
Abstract
Friedreich’s ataxia (FRDA; OMIM 229300), an autosomal recessive neurodegenerative mitochondrial disease, is the most prevalent hereditary ataxia. In addition, FRDA patients have shown additional non-neurological features such as scoliosis, diabetes, and cardiac complications. Hypertrophic cardiomyopathy, which is found in two thirds of patients at the time of diagnosis, is the primary cause of death in these patients. Here, we used small RNA-seq of microRNAs (miRNAs) purified from plasma samples of FRDA patients and controls. Furthermore, we present the rationale, experimental methodology, and analytical procedures for dataset analysis. This dataset will facilitate the identification of miRNA signatures and provide new molecular explanation for pathological mechanisms occurring during the natural history of FRDA. Since miRNA levels change with disease progression and pharmacological interventions, miRNAs will contribute to the design of new therapeutic strategies and will improve clinical decisions.
Design Type(s) | disease state design • microRNA profiling by high throughput sequencing assay |
Measurement Type(s) | microRNA profiling assay |
Technology Type(s) | RNA sequencing |
Factor Type(s) | diagnosis |
Sample Characteristic(s) | Homo sapiens • blood plasma |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
Friedreich’s ataxia (FRDA) is the most common hereditary ataxia, with elevated differences in symptomatology between individuals and within families. Instability of the GAA expansion size is responsible for approximately 50% of the variability found in disease onset1. Ataxia stems from spinocerebellar degeneration, peripheral sensory neuropathy, and cerebellar and vestibular pathology, and pyramidal disabilities begin to appear after its onset2. Other non-neurological features are related to Friedreich’s ataxia. For example, hypertrophic cardiomyopathy, which is the primary cause of death in these patients, is found very often in FRDA patients. Also, diabetes mellitus3 and scoliosis are associated with FRDA4,5.
MicroRNAs (miRNAs) are made up of about 22 nucleotides and are the best-characterized small non-coding RNAs (sncRNAs). There is a continuously increasing number of human transcripts corresponding to miRNAs, and their sequences and annotations have been published6. miRNA can target mRNAs and control their degradation when there is enough complementarity between the two or when translational repression of protein expression ocurrs7,8. Some miRNAs are released from cells in membrane-bound vesicles which protect them from RNase activity9, and for this reason miRNAs can be detected in circulating fluids such as plasma, serum, urine and saliva10–12. Besides their role in specific tissues, and recently as a stable molecule in circulating fluids13, miRNAs have been proposed as biomarkers for some diseases such as cancer14,15, diabetes16, neurodegenerative diseases17, etc.
A small RNA profiling dataset for FRDA patients and healthy controls was generated to identify different miRNA signatures that could explain physiological and molecular pathways underlying this disease and to help determine the phenotypic variability of patients18. We found different expression profiles of miRNAs (hsa-miR-128-3p, hsa-miR-625-3p, hsa-miR-130b-5p, hsa-miR-151a-5p, hsa-miR-330-3p, hsa-miR-323a-3p, and hsa-miR-142-3p) between samples from patients and samples from healthy subjects. In addition, we found that hsa-miR323a-3p is a biomarker for phenotypic differentiation in FRDA patients suffering from cardiomyopathy. To the best of our knowledge, this data set represents the largest public small RNA-seq data on a cohort of FRDA patients. The potential for identifying miRNA signatures in FRDA goes beyond the discovery of physiological and molecular pathways underlying this disease. Understanding the phenotypic variability of patients is also necessary for designing the most appropriate therapy for each of them, according to their specific pattern of disease progression.
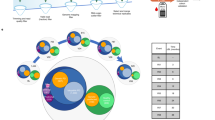
In this study, blood samples of FRDA patients (e.g.; FRDA patients with cardiomyopathy “N+C”, FRDA patients with diabetes “N+D”, FRDA patients with only neurological features “N”) and healthy controls were processed and plasma samples were obtained. Plasma samples were used to purify small RNA fractions, then were used to construct the small RNA-seq libraries, and finally were sequenced using the Illumina HiScanSQ platform (Figure 1). The sequence reads were mapped against the human Hg38 build (UCSC Genome Browser). After that, the intersection between the aligned position of the reads and the miRNA coordinates taken from miRBase v21 was performed. In short, we have provided a small RNA-seq data resource on FRDA patients, which is useful to understand phenotypical variability of the disease. Furthermore, the data resource provides an opportunity to identify the biomarkers of diagnosis, prognosis, and treatment monitoring in FRDA.

Plasma samples from Friedreich’s Ataxia (n=25) and healthy subjects (n=17) were analyzed. FRDA patients were classified into 3 groups: only neurological disorder (N; n=11), neurological disorder plus cardiomyopathy (N+C; n=9), and neurological disorder plus diabetes (N+D; n=6). One FRDA patient with neurologic symptoms additionally showed both comorbidities, cariomyopahty and diabetes, and thus this patient was classified in both neurological disorder plus cardiomyopathy (N+C), and neurological disorder plus diabetes (N+D) groups. Small RNA from the plasma samples of each FRDA patient and healthy subject was isolated and sequenced to obtain a miRNA expression profile. Next, mapping of the sequencing reads provided whole miRNome status of individual samples. Differential miRNA expression between FRDA patients and healthy subjects and within patients was performed to identify the miRNA signatures of FRDA patients and their concomitant diseases.
Methods
We have already presented some of the methods and tools used in our primary publication18. With this paper, we want to expand our previous descriptions and provide a comprehensive resource for reproducing both the experimental and computational analyses. The experimental and analytical procedure we used is described in Figure 1.
Patient and healthy subject recruitment and clinical features
Patients diagnosed with FRDA without neoplastic diseases or active infection were recruited. Data about age, sex, history of diabetes, cardiopathy, and number of GAA repeats in both alleles were recorded (Table 1; n=25). The scale for assessment and rating of ataxia (SARA), a neurological examination-based method to assess the disease, was used to measure the clinical severity of the disease19. FRDA patients were enrolled in the study following study approval by the Biomedical Research Ethics Committee (CEIB) of Hospital La Paz (Madrid). Plasma biospecimens from FRDA patients were preserved in a public repository of FRDA in the CIBERER Biobank (www.ciberer-biobank.es; Spanish Biobank Registry number: 000161X02). Healthy volunteers (n=25) with no neoplastic diseases, active infection, cardiomyopathy, heart problems, hypertension, or diabetes were enrolled by the Basque Biobank for Research-OEHUN (www.biobancovasco.org) and by the Biobank for Biomedical Research and Public Health of the Valencian Community (IBSP-CV) through the Spanish National Biobank Network (RNBB 2013/12). The subjects of both groups (healthy volunteers and FRDA patients) were matched by sex and age and were processed identically. The selection process and all experimental methods were carried out in accordance with the relevant clinical guidelines, following standard operation procedures, and with the approval of the ethics and scientific committees. All experimental protocols were approved by the Biomedical Research Ethics Committee (CEIB) of Hospital La Paz (Madrid) and the ethics and scientific committees of the IBSP-CV. Informed consent was obtained from all participants.
Sample collection and small RNA extraction and quantification
Plasma samples were extracted from FRDA patients and healthy participants. For that, blood samples were collected in EDTA tubes and centrifuged at 2500 rpm for 10 min. Once plasma was obtained, each sample was stored at −80 °C until RNA extraction. 500 μL of plasma were used to isolate cell-free total RNA (including miRNAs) using the miRNeasy Serum/Plasma kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol. The RNA was eluted with 25 μL of RNase-free water. The concentration of cell-free total RNA (including miRNAs) was quantified using NanoDrop ND 2000 UV-spectrophotometer (Thermo Scientific, Wilmington, DE, USA).
Small RNA-seq library preparation and sequencing
Small RNA samples were converted to Illumina sequencing libraries using the NEBNext Multiplex Small RNA Library Prep Set for Illumina (Set 1&2) (New England Biolabs, MA, USA), following the manufacturer’s protocol. Briefly, 5′ and 3′ adapters were ligated with small RNA molecules purified from plasma, followed by a cDNA library construction and incorporation of index tags by reverse transcription-PCR (RT-PCR). The products of this RT-PCR were purified using 6% non-denaturing polyacrylamide gel electrophoresis, and then size selection of 145–160 bp fraction was performed. The cDNA library samples were hybridized to a paired end flow cell and individual fragments were clonally amplified by bridge amplification on the Illumina cBot cluster generation. Then, the flow cell was loaded on the HiScanSQ platform and sequenced using Illumina’s sequencing by synthesis chemistry, generating 50 bp single end reads.
Pre-processing and processing of the reads
The quality of the small RNA libraries was first evaluated using FastQC v0.11.5 software (Figure 2). The most important metrics checked were the overall sequence quality: mean of phred quality per base and per read greater than 30; the GC percentage distribution per read: the data (red curve) is expected to approximately follow the theoretical distribution (blue curve) (Figure 2c). The peaks on the left or on the right side are an indicator of the presence of adapters in the reads. We also checked the presence/absence of overrepresented sequences. Based on the results obtained, the sequence reads were trimmed to remove the following adapter: 5′ AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC- NNNNNN- ATCTCGTATGCCGTCTTCTGCTTG 3′ from each sample. The 6 N’s sequence is unique per sample and allows it to be barcoded. After this step, the bases at the end of the sequences with a quality less than 20 were removed. Finally, all the sequences with length less than 18 nucleotides were discarded. These operations were performed using the tool Cutadapt20. The remaining sequences were aligned to the human genome reference (Hg38) from the UCSC Genome Browser. The expression of every miRNA per sample was measured using an annotation file from miRBase (v21). It contained all the mature miRNAs known in humans so far. The alignment and quantification steps were performed using the Subread21 and RSubread22 packages (http://subread.sourceforge.net/), respectively.

(a) Phred quality score distribution over all reads in each base. (b) Quality score distribution over all sequences. (c) GC content (%) distribution over all sequences.
Differential expression analysis
Differential expression analysis between FRDA patients and healthy subjects
Firstly, a differential expression analysis of the miRNAs was performed between patients (n=25) and controls (n=17). In order to do it, we filtered the miRNAs with less than 1 cpm (counts per million) in 17 samples (the size of the smallest group). Subsequently, we performed a correction of factors using the TMM method23 and calculated the effective library sizes. We also estimated the specific dispersions per miRNA with the quantile-adjusted conditional maximum likelihood (qCML) method24. The differential expression analysis was executed using the exact test24,25. In addition, we carried out a normalization of the original counts estimating the effective library sizes using the geometric mean. The normalized values of the most significant miRNAs (FDR <1e-4) obtained from the differential expression test were used to assess their correlation. Those miRNAs with a level of correlation lower than 0.7 were used to fit a logistic regression model with LASSO penalty26. In order to select the most important miRNAs of the model, a leave one out cross validation was performed. Those miRNAs that had non-zero coefficients at the value of λ and that gave minimum mean cross-validated error were selected.
Differential expression analysis between FRDA patients grouped by phenotype
We divided the FRDA patients into 3 subgroups according to their phenotype, considering the features described in Table 1. Thus, patients were grouped as 1) patients showing only neurological symptoms (n=11); 2) patients showing neurological symptoms “N” and suffering cardiomyopathy “N+C” (n=9), and 3) patients showing neurological symptoms and diabetes “N+D” (n=6). One FRDA patient with neurologic symptoms additionally showed both comorbidities, cardiomyopathy and diabetes, and thus this patient was classified in both the neurological disorder plus cardiomyopathy (N+C), and neurological disorder plus diabetes (N+D) groups. After this stratification, those miRNAs which did not reach 1 cpm (count per million) in at least 5 samples (size of the smallest group) were filtered out. The data were normalized using the TMM method. Afterwards, a Cox-Reid dispersion27 per miRNA was estimated. To find the differentially expressed miRNAs among the three groups compared, the GLM (generalized linear model) 25 approach was used. Additionally, we performed new analyses taking into account other variables such as age, sex and disease onset. The last variables were organized as a dichotomy variable according to median values: 37 and 13 years, respectively. Finally, we carried out every comparison between the different groups using the GLM approach. All statistical analyses were performed using R software (version 3.2.2) and the following packages: edgeR (version 3.12.0), DESeq (version 1.22.0), caret (version 6.0–58), glmnet (version 2.0-2), ROCR (version 1.0-7).
Data Records
RNA-seq data files in FastQ format were deposited at NCBI Sequence Read Archive (Data Citation 1). This accession contains a total of 42 FastQ files resulting from the single end runs for each of the 42 samples. The FastQ format data serves as the raw data from sequencing, which are subjected to further downstream processing. The processed data were deposited at NCBI Gene Expression Omnibus (Data Citation 2).
Technical Validation
Sequencing quality control
We used FastQC v0.11.5 software to perform quality control assessments of the FastQ files before and after the pre-processing steps (filtering, quality trimming and adapter removal). We analysed several measurements, including the overall sequence quality, the GC percentage distribution (i.e. the proportion of guanine and cytosine bp across the reads) and the presence/absence of overrepresented sequences. A representative summary plot after the pre-processing steps is shown (11_TAGCTT_L005_R1_001). Here, the quality scores per base were high, with a median quality score above 30 suggesting high quality sequences across all bases (Figure 2a). The quality score distribution over all sequences was analyzed to see if a subset of sequences had universally poor quality. The average quality for most sequences was high, with scores above 37, which indicated that a significant proportion of the sequences in a run had overall high quality (Figure 2b). The GC distribution per base over all sequences was examined. Despite the GC composition pattern being more similar to the theoretical distribution after the pre-processing steps, it still had a bias (Figure 2c). In addition, overrepresented sequences were examined. Before the adapter removal, some of them were identified as the Illumina indexed adapters used in the sequencing process. After this step, the adapters were no longer identified but we still had overrepresented sequences, possibly because of highly expressed miRNAs (Table 2). All other FastQC files were shown to have similar quality metrics compared to sample (11_TAGCTT_L005_R1_001).
Real-time qPCR validation of selected miRNAS from Small RNA-seq
Reverse transcription reactions were performed using the TaqMan miRNA Reverse Transcription kit and miRNA-specific stem-loop primers (Part No. 4366597, Applied Biosystems. Inc, CA; USA) and 100 ng of input cell-free RNA in a 20 μL RT reaction. Real-time PCR reactions were performed in triplicate, in scaled-down 10 μL reaction volumes using 5 μL TaqMan 2x Universal PCR Master Mix (Applied Biosystems. Inc, CA; USA) with No UNG, 0.5 μL TaqMan Small RNA assay (20x) (Applied Biosystems. Inc, CA; USA) [hsa-miR-128-3p (002216), hsa-miR-625-3p (002432), hsa-miR-130b-5p (002114), hsa-miR-151a-5p (002642), hsa-miR-330-3p (000544), hsa-miR-323a-3p (002227), hsa-miR-142-3p (000464), hsa-miR-16-5p (000391)], 3.5 μL of nuclease free water and 1 μL of RT product. Real-time PCR was carried out on an Applied BioSystems 7900HT thermocycler (Applied Biosystems. Inc, CA; USA) programmed as follows: 50 °C for 2 min, 95 °C for 10 min followed by 45 cycles of 95 °C for 15 s and 60 °C for 1 min. We used hsa-miR-16-5p (000391), one of the most stable miRNAs in terms of read counts, and which has been used previously as an endogenous control27, to normalize the expression in plasma samples. RNU48 (001006), meanwhile, was used to normalize the expression in cell-line samples. All the fold-change data were obtained using the delta-delta CT method (2−ΔΔCT)28. The seven differentially expressed miRNAS detected after small RNA-seq were validated using RT-qPCR. All miRNAs were present in plasma at higher levels in patients (n=25) compared to healthy controls (n=25), in agreement with the results obtained by small RNA-seq. Relative expression levels of the miRNAs in plasma from FRDA patients compared to healthy subjects were shown in Seco-Cervera et al.18
Usage Notes
Before processing the raw reads (Data Citation 1) we performed a visual exploration of them by looking for the adapter used in the sequencing process. We saw adapters ligated to the 5′ end for some reads and to the 3′ end in other reads. Despite expecting to always find it at the 3′ end, the opposite situation can sometimes occur. Therefore, we removed the adapter specifying the -b option in Cutadapt. It indicates to the program that the adapter may appear at the beginning (even degraded), within the read, or at the end of the read (even partially).
The alignment can be performed using standard tools, such as Bowtie229, STAR30, or Burrows-Wheeler Aligner (BWA)31. In our study, we selected the Subread aligner because it is more accurate and faster than previous aligners (nearly four times as fast as the nearest competitor, Bowtie2)21. Additionally, the parameters needed when mapping miRNA-seq reads have been well documented. On the other hand, although known miRNA sequences from miRbase can be used as a reference, we suggest using the whole human genome. In this way, the reads aligning to miRNA sequences and to many other features in the genome at the same time can be discarded. The quantification of microRNA expression can be performed using tools like bedtools intersect32 or featureCounts22. In this step, it is important to allow multiple hits of each read when mapping, since there are multiple copies of some microRNAs in the genome and if it is not allowed, the results might be misleading, or wrong.
Regarding differential expression analysis, we recommend using the popular R packages EdgeR25 and DESeq33. In the case of using the EdgeR package, it is necessary to filter miRNAs which are not expressed in any condition since they can add some noise to the analysis. Another important aspect to note is the use of an appropriate method according to the different types of comparisons performed. When considering a single study factor, qCML is a good method to estimate the dispersions per miRNA and the exact test to do the differential expression analysis. However, when two or more study factors are included in the analysis, it is highly recommended to estimate dispersions per miRNA with the CR method and to use the likelihood ratio test GLM for differential expression.
Additional information
How to cite this article: Seco-Cervera, M. et al. Small RNA-seq analysis of circulating miRNAs to identify phenotypic variability in Friedreich’s ataxia patients. Sci. Data 5:180021 doi: 10.1038/sdata.2018.21 (2018).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Filla, A. et al. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am J Hum Genet 59, 554–560 (1996).
Delatycki, M. B. & Corben, L. A. Clinical features of Friedreich ataxia. Journal of child neurology 27, 1133–1137 (2012).
Finocchiaro, G., Baio, G., Micossi, P., Pozza, G. & di Donato, S. Glucose metabolism alterations in Friedreich's ataxia. Neurology 38, 1292–1296 (1988).
McCabe, D. J. et al. Typical Friedreich's ataxia without GAA expansions and GAA expansion without typical Friedreich's ataxia. Journal of neurology 247, 346–355 (2000).
Parkinson, M. H., Boesch, S., Nachbauer, W., Mariotti, C. & Giunti, P. Clinical features of Friedreich's ataxia: classical and atypical phenotypes. Journal of neurochemistry 126 (Suppl 1): 103–117 (2013).
Kozomara, A. & Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic acids research 42, D68–D73 (2014).
Peters, L. & Meister, G. Argonaute proteins: mediators of RNA silencing. Molecular cell 26, 611–623 (2007).
Hammond, S. M., Bernstein, E., Beach, D. & Hannon, G. J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404, 293–296 (2000).
Fevrier, B. & Raposo, G. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol 16, 415–421 (2004).
Mitchell, P. S. et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proceedings of the National Academy of Sciences of the United States of America 105, 10513–10518 (2008).
Park, N. J. et al. Salivary microRNA: discovery, characterization, and clinical utility for oral cancer detection. Clin Cancer Res 15, 5473–5477 (2009).
Hanke, M. et al. A robust methodology to study urine microRNA as tumor marker: microRNA-126 and microRNA-182 are related to urinary bladder cancer. Urol Oncol 28, 655–661 (2010).
Russo, F. et al. in Epigenetic Biomarkers and Diagnostics, 239–258 (Academic Press, 2016).
Lopez-Serra, P. & Sandoval, J. in Epigenetic Biomarkers and Diagnostics, 539–555 (Academic Press, 2016).
Stefansson, O. A in Epigenetic Biomarkers and Diagnostics, (587–612 (Academic Press, 2016).
Brennan, E., McClelland, A., Hagiwara, S., Godson, C. & Kantharidis, P in Epigenetic Biomarkers and Diagnostics, 643–661 (Academic Press, (2016).
Maciotta, S., Meregalli, M. & Torrente, Y. The involvement of microRNAs in neurodegenerative diseases. Frontiers in cellular neuroscience 7, 265 (2013).
Seco-Cervera, M. et al. Circulating miR-323-3p is a biomarker for cardiomyopathy and an indicator of phenotypic variability in Friedreich's ataxia patients. Sci Rep 7, 5237 (2017).
Schmitz-Hubsch, T. et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 66, 1717–1720 (2006).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 17, 10–12 (2011).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41, e108 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11, R25 (2010).
Robinson, M. D. & Smyth, G. K. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 23, 2881–2887 (2007).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw 33, 1–22 (2010).
McCarthy, D. J., Chen, Y. & Smyth, G. K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40, 4288–4297 (2012).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol 11, R106 (2010).
Data Citations
Acknowledgements
This research was supported by FIS (PI12/02263), FIS (PI16/01031) from ISCIII and co-financed by the European Regional Development Funds (ERDF) and ACCI2014 (CIBERer-ISCIII). J.L. García-Giménez and F.V.Pallardó are supported by the Ministerio de Economía y Competitividad, Instituto de Salud Carlos III through CIBERer (Biomedical Network Research Center for Rare Diseases and INGENIO2010). M. Seco-Cervera is an ISCIII's fellow (reference number: FI14/00433) co-financed by the European Regional Development Funds (ERDF) at Fundación INCLIVA. J.S Ibáñez-Cabellos is an “Atracció de Talent” fellow (University of Valencia). We would like to give a special thanks to all the participants enrolled in this study for their participation and to the Basque Biobank for Research-OEHUN, the Biobank for Biomedical Research and Public Health of the Valencian Community and CIBERER’s Biobank for their collaboration. L. Peiró-Chova would like to offer thanks for the financial support provided by the Ministerio de Economía y Competitividad, ISCIII, FEDER, under grant PT13/0010/0004. We would like to thank Dr. Javier Chaves and the Unidad de Genotipado y Diagnóstico Genético del Instituto de Investigación Sanitaria INCLIVA for library construction and small RNA sequencing.
Author information
Authors and Affiliations
Contributions
Study design: J.L.G.G., M.S.C. and F.V.P. Study conduct: J.L.G.G. and F.V.P. miRNA extraction, small RNA-seq and qRT-PCR validation: M.S.C., J.L.G.G., J.S.I.C., L.P.C. Bioinformatics and biostatistics analyses: D.G.R., and M.S.C. Data analysis: J.L.G.G., M.S.C., D.G.R., and L.P.C. Data interpretation: J.L.G.G., M.S.C., and F.V.P. Manuscript drafting: M.S.C. and J.L.G.G. Review of manuscript content: M.S.C. and J.L.G.G. Approval of final version of manuscript: all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Seco-Cervera, M., González-Rodríguez, D., Ibáñez-Cabellos, J. et al. Small RNA-seq analysis of circulating miRNAs to identify phenotypic variability in Friedreich’s ataxia patients. Sci Data 5, 180021 (2018). https://doi.org/10.1038/sdata.2018.21
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2018.21
This article is cited by
-
An RNA-seq study in Friedreich ataxia patients identified hsa-miR-148a-3p as a putative prognostic biomarker of the disease
Human Genomics (2024)
-
Colonization of the Southern Hemisphere by Sagina and Colobanthus (Caryophyllaceae)
Plant Systematics and Evolution (2022)
-
COMPSRA: a COMprehensive Platform for Small RNA-Seq data Analysis
Scientific Reports (2020)
-
A Comprehensive Transcriptome Analysis Identifies FXN and BDNF as Novel Targets of miRNAs in Friedreich’s Ataxia Patients
Molecular Neurobiology (2020)
-
Whole-exome and whole-transcriptome sequencing of canine mammary gland tumors
Scientific Data (2019)
