter, augustus_species = AUGUSTUS training parameter; 17) SNAP: release-2013-11-29; 18) AUGUSTUS: version 3.3.1; 18) KEGG: version 84.0; 19) SwissProt and TrEMBL: release 2017-09; 20) InterProscan: version 5.16-55.0, with parameters -goterms -f tsv -appl ProDom -appl PRINTS-appl Pfam -appl PIRSF -appl PANTHER -appl TIGRFAM -appl SUPERFAMILY -appl ProSitePatterns -appl ProSiteProfiles -appl Coils -appl SMART; 20) BLASTP: version 2.6.0; 21) MUSCLE: v3.8.31; 22) CODEML version 4.8; 23) KaKs Calculator version 2.0.
Data Records
The raw data of the whole genome and the RNA-seq sequencing was submitted to the National Center for Biotechnology Information (NCBI) (Data Citations 1), and more detailed information about the reads is shown in Data_Descriptor_Worksheets.xlsx. The final assembly was deposited at NCBI GenBank (Data Citations 2). The other files, such as the assembled contigs, scaffold, de novo RNA-seq assembly, repeat annotation, gene prediction and gene function annotation were uploaded to Figshare (Data Citations 3), and the file with descriptions is presented in Supplementary Table 5.
Technical Validation
Genome assembly and gene prediction quality assessment
The completeness of the genome assembly and gene prediction was assessed using BUSCO18 based on evolutionarily informed expectations of gene content from near-universal single-copy orthologues selected from OrthoDBv964. The completeness of the resulting assembly was comparable to the other arthropods; BUSCO analysis showed that 96.2 and 0.8% of the 1066 arthropod datasets (arthropoda_odb9, http://busco.ezlab.org/) were identified as complete and fragmented separately, whereas 3% of the BUSCO genes were missing from the assembled genome sequence (Supplementary Table 6). The completeness of the current gene prediction is also comparable to the other arthropod, 95.4% and 2.3% of the 1066 arthropod datasets (arthropoda_odb9, http://busco.ezlab.org/) were identified as complete and fragmented separately, whereas 2.3% of the BUSCO genes were missing from the current annotation (Supplementary Table 7). The de novo transcriptome assembly of three larval stages was aligned to the genome using BLAT65 with default parameters to evaluate the coverage of the gene region. The alignment results of the Unigenes indicated that the assembled genome of T. tridentatus covered 95.59~98.01% of the Unigenes, 89.02~93.55% of the Unigenes with at least 90% coverage in one scaffold, and 93.62~98.81% of the Unigenes with at least 50% coverage in one scaffold (Supplementary Table 8), suggesting that the gene regions were mostly included in the current assembly.
Comparison with other horseshoe crab genomes
The current assembly of the T. tridentatus genome may be the most complete version of horseshoe crab. The contig and scaffold N50 of the current assembly were much longer than any other published horseshoe crab databases (Supplementary Table 9), the sequence depths and assembled sizes were also much larger than other sources. The longest scaffold of T. tridentatus measures 18,230,544 bp, which is three times more than L. polyphemus (PRJNA20489). The numbers of ≥1 Mb scaffold in T. tridentatus are 446, while the numbers of ≥ 1 Mb scaffold in L. polyphemus only are 147 (Table 3). The Ks distribution of L. polyphemus exhibited peak centred at 0.14 and 0.84, whereas the Ks distribution of T. tridentatus showed peaks centred at 0.16 and 1.16 (Fig. 3). The evidence showed the lineage of the horseshoe crabs had undergone two rounds of WGDs, which was consistent with the previous result9 but with a variant peak. Meanwhile, the Ks distribution of the orthologous genes between T. tridentatus and L. polyphemus showed a peak centred at 0.20 (Fig. 3), which suggests the last common ancestor of the extant horseshoe carb was posterior to the ancient duplication but predated the recent duplication.

The ‘*’ indicates the peak derived from the whole-genome duplication event, whereas the ‘#’ indicates the peak derived from small-scale duplications events (SSDs, tandem duplications) in the Chinese horseshoe crab.
Usage notes
All analyses were run on Linux systems, and the optimal parameters are given in the Code availability section.
Additional information
How to cite this article: Liao, Y. Y. et al. Draft genomic and transcriptome resources for marine chelicerate Tachypleus tridentatus. Sci. Data. 6:190029 https://doi.org/10.1038/sdata.2019.29 (2019).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Sekiguchi, K. & Nakamura, K. Ecology of the extant horseshoe crabs. Progress In Clinical & Biological Research 29, 37–45 (1979).
Tanacredi, J. T., Botton, M. L . & Smith, D. R. Biology And Conservation Of Horseshoe Crabs. (Springer: US, 2009).
Cooper, J. & Pearson, S. M. Detection of endotoxin in biological products by the limulus test. Developments In Biological Standardization 34, 7–13 (1977).
Walls, E. A., Berkson, J. & Smith, S. A. The Horseshoe Crab, Limulus polyphemus: 200 Million Years of Existence, 100 Years of Study. Reviews In Fisheries Science 10, 39–73 (2002).
Kamaruzzaman, B. Y., John, A. B., Zaleha, K. & Jalal, K. Molecular phylogeny of horseshoe crab. Asian Journal Of Biotechnology 3, 302–309 (2011).
Chen, C. P. et al. Conservation of the horseshoe crab at Kinmen, Taiwan: strategies and practices Biodiversity And Conservation 13, 1889–1904 (2004).
Rudkin, D. M., Young, G. A. & Nowlan, G. S. The oldest horseshoe crab: a new Xiphosurid from Late Ordovician Konservat‐Lagerstätten Deposits, Manitoba, Canada. Palaeontology 51, 1–9 (2008).
Simpson, S. D., Ramsdell, J. S., Watson Iii, W. H. & Chabot, C. C. The Draft Genome and Transcriptome of the Atlantic Horseshoe Crab,Limulus polyphemus. International Journal Of Genomics 2017, 1–14 (2017).
Nossa, C. W. et al. Joint assembly and genetic mapping of the Atlantic horseshoe crab genome reveals ancient whole genome duplication. GigaScience 3, 9 (2014).
Chesmore, K. N., Watson, W. H. & Chabot, C. C. Identification of putative circadian clock genes in the American horseshoe crab, Limulus polyphemus. Comparative Biochemistry And Physiology Part D: Genomics And Proteomics 19, 45–61 (2016).
Kenny, N. et al. Ancestral whole-genome duplication in the marine chelicerate horseshoe crabs. Heredity 116, 190 (2016).
Chen, M. et al. De NovoAssembly and Characterization of Early Embryonic Transcriptome of the Horseshoe CrabTachypleus tridentatus. Plos One 11, e0145825 (2016).
Luo, Y. J. et al. The Lingula genome provides insights into brachiopod evolution and the origin of phosphate biomineralization. Nature Communications 6, 8301 (2015).
Amemiya, C. T. et al. The African coelacanth genome provides insights into tetrapod evolution. Nature 496, 311–316 (2013).
Luo, R. et al. SOAPdenovo2: an empirically improved memory-efficient short-readde novoassembler. GigaScience 1, 18 (2012).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764 (2011).
Kajitani, R. et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Research 24, 1384–1395 (2014).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2016).
Smith Dvm, S. A. & Berkson, J. Laboratory culture and maintenance of the horseshoe crab (Limulus polyphemus). Lab Animal 34, 27 (2005).
Sekiguchi, K. Biology of horseshoe crabs. (International Specialized Book Service Incorporated, 1988).
Chen, Y. et al. SOAPnuke: A MapReduce Acceleration supported Software for integrated Quality Control and Preprocessing of High-Throughput Sequencing Data. GigaScience 7, 1–6 (2018).
Vurture, G. W. et al. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Goldberg, R. B. et al. DNA sequence organization in the genomes of five marine invertebrates. Chromosoma 51, 225–251 (1975).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29, 644 (2011).
Pertea, G. et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics 19, 651–652 (2003).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360 (2015).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature Protocols 11, 1650 (2016).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 33, 290–295 (2015).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 5, 4.10.1–4.10.14 (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268 (2007).
Smit, A. F. A. & Hubley, R. RepeatModeler Open-1.0 (2008).
Campbell, M. S., Holt, C., Moore, B. & Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Current Protocols In Bioinformatics 48, 4.11.1–4.11.39 (2014).
Battelle, B. A. et al. Opsin repertoire and expression patterns in horseshoe crabs: evidence from the genome of Limulus polyphemus (Arthropoda: Chelicerata). Genome Biology And Evolution 8, 1571–1589 (2016).
Cao, Z. et al. The genome of Mesobuthus martensii reveals a unique adaptation model of arthropods. Nature Communications 4, 2602 (2013).
Sanggaard, K. W. et al. Spider genomes provide insight into composition and evolution of venom and silk. Nature Communications 5, 3765 (2014).
Gulia-Nuss, M. et al. Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nature Communications 7, 10507 (2016).
Korf, I. & Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research 28, 27–30 (2000).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research 28, 45–48 (2000).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Bateman, A. et al. The Pfam protein families database. Nucleic Acids Research 32, D138–D141 (2004).
Attwood, T. K. et al. PRINTS-S: the database formerly known as PRINTS. Nucleic Acids Research 28, 225–227 (2000).
Corpet, F., Gouzy, J. r. m. & Kahn, D. Recent improvements of the ProDom database of protein domain families. Nucleic Acids Research 27, 263–267 (1999).
Nikolskaya, A. N., Arighi, C. N., Huang, H., Barker, W. C. & Wu, C. H. PIRSF family classification system for protein functional and evolutionary analysis. Evolutionary Bioinformatics Online 2, 197 (2006).
Mi, H. et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Research 33, D284 (2005).
Haft, D. H. et al. TIGRFAMs and Genome Properties in 2013. Nucleic Acids Research 41, 387–395 (2013).
Gough, J., Karplus, K., Hughey, R. & Chothia, C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. Journal Of Molecular Biology 313, 903–919 (2001).
Sigrist, C. J. et al. New and continuing developments at PROSITE. Nucleic Acids Research 41, 344–347 (2013).
Hulo, N et al. The 20 years of PROSITE. Nucleic Acids Research 36, D245 (2008).
Burkhard, P., Stetefeld, J. & Strelkov, S. V. Coiled coils: a highly versatile protein folding motif. Trends In Cell Biology 11, 82–88 (2001).
Schultz, J. R., Copley, R. R., Doerks, T., Ponting, C. P. & Bork, P. SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Research 28, 231–234 (2000).
Adams, K. L. & Wendel, J. F. Polyploidy and genome evolution in plants. Current Opinion In Plant Biology 8, 135–141 (2005).
Mclysaght, A. & Hokamp KWolfe, K. H. Extensive genomic duplication during early chordate evolution. Nature Genetics 31, 200–204 (2002).
Kevin, B. et al. BLAST+ : architecture and applications. BMC Bioinformatics 10, 421 (2009).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32, 1792–1797 (2004).
Yang, Z. & Yang, Z. H. PAML 4: Phylogenetic analysis by maximum likelihood. Molecular Biology And Evolution 24, 1586–1591 (2007).
Maere, S. et al. Modeling Gene and Genome Duplications in Eukaryotes. Proceedings of The National Academy of Sciences of the United States of America 102, 5454 (2005).
Blanc, G. & Wolfe, K. H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16, 1667–1678 (2004).
Zhang, Z. et al. KaKs_Calculator: Calculating Ka and Ks Through Model Selection and Model Averaging. Genomics,Proteomics & Bioinformatics 4, 259–263 (2006).
Muse, S. V. Estimating synonymous and nonsynonymous substitution rates. Molecular Biology And Evolution 13, 105 (1996).
Zdobnov, E. M. et al. OrthoDB v9. 1: cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Research 45 , D744-D749 (2016).
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome Research 12, 656–664 (2002).
Data Citations
NCBI Sequence Read Archive SRP149385 (2018)
GenBank QXHF00000000 (2018)
Liao, Y. Y. figshare https://doi.org/10.6084/m9.figshare.7380866 (2018)
Acknowledgements
This project was funded by National Natural Science Foundation of China (41466003, 41706183), Natural Science Foundation of Guangxi (2015GXNSFDA13901, 2017GXNSFBA198181) and Guangxi Key Laboratory of Beibu Gulf Marine Biodiversity Conservation (2018ZA01, 2015KA04). We are grateful to the Guangxi Colleges and Universities Innovation Research Team for their helpful discussions on this project.
Author information
Authors and Affiliations
Contributions
J.F. Han, J.X. Liu, C. Zhang and Y.Y. Liao designed the research; Y.Y. Liao, P.W. Xu, K.Y. Kwan, Z.Y. Ma and H.Y. Fang analysed the data and wrote the manuscript; P.L. Wang, S.Y. Yang, S.Q. Xu, W.F. Li and Y.Y. Liao provided materials for genome sequencing; L.R. Bai, Y.Q. Zhang, J. Lei, K. Liu, F. Li, J. Li, P. Zhu and Y.J. Wang provided materials for transcriptome sequencing; D. Qian, S.B. Xie, D.J. Zhou, H.P. Wu, Y.H. Xu and H. Huang performed sample preparation and total RNA extraction; and J.X. Liu, J.Y. Xu and J.F. Han revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information accompanies this paper at
ISA-Tab metadata
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Liao, Y., Xu, P., Kwan, K. et al. Draft genomic and transcriptome resources for marine chelicerate Tachypleus tridentatus. Sci Data 6, 190029 (2019). https://doi.org/10.1038/sdata.2019.29
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2019.29
This article is cited by
-
Transcriptome Analysis of the Digestive Tract of Tachypleus tridentatus and Carcinoscorpius rotundicauda
Journal of Ocean University of China (2022)
-
Horseshoe crab genomes reveal the evolution of genes and microRNAs after three rounds of whole genome duplication
Communications Biology (2021)
-
The draft genome of horseshoe crab Tachypleus tridentatus reveals its evolutionary scenario and well-developed innate immunity
BMC Genomics (2020)
-
Chromosome-level assembly of the horseshoe crab genome provides insights into its genome evolution
Nature Communications (2020)
-
Developmental gene expression as a phylogenetic data class: support for the monophyly of Arachnopulmonata
Development Genes and Evolution (2020)
