
Abstract
The scarcity of long-term data on soil microbial communities in the decades following timber harvesting limits current understanding of the ecological problems associated with maintaining the productivity of managed forests. The high complexity of soil communities and the heterogeneity of forest and soil necessitates a comprehensive approach to understand the role of microbial processes in managed forest ecosystems. Here, we describe a curated collection of well replicated, multi-faceted data from eighteen reforested sites in six different North American ecozones within the Long-term Soil Productivity (LTSP) Study, without detailed analysis of results or discussion. The experiments were designed to contrast microbial community composition and function among forest soils from harvested treatment plots with varying intensities of organic matter removal. The collection includes 724 bacterial (16S) and 658 fungal (ITS2) amplicon libraries, 133 shotgun metagenomic libraries as well as stable isotope probing amplicon libraries capturing the effects of harvesting on hemicellulolytic and cellulolytic populations. This collection serves as a foundation for the LTSP Study and other studies of the ecology of forest soil and forest disturbance.
Design Type(s) | observation design • longitudinal data collection method |
Measurement Type(s) | GeneSequence • whole genome sequencing • metagenomics analysis |
Technology Type(s) | amplicon sequencing • Whole-Genome Shotgun Sequencing • radioisotope labeled DNA probe |
Factor Type(s) | soil • temperature • hydrological precipitation process • environmental factor • experimental condition • isotopic labelling |
Sample Characteristic(s) | soil metagenome • Province of Ontario • State of California • State of Texas • Province of British Columbia • forest soil |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
The Long-term Soil Productivity Study (LTSP) was initiated in 1989 to track changes in soil quality and productivity of managed forests. Foresters lacked data on the impact of growing trends in intensified forest management, such as shorter crop cycles, greater biomass extraction, and use of highly mechanized equipment1. Partners in the LTSP Study have since collected longitudinal data from a range of North America’s most-productive forests (>100 sites) to assess the long-term effects of soil compaction and organic matter (OM) removal. The objective is to develop indices for monitoring soil quality based in physical, chemical, and biological properties of soil. Each LTSP site has replicated experimental plots for harvested treatments with three intensities of OM removal as well as unharvested reference plots. OM removal intensity corresponded to common harvesting strategies, such as debranching in situ (OM1) or removal of both trunks and branches (OM2), or the less common, and extreme, practice of removing trunks, branches and top soil (OM3). In the ensuing years, these treatments produced a consistent gradient in soil properties according to harvesting intensity, such as decreasing amounts of total carbon and nitrogen as well as increased dryness, mean daily temperature and temperature fluctuation. While the latest LTSP studies show that the effects of varying degrees of OM removal appear minor on net primary productivity2,3,4, the metagenomic datasets presented here show consistent changes in bacterial and fungal populations that reflected harvesting intensity5,6,7,8,9.
We collected data on soil microbial communities from all treatment plots at eighteen LTSP sites in six different ecozones between 2008 and 2014 when reforested stands were 11 to 17 years old (Fig. 1; Table 1). To capture the extent of harvesting impacts throughout the soil profile, corresponding samples from organic and mineral layers were included in all datasets. The goal was to identify changes in community composition, diversity, and functional potential resulting from the intensity of OM removal. We surveyed soil microbial communities in treatment plots using amplicon sequencing (Table 2 (available online only)) of bacteria (Data Citation 1) and fungi (Data Citation 2), along with shotgun metagenomes from all treatment plots at a single site within each ecozone (Data Citation 3; Table 3 (available online only)). Shotgun metagenomes revealed impacts on the functional potential of communities, such as decomposers involved in carbon cycling5, which led to further targeted studies of hemicellulolytic7 (Data Citations 4,5) and cellulolytic8 populations (Data Citation 6) using 13C-stable isotope probing (Table 4 (available online only)). Stable isotope probing (SIP) can be used to track populations that assimilate 13C-label into their biomass by recovering and sequencing the resultant ‘heavier’13C-enriched DNA10.

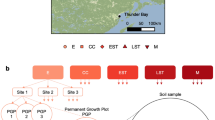
The locations and names of eighteen North American sampling sites are shown grouped into six ecozones. The term ‘ecozone’ is used to refer to the distinct local assemblages of organisms and climatic factors between groupings of sites. An overview of the design for soil sampling along with data collection are superimposed on the map. OM removal treatments are shaded according to the intensity of OM removal.
Here, we provide an overview of the data collection, without detailed analysis of results or discussion, to draw attention to its unparalleled comprehensive and multi-faceted view into forest soil microbial communities. The collection is the first high-throughput sequencing-based survey of LTSP sites, and, as such, provides base-line data for future LTSP investigations at an important, stage of forest regeneration just prior to canopy closure. Researchers can revisit this collection as our understanding of the ecology of forest soils advances. This will be important given the substantial proportion of unknown taxa in these collections affected by timber harvesting9. SIP data offers unique insights into the effects of timber harvesting on decomposer populations, including detailed information on uncultured taxa provided by the ten partial genomes recovered from SIP-cellulose shotgun metagenomes (Data Citation 6). The consistency in experimental design, sequencing methodology and sample sources ensures the value of this collection for on-going studies of forest soil microbial communities, in particular those pertaining to biogeography, soil strata and forest disturbance.
Methods
Experimental design
Soil samples were collected from reforested experimental plots within the Long-Term Soil Productivity (LTSP) Study from eighteen different sites across six conifer-dominated North American ecozones named after the predominant tree species (Fig. 1; Table 1): IDFBC (interior Douglas-fir), SBSBC (sub-boreal spruce), PPCA (ponderosa pine), BSON (black spruce), JPON (jack pine) and LPTX (loblolly pine). Ecozones were chosen to exemplify a broad range of climates and regions in North America where forestry is a major industry. These differed by several factors, including soil type, mean annual temperature and precipitation, tree species and bulk soil chemistry, such as carbon and nitrogen content and pH (Table 1). Each ecozone contained three sites with four treatments: REF (or OM0), a neighbouring unharvested reference plot; and three harvested treatments: OM1, where only tree boles (stems) were removed and woody debris was left in place; OM2, where whole trees including branches were removed and; OM3, where whole trees were removed and the upper organic layer of forest floor scraped away (Fig. 2). Compaction was controlled and, in all cases, plots with minimum compaction (‘C0’) were sampled. Moderate (C1) and severe (C2) compaction treatments were included in 16S rRNA gene and ITS pyrotag libraries in SBSBC and IDFBC (Table 2 (available online only)). Similarly, additional amplicon libraries were prepared from soils in JPON and LPTX which had been exposed to glyphosate (Table 2 (available online only)). Samples were collected from triplicate plots at each of the three sites in BSON and JPON, while at sites in the other four ecozones triplicate samples were collected from a single, larger plot. Each sample corresponds to a composite from between three to five sampling points (consistent within a given ecozone) in a plot which helped account for soil heterogeneity. Organic layer samples (O-horizons) were first sampled with a trowel and then the top 20 cm of mineral layer soil (A and upper B-horizon) were collected using a Stoney auger (5 cm diameter). For several 16S rRNA gene amplicon and whole shotgun sequencing libraries from Skulow Lake (denoted in Tables 2 and 3 (available online only)), samples were collected from five soil horizons: LFH and mineral horizons (Ahe, Ae, AB and Bt), distinguished using criteria from the Canadian System of Soil Classification. Samples were stored at 4 °C during transport and until each sample was sieved through 2-mm mesh to remove roots, then stored at −80 °C until DNA was extracted within three months of sampling. Soil samples used in metatranscriptomics were flash frozen in liquid nitrogen, transported on dry ice and subsequently stored at −80 °C until DNA and RNA was extracted within 12 months of sampling.

These photographs capture the initial variation in the amount of organic matter (OM) removal at plots which were sampled 11–17 years for this study. Plots like these were replicated at three sites within every ecozone [photo credit: Dr Matt Busse; mbusse@fs.fed.us
Amplicon and shotgun metagenome sequencing
DNA was extracted from field samples (0.5 g of soil) using the manufacturer’s recommended protocol for the FastDNA Spin Kit for Soil (MPBio, Santa Ana, CA). Amplicon libraries were prepared for the 16S rRNA gene (V1-V3 regions) and fungal internal transcribed spacer region (ITS2) according to the procedure of Hartmann et al.5. The region spanning V1–V3 was amplified using barcoded universal primers: 27F (5′- AGA GTT TGA TCM TGG CTC AG–3′) and 519R (5′- GWA TTA CCG CGG CKG CTG–3′)11,12 and the fungal internal transcribed spacer (ITS2) region was amplified using barcoded primers: ITS3 (5′- GCA TCG ATG AAG AAC GCA GC–3′) and ITS4 (5′- TCC TCC GCT TAT TGA TAT GC–3′)13. Amplicons were generated via polymerase chain reaction (PCR) in triplicate for each sample and pooled prior to purification and quantification. DNA quantitation was performed using Pico-Green fluorescent dye (ThermoFisher, MA, USA). Samples were sequenced using the Roche 454 Titanium platform at the McGill University and at the Genome Québec Innovation Centre with a maximum of 40 samples multiplexed on each quarter plate. Table 2 (available online only) contains information on all amplicon libraries created from field soil samples, including 16S rRNA gene amplicon (Data Citation 1) and ITS amplicon libraries (Data Citation 2). This includes a second set of samples from Skulow Lake with a narrower focus on five soil depths in only REF and OM3 (Table 2 (available online only)). These amplicon libraries were made from primers targeting the V6-V8 region of the 16S rRNA gene and were amplified according to Gies et al.14 using barcoded universal primers: 926F (5′- CC TAT CCC CTG TGT GCC TTG GCA GTC TCA GAA ACT YAA AKG AAT TGR CGG-3′) and 1392R (5′‐ CGT ATC GCC TCC CTC GCG CCA TCA GAC GGG CGG TGT GTR C-3′). PCR product was pooled, purified, quantified and sequenced as for other libraries.
Whole shotgun metagenomes were generated for a single site in each of the six ecozones resulting in three replicates for each treatment and each soil horizon for each ecozone. At Skulow Lake (SBSBC), sampling for metagenomes focused on changes along the soil profile and, thus, did not cover all four OM harvesting treatments, only REF versus OM3. Unlike in pyrotag libraries, a second complete set of shotgun metagenomes from Skulow Lake does not exist. Insufficient organic layer soil was available from OM3 to prepare shotgun metagenomes for BSON, IDFBC, JPON, and PPCA. The same soil samples were used for shotgun and amplicon metagenomes, but from separate DNA extractions. After quantification, triplicate samples were multiplexed in each Illumina HiSeq lane for sequencing. Samples from ecozones BSON, JPON, PPCA and LPTX were sequenced at the US DOE Joint Genome Institute (Walnut Creek, CA) producing paired-end, 150-bp Illumina libraries while samples for the IDFBC and SBSBC ecozones were sequenced at the Michael Smith Genome Sciences Centre (Vancouver, Canada) resulting in paired-end, 75-bp and 100-bp Illumina libraries, respectively.
Stable isotope probing amplicon and shotgun metagenome sequencing
Microcosms were prepared by adding between 0.75 and 2.0 g of 2 mm sieved organic or mineral layer soil to 30-mL serum vials. Larger quantities of mineral soil were necessary because of lower microbial biomass, requiring two DNA extractions per mineral soil to obtain the necessary 5 μg of DNA. Moisture content was standardized to 60% w/v for mineral soil, due to lower absorptive capacity, and 125% w/v for organic soil and pre-incubating at 20 °C for one week. Microcosms were then amended with either 1.0% w/w of 13C-labeled hemicellulose (97 atom %; IsoLife; U-10509, Lot: 0901-0273) or custom prepared bacterial cellulose (99 atom % 13C). Each soil sample was paired with a microcosm containing the same amount of unlabelled (~1.1 atom % 13C) substrate. Separation between 13C-enriched DNA and unlabelled DNA is never complete, in part, due to variation in GC content15. Thus, 13C-libraries are always compared to identically prepared sequencing libraries from samples amended with unlabelled substrate. Following either 2-day (hemicellulose), 11-day (cellulose—organic layer) or 14-day (cellulose—mineral layer) incubations, soil was lyophilized and stored at −80 °C until DNA was extracted as previously described. DNA extracts from replicates within each site were pooled in equal amounts and unlabelled controls were processed identically. Harvested treatment OM2 was not included in SIP-hemicellulose libraries.
High purity 13C-labled cellulose was necessary to ensure that only organisms possessing the necessary metabolic capability could assimilate the 13C. Bacterial cellulose was utilized due to irremediable impurity in plant-derived cellulose from IsoLife (58% glucose+4.4% lignin+remainder of sugars from hemicellulose). Bacterial cellulose was produced by cultivating Gluconacetobacter xylinus str. KCCM 10,100 with either 13C-labeled glucose (99 atom % 13C, Cambridge Isotope Laboratories, MA, USA), or unlabelled glucose, as sole carbon source in Yamanaka medium16. Cellulose pellicules were purified by boiling in sodium hydroxide per previously described methods17, with the addition of a second boiling step and an increase in boiling time to 4 h. While IsoLife hemicellulose was also impure (53% hemicellulose sugars), the sources of impurity were more recalcitrant forms of carbon, such as cellulose and lignin, which were not substantially degraded during the 2-day incubation.
13C-enriched DNA was separated and recovered using cesium chloride density gradient ultracentrifugation10. The atom % 13C was measured before and after density separation using UHPLC-MS/MS18. Amplicon libraries were prepared and sequenced from 13C-enriched DNA for SIP-hemicellulose (Data Citations 4,5) and SIP-cellulose (Data Citation 6) experiments targeting both bacterial and fungal phylogenetic markers as previously described (Table 4 (available online only)). Four shotgun metagenome libraries were generated from 13C-enriched DNA from SIP-cellulose incubations for REF, OM1 and OM3 treatments and one unlabelled control sample (REF) from PPCA. Sufficient DNA for metagenome library preparation was achieved by pooling the corresponding DNA extracts from mineral layer samples at all three sites in PPCA. Shotgun metagenomes were prepared from 40–50 ng of enriched DNA using the Nextera DNA Sample Preparation Kit (Illumina Inc., CA, USA) and were multiplexed on two lanes of Illumina HiSeq (2×100-bp), yielding 285 million paired-end reads (Data Citation 6). There was insufficient 13C-enriched DNA to generate metagenomes from the organic layer samples. Raw sequences were quality-controlled, assembled and binned into partial genomes (Table 4 (available online only); Data Citation 6) according to methods described in Wilhelm et al.8.
Data Records
The raw pyrosequencing output (~400-bp reads) for all 16S rRNA gene (n=724 samples) and ITS (n=658 samples) amplicon libraries that were generated from field soil samples averaged 8,900 and 8,400 reads per library (post-QC), respectively, and are archived at the European Sequencing Archive (Table 2 (available online only)). The raw SFF files for 16S rRNA amplicon libraries can be found with the study accession PRJEB8599 (Data Citation 1), except for all libraries from British Columbia which were archived in study accession PRJEB12501 in ‘fastq’ format (Data Citation 2). The latter contains the entire collection of ITS amplicon libraries in ‘fastq’ format. All libraries were extracted from raw SFF files using the mothur command ‘sffinfo’ and either uploaded in standard flowgram format (SFF) or converted to ‘fastq’ format19 using ‘sffinfo’ to produce paired ‘fasta’ and ‘qual’ files, which were merged into ‘fastq’ format using ‘PairedFastaQualIterator’ from the SeqIO module in BioPython20.
Raw shotgun metagenomic data for all ecozones can be downloaded in ‘fastq’ format with the study accession PRJEB8420 from the European Nucleotide Archive (Table 3 (available online only), Data Citation 3). After our quality filtering, libraries had a median count of 59.3 million sequences for 150-bp read libraries, while shorter read libraries (75-bp) had higher median counts (115 million). These numbers are provided as an estimate of the number of high quality reads obtainable from our raw data, but will change depending on the parameters selected during quality processing.
Due to cost of 13C-labeled materials and additional labour required to process SIP samples, only a subset of ecozones were selected for SIP-hemicellulose (PPCA & IDFBC) and SIP-Cellulose (PPCA) characterizations. The raw pyrosequencing output (~400-bp reads) for SIP-hemicellulose 16S rRNA (Data Citation 4) and ITS (Data Citation 5) amplicon libraries, which averaged 4,200 and 3,800 reads per library (post-QC), respectively, are available in SFF format from the European Nucleotide Archive (Table 4 (available online only)). These datasets include both 13C- and 12C-libraries for 16S rRNA genes, n=35 and 15, respectively, and for ITS, 18 and 2, respectively. Similarly, the raw sequencing data for all SIP-cellulose 16S rRNA amplicon and ITS amplicon libraries, averaging 11,800 and 10,300 reads per library, respectively, are available in ‘fastq’ format from the European Nucleotide Archive (Table 4 (available online only)). The 13C- and 12C-libraries from these data are more balanced, with a total of 2416S rRNA gene libraries for both, and 24 and 23, respectively, for ITS libraries. Raw Illumina, paired-end, 100-bp shotgun metagenome sequencing libraries for pooled, SIP-cellulose mineral soil incubations were archived in ‘fastq’ format (Data Citation 6; Table 4 (available online only)), along with 10 partial genomes in ‘fasta’ format comprised of assembled scaffolds (Data Citation 6; Sample accessions: ERZ288956—ERZ288966).
Technical Validation
The recovery of 13C-enriched DNA was validated by quantifying 13C-content of nucleic acids18 (Fig. 3). The completeness of draft genomes recovered from SIP-Cellulose metagenomes was assessed by scanning for essential single-copy, house-keeping genes with hidden Markov models21.

The assimilation of 13C by functional guilds during stable isotope probing experiments was evident in (a) the total 13C-enrichment of soil DNA extract and (b) recovery of DNA from the densest CsCl gradient fractions (F1-F7). These differences were evident in comparing soils amended with either 12C- (unlabelled) or 13C-cellulose. These trends were apparent in both cellulose and hemicellulose SIP experiments. Boxplots depict the average (centre line) and spread (from 25th to 75th percentile) of data, while the whiskers extend to the extrema. A total of twelve samples were averaged for each factor.
Usage Notes
Metagenomes for all ecozones, except IDFBC, can be also found at the DOE JGI portal (http://genome.jgi.doe.gov/) under proposal ID 543. This site provides the raw sequencing data and annotation for the assembled and unassembled metagenomes.
Additional Information
Tables 2–4 are only available in the online version of this paper.
How to cite this article: Wilhelm, R. C. et al. A metagenomic survey of forest soil microbial communities more than a decade after timber harvesting. Sci. Data 4:170092 doi: 10.1038/sdata.2017.92 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Powers, R. F. et al. The North American long-term soil productivity experiment: Findings from the first decade of research. Forest Ecol. Manag. 220, 31–50 (2005).
Thiffault, E. et al. Effects of forest biomass harvesting on soil productivity in boreal and temperate forests—A review. Environ. Rev. 19, 278–309 (2011).
Ponder, F. et al. Effects of organic matter removal, soil compaction and vegetation control on 10th year biomass and foliar nutrition: LTSP continent-wide comparisons. Forest. Ecol. Manag. 278, 35–54 (2012).
Holub, S. M., Terry, T. A., Harrington, C. A., Harrison, R. B. & Meade, R. Tree growth ten years after residual biomass removal, soil compaction, tillage, and competing vegetation control in a highly-productive Douglas-fir plantation. Forest Ecol. Manag. 305, 60–66 (2013).
Cardenas, E. et al. Forest harvesting reduces the soil metagenomic potential for biomass decomposition. ISME J. 9, 1–12 (2015).
Hartmann, M. et al. Significant and persistent impact of timber harvesting on soil microbial communities in Northern coniferous forests. ISME J. 6, 2199–2218 (2012).
Leung, H. T., Maas, K. R., Wilhelm, R. C. & Mohn, W. W. Long-term effects of timber harvesting on hemicellulolytic microbial populations in coniferous forest soils. ISME J. 10, 363–375 (2016).
Wilhelm, R. et al. Long-term Enrichment of Stress-tolerant Cellulolytic Soil Populations following Timber Harvesting Revealed by Multi-omic Stable Isotope Probing. Front. Microbiol 8, 537 (2017).
Wilhelm, R. et al. Biogeography and Organic Matter Retention Shape Effects of Timber Harvesting on Forest Soil Microbial Communities in Long-term Soil Productivity Study. ISME J. 10.1038/ismej.2017.109 (2017).
Neufeld, J. D. et al. DNA stable-isotope probing. Nat. Protoc. 2, 860–866 (2007).
Lane, D. J. in Nucleic Acid Techniques in Bacterial Systematics (eds Stackebrandt, E. & Goodfellow, M. ) 115–175 (John Wiley and Sons, 1991).
Amann, R. I., Ludwig, W. & Schleifer, K. H. Phylogenetic identification and in-situ detection of individual microbial-cells without cultivation. Microbiol. Rev. 59, 143–169 (1995).
White, T. J., Bruns, T., Lee, S., & Taylor, J. in PCR Protocols: a Guide to Methods and Applications (eds Innis, M. A., Gelfand, D. H., Sninsko, J. J. & White, T. J. ) 315–322 (Academic Press, 1990).
Gies, E. A., Konwar, K. M., Beatty, T. & Hallam, S. J. Illuminating Microbial Dark Matter in Meromictic Sakinaw Lake. Appl. Env. Microbiol 80, 6807–6818 (2014).
Youngblut, N. D. & Buckley, D. H. Intra-genomic variation in G+C content and its implications for DNA stable isotope probing. Environ. Microbiol. Rep 6, 767–775 (2014).
Ruka, D. R., Simon, G. P. & Dean, K. M. Altering the growth conditions of Gluconacetobacter xylinus to maximize the yield of bacterial cellulose. Carbohyd. Polym. 89, 613–622 (2012).
Pinnell, L. J., Dunford, E. A., Ronan, P., Hausner, M. & Neufeld, J. Recovering glycoside hydrolase genes from active tundra cellulolytic bacteria. Can. J. Microbiol. 60, 469–476 (2014).
Wilhelm, R., Szeitz, A., Klassen, T. L. & Mohn, W. W. Sensitive, Efficient Quantitation of 13C-Enriched Nucleic Acids via Ultrahigh-Performance Liquid Chromatography–Tandem Mass Spectrometry for Applications in Stable Isotope Probing. Appl. Environ. Microbiol. 80, 7206–7211 (2014).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Cock, P. J. A. et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423 (2009).
Albertsen, M. et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31, 533–538 (2013).
Data Citations
Acknowledgements
This study was supported by grants from Genome Canada and Genome BC as well as an NSERC Strategic Project Grant. Sampling and data collection were contributed by collaborators at the Canadian Forestry Services (Drs Paul Hazlett, Kara Webster, Robert L. Fleming), the Ontario Ministry of Natural Resources (Dr David Morris), BC Ministry of Forests, Lands and Natural Resources Operations (Drs Shannon Berch, Chuck Bulmer, Bill Chapman, Stephane Dubé, Graeme Hope and Marty Kranabetter) and the U.S. Forest Service (Drs Matt Busse and Andy Scott). Drs E.C., M.H. and K.M. were supported by postdoctoral fellowships from the Tula Foundation and both R.C.W. and A.H. by NSERC graduate scholarships. Sequencing conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We also acknowledge the contribution of scientists at the McGill University and Génome Québec Innovation Center, Montréal, Canada.
Author information
Authors and Affiliations
Contributions
R.C.W. contributed to field sampling, processing of soil samples and DNA extractions, all data corresponding to the SIP-cellulose experiment, data curation and data submission, and was the principle author of this manuscript. E.C. contributed shotgun metagenomes, data curation, and data submission. H.L. contributed to field sampling, processing of soil samples and DNA extraction and produced all data corresponding to SIP-hemicellulose experiment. K.M. performed PCR for amplicon libraries from PPCA, LLTX, JPON and BSON field soils and helped to consolidate metadata. A.H. contributed amplicon and shotgun metagenomes at various soil depths from Skulow Lake. M.H. contributed to field sampling and all amplicon data from SBSBC and IDFBC. S.H. designed and initiated the sampling program with W.W.M. and supervised sample processing and sequencing for the project. W.W.M. conceived and supervised the project, contributed to sampling and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Wilhelm, R., Cardenas, E., Leung, H. et al. A metagenomic survey of forest soil microbial communities more than a decade after timber harvesting. Sci Data 4, 170092 (2017). https://doi.org/10.1038/sdata.2017.92
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2017.92
This article is cited by
-
Cultivation-independent genomes greatly expand taxonomic-profiling capabilities of mOTUs across various environments
Microbiome (2022)
-
GlobalFungi, a global database of fungal occurrences from high-throughput-sequencing metabarcoding studies
Scientific Data (2020)
-
Bacterial contributions to delignification and lignocellulose degradation in forest soils with metagenomic and quantitative stable isotope probing
The ISME Journal (2019)
-
Biogeography and organic matter removal shape long-term effects of timber harvesting on forest soil microbial communities
The ISME Journal (2017)
