
Abstract
Neuroblastoma, a pediatric tumor of the sympathetic nervous system, is predominantly driven by copy number aberrations, which predict survival outcome in global neuroblastoma cohorts and in low-risk cases. For high-risk patients there is still a need for better prognostic biomarkers. Via an international collaboration, we collected copy number profiles of 556 high-risk neuroblastomas generated on different array platforms. This manuscript describes the composition of the dataset, the methods used to process the data, including segmentation and aberration calling, and data validation. t-SNE analysis shows that samples cluster according to MYCN status, and shows a difference between array platforms. 97.3% of samples are characterized by the presence of segmental aberrations, in regions frequently affected in neuroblastoma. Focal aberrations affect genes known to be involved in neuroblastoma, such as ALK and LIN28B. To conclude, we compiled a unique large copy number dataset of high-risk neuroblastoma tumors, available via R2 and a Shiny web application. The availability of patient survival data allows to further investigate the prognostic value of copy number aberrations.
Design Type(s) | disease outcome analysis objective • data analysis objective |
Measurement Type(s) | copy number variation |
Technology Type(s) | digital curation |
Factor Type(s) | survival |
Sample Characteristic(s) | Homo sapiens • neuroblastoma |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
Neuroblastoma is a pediatric tumor of the sympathetic nervous system, affecting mainly children under the age of five years1. It is the most common extracranial solid cancer, making up 5% of childhood cancer diagnoses, while accounting for approximately 10% of childhood cancer deaths2,3.
Neuroblastoma is characterized by extensive clinical heterogeneity, illustrated by different clinical evolutions ranging from spontaneous regression to aggressive disease. Risk stratification of neuroblastoma patients is based on clinical parameters, histopathological parameters and genetic parameters including MYCN amplification, 11q loss and the global copy number profile4,5.
While recurrent single nucleotide mutations, such as those targeting ALK and ATRX, are observed infrequently at diagnosis6,7, copy number aberrations occur at much higher frequency and are strongly associated with disease outcome. Tumors with only numerical aberrations have a favorable prognosis, while the presence of at least one segmental aberration is indicative of poor survival outcome8.
Neuroblastoma is a rare disease, therefore it is challenging to collect enough tumor material and data for a sufficiently powered study. Within the Ultra-High-Risk (UHR) working group of the International Neuroblastoma Response Criteria (INRC) consortium5, we collected DNA copy number profiles and clinical data of 556 high-risk neuroblastoma patients, resulting in a unique dataset. The aim of this project was to explore whether specific (combinations of) segmental DNA copy number aberrations allow to better discriminate high-risk patients with fatal outcome from high-risk patients with favorable outcome9.
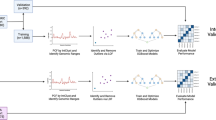
The collected copy number profiles were generated on several aCGH and SNP array platforms. The data were uniformized by converting to the same genome build hg19, and by segmenting with the same method (summarized in Fig. 1). From the segmented data, aberrations were called using specific cutoffs for each array platform. Exploration of the collected and processed data confirmed the presence of segmental and focal aberrations in regions frequently affected in neuroblastoma and that samples cluster according to MYCN status rather than platform and inter-laboratory differences.

Raw/normalized probe-level data were converted to hg19 genome build using liftOver, followed by median normalization and segmentation using SegAnnDB. Data are available in Gene Expression Omnibus (GEO). Segmented data are available in Table 3.
In this data descriptor, we describe in detail the composition of the data with respect to sample inclusion criteria and clinical characteristics and the methods used to process the data. We provide access to normalized probe-level data and segmented data as well as instructions to analyze and visualize the data using either the R2 platform10, the statistical programming language R or a Shiny web application.
Methods
DNA copy number data collection
Copy number profiling data from 556 primary neuroblastoma tumors were collected through the UltraHigh-Risk (UHR) working group of the International Neuroblastoma Response Criteria (INRC) consortium. These tumors originated from high-risk patients enrolled in the SIOPEN, GPOH, COG or Japanese treatment protocol (SIOPEN: European Society for Paediatric Oncology Neuroblastoma, GPOH: Society for Paediatric Oncology and Haematology, COG: Children’s Oncology Group). Published6,11,12 and unpublished copy number profiling data generated on different platforms including aCGH arrays from Agilent (resolution ranging from 44k-1M) and NimbleGen (72k) and SNP arrays from Affymetrix (250k and 2.6 M) and Illumina (550k) were included in this study. Initially, data from 671 high-risk samples were collected, with high-risk defined as either stage 4 and older than 1 year, or MYCN-amplified (any age and stage). MYCN amplification status was determined by FISH or qPCR. Of the 671 samples, 16 samples were discarded due to bad quality profiles and 98 samples were discarded due to suspected contamination with normal cells (49 silent profiles without detected copy number aberrations and 50 with almost undetectable aberrations), resulting in a set of 556 samples. Sample annotations, including among others age at diagnosis, disease stage, MYCN status and overall and event-free survival, are available at Figshare (Table 1: Patient annotations, Data Citation 1). However, event-free survival data are not available for the Japanese dataset. This collection strategy has been reported already in the Supplementary Methods of the research paper originally related to this dataset9. The patient annotations in this publication are the same as Supplementary Table 1 in the related paper9.
DNA copy number data processing
The processing steps performed on the collected data to obtain segmented copy number data is summarized in Fig. 1. Raw or normalized copy number data were collected. Raw data were median-normalized. When necessary, chromosomal coordinates were converted from genome build hg17 or hg18 to genome build hg19 using the “liftOver” function from the R package “rtracklayer”. Only for the COG cohort (Illumina platform), instead of using liftOver, we replaced the probe positions with hg19 positions downloaded from the Illumina website. These normalized, hg19 probe-level copy number data are available at GEO with accession number GSE103123 (Data Citation 2). An overview of the processing steps applied to each of the collected datasets to obtain the data available at GEO is available at Figshare (Table 2: data processing steps, Data Citation 1). Starting from the normalized hg19 data, segmentation of the copy number profiles was done using SegAnnDB13 (http://seganndb.genap.ca/, a video with instructions can be found on the website). This is an interactive web-based tool that combines mathematical modelling with visual annotation to call aberrations, ensuring qualitative breakpoint calling for the majority of samples. Aberrations that were abundantly present across samples profiled on a certain platform were considered as artefacts (usually also a high variability in probe copy number value was observed for these regions) and ignored in the segmentation process. Due to the semi-automatic nature, not all aberrations are detected by this tool, e.g. subclonal aberrations might be missed. Only copy number data were used as input for the segmentation as allele ratios were available only for a limited number of samples. As not for all copy number profiling experiments gender-matched controls were used, data from chromosome Y were omitted. The segmented copy number profiles were downloaded from the SegAnnDB server for further analysis and are available at Figshare (Table 3: Segmented copy number data, Data Citation 1). A shorter version of these processing methods are included in the (Supplementary) Methods section of the related paper9.
Defining cutoffs to call aberrations
Histograms of the segment log2 ratios for each array platform reveal that their distribution is dependent on the array platform. More specifically, aCGH arrays generate log2 ratios with a higher dynamic range than SNP arrays (Fig. 2). Based on this observation, gains and losses were called using platform-specific cutoffs. Following cutoffs were used for gains/losses respectively: +0.15/-0.25 for SNP arrays (Affymetrix and Illumina), and +0.2/-0.3 for aCGH arrays (Agilent and NimbleGen), represented by the vertical lines in Fig. 2. Also for calling of amplifications platform-specific cutoffs were used. We compared copy number values (log2 ratio) of segments encompassing MYCN for MYCN-amplified and nonamplified samples (as determined based on FISH/qPCR analyses) (Fig. 3). Cutoffs were chosen to maximize discrimination between MYCN-amplified and nonamplified samples and exclude high-level gains: 1.5 for Affymetrix and NimbleGen arrays, 2 for Agilent arrays and 0.7 for Illumina arrays. Finally, segments with log2 ratio lower than -2 were called as homozygous deletion. This computational method using cutoffs generates a good general view of the aberrations present in the study population, but occasionally misses some aberrations, e.g. subclonal aberrations will typically not reach the cutoff. The resulting annotations are included in the segmented data available at Figshare (Table 3: Segmented copy number data, Data Citation 1). This paragraph is a more elaborate version of the procedure described in the related paper9.

Cutoffs are determined based on intersecting yellow curves (normal segments) with blue or red curves (lost/gained segments). For SNP arrays (left) lower cutoffs are used compared to aCGH platforms (right) to call copy number aberrations. Figure adapted from Supplementary Figure 1 in related paper9.

MYCN log2 ratios of MYCN-amplified cases are lower for SNP arrays than aCGH arrays, justifying platform-specific cutoffs to call amplifications. Figure adapted from Supplementary Figure 10 in related paper9.
Presence of segmental aberrations of frequently affected chromosome arms
For each patient, we determined the presence of aberrations in any of the frequently affected chromosome arms, namely loss of 1p, 3p, 4p, 11q and 14q, and gain of 1q, 2p and 17q. Aberrations of (partial) chromosome arms were defined as gains and losses larger than 3 Mb, hereby excluding whole chromosome aberrations and amplifications. In case of aberrations spanning the centromere, the aberration is considered to be on the p arm if the part on p is longer and vice versa. Note that this is a computational scoring generating a general image of abundance of aberrations, not aiming to establish a detailed genomic profile for individual patients. The resulting annotations are included in the patient annotations available at Figshare (Table 1: Patient annotations, Data Citation 1). This procedure is also described in the Supplementary Methods of the related paper9.
Code availability
The code to analyze or visualize the data in R can be found on GitHub (https://github.com/padpuydt/copynumber_HR_NB/). See Usage Notes for more information.
Data Records
All tables are available at figshare (Data Citation 1). Clinical and genomic information of the patients (Table 1: Patient annotations, Data Citation 1) include age at diagnosis, disease stage, and overall and event-free survival, MYCN status and the presence of segmental aberrations of chromosome arms frequently affected in neuroblastoma, such as 1p loss and 2p gain. Normalized probe-level copy number data using genome build hg19 can be accessed through GEO, with accession number GSE103123 (Data Citation 2). An overview is provided of the processing steps that were performed on the collected data to obtain the data that is available on GEO (Table 2: data processing steps, Data Citation 1), as well as segmented copy number data (Table 3: Segmented copy number data, Data Citation 1), in which the column “annotation” reports on the presence of an aberration considering the platform-specific cutoffs (see Methods).
Technical Validation
Bias
To check for possible bias, we performed t-SNE analysis using the R2 platform10, for several genomic (top), clinical (middle) and technical (bottom) variables (Fig. 4). This analysis reveals a clear clustering of samples according to MYCN status (Fig. 4a). Samples with only numerical aberrations are somewhat clustering together and situated in between the MYCN-amplified and MYCN-nonamplified group (Fig. 4b). Samples slightly cluster according to the clinical variables age at diagnosis (in three categories) and stage of the disease, but this mainly reflects the fact that young ( < 1 year) and non-stage 4 patients were only included in the study when presenting with MYCN amplification (Fig. 4c and d). In Fig. 4e, a slight bias is observed for different array platforms, with samples analyzed on CGH arrays (especially Agilent) being on the outer rim, and samples analyzed on SNP arrays (especially Illumina) situated closer to the center. This can be explained by the fact that CGH arrays generate copy number log2 ratios with a higher dynamic range than SNP arrays, as also observed in the histograms in Fig. 2. This effect is demonstrated in the simulations by Wattenberg et al. (section 6)14 . In Fig. 4f, we demonstrate that there is no bias across labs that use the same platform.

Samples are colored according to genomic (top), clinical (middle) and technical (bottom) variables, indicating that technical bias is limited to variable dynamic ranges among platforms and that samples cluster according to MYCN status. cog: Children’s Oncology Group, USA; fr-clb: Centre Léon Bérard, France; fr-ic: Institut Curie, France; gh: Ghent, Belgium; it: Genova, Italy; jap: Chiba, Japan; sp: Valencia, Spain; col: Cologne, Germany; vi: Vienna, Austria.
Detection of typical segmental and focal aberrations
Of the 556 collected high-risk samples, 97.3% contain segmental aberrations, corresponding to regions frequently affected in neuroblastoma. The heatmap of chromosomal gains and losses of all tumors (Fig. 5) clearly shows recurrent loss of 1p, 3p, 4p, and 11q and recurrent gain of 1q, 2p, 7 and 17q. This visualization confirms previous reports15 that most tumors with MYCN amplification also have 1p loss and 17q gain and that many tumors with 11q loss also present with 3p loss and 17q gain. A frequency plot of the gains and losses can be found in Fig. 1b of the related research paper9. Next to larger segmental aberrations, also focal (<5 Mb) aberrations were observed that have previously been described in neuroblastoma, including focal gains or amplifications of ALK and LIN28B.

Heatmap of aberrations in each individual sample, illustrating patterns of co-occurring aberrations, e.g. 11q and 3p loss, or MYCN amplification (MNA) and 1p deletion. WCA: whole-chromosome aberrations. Figure adapted from Supplementary Figure 5 in related paper9.
Detection of amplifications
In order to set a good cut-off to determine amplifications, log2 copy number ratios of the MYCN locus were compared in samples with or without MYCN amplification according to FISH and qPCR. This analysis (Fig. 3) reveals that the dynamic range of log2 ratios is highly variable between platforms and suggests that cutoffs used to call amplifications should be determined per platform. Using these cutoffs we can correctly determine MYCN amplification status (as previously established by FISH/qPCR) in 97% of Affymetrix samples, 95% of Illumina samples, 99% of Agilent samples and 100% of NimbleGen samples. As described in the related paper9 and by Curtis et al.16, detection of amplifications is more cumbersome in data from Illumina platforms and should be interpreted with caution.
Usage Notes
Probe-level data can be accessed at GEO with the accession number GSE103123 (Data Citation 2). Sample annotations are provided in the GEO series metadata and at Figshare (Table 1: Patient annotations, Data Citation 1). Instead of downloading probe-level data, it is also possible to directly use the segmented data, also available at Figshare (Table 3: Segmented copy number data, Data Citation 1). The segmented profiles have been manually inspected to ensure confidence of the called aberrations. The column “annotation” represents the presence of an aberration considering the platform-specific cutoffs (see Methods for details), but also the segment log2 ratios can be used.
Data can be analyzed and visualized using either the R statistical program, the R2 web-based platform10, or a Shiny web application17. For R users we recommend the R package “copynumber”18, which provides functions for visualization, including frequency plots and heatmaps with chromosome ideograms. The R package CNTools19 can be used to transform the segmented data into a window- or gene-based matrix. A script is provided on GitHub, including functions to select samples with an aberration for a certain gene/region (https://github.com/padpuydt/copynumber_HR_NB/).
Using the R2 platform10, one can perform multiple data analysis procedures and create several types of plots. A few examples are plotting a gene’s copy number in groups of samples, associations between sample annotations, survival analysis and viewing samples in the genome browser (Fig. 6). To create these plots, go to the dataset home page on R2 (https://hgserver1.amc.nl/cgi-bin/r2/main.cgi?&dscope=NB556HR&option=about_dscope). The R2 genome browser is directly linked there. The other three analyses/plots can be found under “Other Analyses”. There we selected “View a gene in groups”, “Relate 2 tracks” and “Kaplan Meier by annotated parameter” in step 3 (Select type of analysis). In addition, R2 allows to perform correlation analysis between genes, find genes with differential copy number between groups, perform dimensionality reduction with t-SNE maps, or generate a heatmap of a specific (grouped) subset of samples (CGH Sample Sorter on dataset home page). More detailed documentation can be found on the R2 website (http://r2.amc.nl).

(a) Copy number values of CHD5 (1p36.31), comparing samples with MYCN amplification (left) and without MYCN amplification (right). Other sample annotations including age at diagnosis can be read in the heatmap below the plot. (b) Comparing occurrence of two tracks, illustrating that 11q and 3p loss significantly co-occur. (c) Genome browser zooming in on a sample with amplifications for ODC1 (left amplified region) and MYCN (right amplified region). (d) Kaplan-Meier analysis comparing survival of cases with and without 6q loss.
Finally, we developed a Shiny web application17 to interrogate the copy number status and association with survival of any gene or genomic region of interest. After filling in the gene name or cytoband, the samples with an aberration overlapping with that region will be plotted in a heatmap. One can also plot survival curves of samples with versus without an aberration in the region of interest. A preliminary version can be accessed through https://padpuydt.shinyapps.io/check_cn_in_hr_nb/.
Additional information
How to cite this article: Depuydt. P. et al. Meta-mining of copy number profiles of high-risk neuroblastoma tumors. Sci. Data. 5:180240 doi: 10.1038/sdata.2018.240 (2018).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Westermann, F. & Schwab, M. Genetic parameters of neuroblastomas. Cancer lett. 184, 127–147 (2002).
Smith, M. A. et al. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J. Clin. Oncol. 28, 2625–2634 (2010).
Bosse, K. R. & Maris, J. M. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 122, 20–33 (2016).
Cohn, S. L. et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J. Clin. Oncol. 27, 289–297 (2009).
Park, J. R. et al. Revisions to the International Neuroblastoma Response Criteria: A Consensus Statement From the National Cancer Institute Clinical Trials Planning Meeting. J. Clin. Oncol. 35, 2580–2587 (2017).
Chen, Y. et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455, 971–974 (2008).
Cheung, N.-K. V. K. et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 307, 1062–1071 (2012).
Janoueix-Lerosey, I. et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J. Clin. Oncol. 27, 1026–1033 (2009).
Depuydt, P. et al. Genomic Amplifications and Distal 6q Loss: Novel Markers for Poor Survival in High-risk Neuroblastoma Patients. J. Natl. Cancer Inst. https://doi.org/10.1093/jnci/djy022 (2018).
Koster, J. R2: Genomics Analysis and Visualization Platform http://r2.amc.nl (2011).
Fieuw, A. et al. Identification of a novel recurrent 1q42.2-1qter deletion in high risk MYCN single copy 11q deleted neuroblastomas. Int. J. Cancer 130, 2599–2606 (2012).
Coco, S. et al. Age-dependent accumulation of genomic aberrations and deregulation of cell cycle and telomerase genes in metastatic neuroblastoma. Int. J. Cancer 131, 1591–1600 (2012).
Hocking, T. D. et al. SegAnnDB: interactive Web-based genomic segmentation. Bioinformatics 30, 1539–1546 (2014).
Wattenberg, M., Viégas, F. & Johnson, I. How to Use t-SNE Effectively. Distill https://doi.org/10.23915/distill.00002 (2016).
Vandesompele, J. et al. Unequivocal delineation of clinicogenetic subgroups and development of a new model for improved outcome prediction in neuroblastoma. J. Clin. Oncol. 23, 2280–2299 (2005).
Curtis, C. et al. The pitfalls of platform comparison: DNA copy number array technologies assessed. BMC Genomics 10, 588 (2009).
Chang, W. shiny: Web Application Framework for R (2017).
Nilsen, G. et al. Copynumber: Efficient algorithms for single- and multi-track copy number segmentation. BMC Genomics 13, 591 (2012).
Zhang, J. CNTools: Convert segment data into a region by sample matrix to allow for other high level computational analyses (2017).
Data Citations
Depuydt, P. et al. figshare https://doi.org/10.6084/m9.figshare.c.4169864 (2018)
Depuydt, P. et al. NCBI Gene Expression Omnibus GSE103123 (2018)
Acknowledgements
The authors thank Robrecht Cannoodt, Inge M. Ambros, Peter F. Ambros, Shahab Asgharzadeh, Edward F. Attiyeh, Valérie Combaret, Raffaella Defferrari, Matthias Fischer, Barbara Hero, Michael D. Hogarty, Meredith S. Irwin, Susan Kreissman, Ruth Ladenstein, Eve Lapouble, Geneviève Laureys, Wendy B. London, Katia Mazzocco, Akira Nakagawara, Rosa Noguera, Miki Ohira, Julie R. Park, Ulrike Pötschger, Jessica Theissen, Gian Paolo Tonini, Dominique Valteau-Couanet, Luigi Varesio, Rogier Versteeg and John M. Maris for their contributions to the UHR project.
Author information
Authors and Affiliations
Contributions
P.D. performed the data analysis, prepared the script and shiny application and wrote the manuscript. J.K. provided data integration and visualisation/analysis options in the R2 web platform. V.B. and T.D.H. contributed to data processing. F.S. and G.S. designed and supervised the study. K.D.P. designed and supervised the study and wrote the manuscript. All authors contributed to the preparation of the manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Depuydt, P., Koster, J., Boeva, V. et al. Meta-mining of copy number profiles of high-risk neuroblastoma tumors. Sci Data 5, 180240 (2018). https://doi.org/10.1038/sdata.2018.240
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.240
This article is cited by
-
Neuroblastoma arises in early fetal development and its evolutionary duration predicts outcome
Nature Genetics (2023)
-
Precise identification of cancer cells from allelic imbalances in single cell transcriptomes
Communications Biology (2022)
-
resistancebank.org, an open-access repository for surveys of antimicrobial resistance in animals
Scientific Data (2021)
-
The expression of ELOVL4, repressed by MYCN, defines neuroblastoma patients with good outcome
Oncogene (2021)
-
19p loss is significantly enriched in older age neuroblastoma patients and correlates with poor prognosis
npj Genomic Medicine (2020)
