
Abstract
Venoms are a rich source for the discovery of molecules with biotechnological applications, but their analysis is challenging even for state-of-the-art proteomics. Here we report on a large-scale proteomic assessment of the venom of Loxosceles intermedia, the so-called brown spider. Venom was extracted from 200 spiders and fractioned into two aliquots relative to a 10 kDa cutoff mass. Each of these was further fractioned and digested with trypsin (4 h), trypsin (18 h), pepsin (18 h), and chymotrypsin (18 h), then analyzed by MudPIT on an LTQ-Orbitrap XL ETD mass spectrometer fragmenting precursors by CID, HCD, and ETD. Aliquots of undigested samples were also analyzed. Our experimental design allowed us to apply spectral networks, thus enabling us to obtain meta-contig assemblies, and consequently de novo sequencing of practically complete proteins, culminating in a deep proteome assessment of the venom. Data are available via ProteomeXchange, with identifier PXD005523.
Design Type(s) | parallel group design |
Measurement Type(s) | Proteomic Profiling |
Technology Type(s) | liquid chromatography-tandem mass spectrometry |
Factor Type(s) | mass • protease • sample resting period • fractionation • matrix solution concentration |
Sample Characteristic(s) | Loxosceles intermedia • venom |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others
Background & Summary
Scientists have long enlisted venoms in their quest to characterize novel molecules with biotechnological applications1,2. The literature provides innumerous examples of venom-derived applications, ranging from biopesticides to medical applications. In particular, works on serpent venom are, unarguably, success stories. Some examples are: Batroxobin, a widely used thrombin-like enzyme and commonly extracted from the venom of Bothrops atrox and Bothrops moojeni, has been used as a replacement for thrombin in bleeding injuries3; Ecarin, from Echis carinatus, as the primary reagent for laboratorial tests that monitor anticoagulation4; and Captopril, developed from peptides of the Bothrops jararaca venom, as a widely adopted inhibitor of the angiotensin converting enzyme (ACE). Other examples of venom-derived drugs include: Aggrastat, for myocardial infarct and ischemia; Ancrod, for stroke; Defibrase, for acute cerebral infarction and angina pectoris; Exanta, used as an anti-coagulant; Hemocoagulase, for hemorrhage; and Integrilin, for acute coronary syndrome5. Venoms have also been used to search for inhibitors derived from other species (e.g., Didelphis marsupialis)6,7.
Motivated by all the successful research on snake venoms, efforts have been geared towards spider toxins. In particular, those from the Loxosceles genus are already being used in at least four general application fronts, viz.: as therapeutic anti-venom sera8; as tools in molecular and cellular biology research; and as aids in drug development and production of selective and environmentally friendly bioinsecticides5. Peptides originating from the venom of Thrixopelma pruriens have been used in the treatment of pain and inflammation9; the T×2–5 and T×2–6 neuropeptides from the Phoneutria nigriventer venom, for treating erectile dysfunctions10; and distinct bioactive peptides from spider venoms, in the treatment of diverse diseases, such as cancer11. Taken together, toxins have served as an endless treasure trove for biotechnological applications.
Spider venoms, in particular, comprising mainly proteins and peptides2,5,12,13 and displaying great diversity in their toxins, have drawn considerable attention. Yet, characterizing venoms poses great challenges even for state-of-the-art proteomic strategies: in fact, most species lack a reference sequence genome14 and the post-translational modifications of venoms vary greatly. Moreover, current mainstream strategies are not tailored towards performing de novo sequencing of the large (i.e., greater than tryptic), biologically active peptides that abound in venoms. Indeed, peptide-centric approaches are oblivious to whether a sequenced peptide originates from a larger peptide or a full protein, but obtaining the complete sequence of these larger molecules will undoubtedly fuel a great diversity of biotechnological applications. In this regard, it is our view that widely adopted proteomic strategies such as peptide spectrum matching (PSM)15,16 and mainstream de novo sequencing17 only reveal the tip of the iceberg in terms of what can be unveiled from venoms.
One of our goals has been to characterize the venom of the so-called brown spiders (the Loxosceles genus). Altogether, their venom is composed of a complex cocktail of biologically active compounds, with toxins ranging up to 40 kDa and over18. To the best of our knowledge, an in-depth, comprehensive proteomic profiling of the Loxosceles venom tailored towards the discovery of new molecules has so far remained elusive. Currently, there are several descriptions of enzymatic and non-enzymatic proteins from distinct Loxosceles species19,20. In 2003, a study aimed to investigate whether venoms of phylogenetically-related groups of Haplogyne spiders possess sphingomyelinase-D (SMD) toxins21. The study included 10 Loxosceles species and 2 Sicarius species, among other spider genera. The Amplex Red Phospholipase-D assay kit indicated SMD activity and these results were further supported by a Surface-Enhanced Laser Desorption/Ionization (SELDI) Time-of-Flight (TOF) analysis showing mass spectral peaks with m/z’s corresponding to those of SMD. Loxosceles SMDs, later referred to as phospholipases-D (PLDs), are known to be the major component of Loxosceles venoms and are the most well characterized toxin family in brown spider venoms. In 2005, two-dimensional protein profiles of the L. intermedia, L. laeta, and L. gaucho venoms were determined, but protein identification was focused only on the SMD toxins of the L. gaucho venom22. The identification of seven spots of interest was first attempted using data from Matrix-Assisted Laser Desorption/Ionization (MALDI) Time-of-Flight (TOF) Mass Spectrometry (MS) and Electrospray Ionization (ESI) quadrupole-time-of-flight Tandem Mass Spectrometry (MS/MS) for direct search of raw data using MASCOT22. Since the searches retrieved no significant match, de novo sequencing was performed and the resulting sequences were BLASTed against the non-redundant sequences, allowing SMD identification for all analyzed spots22. Only in 2009 was a proteomic study described that targeted the total protein content of the Loxosceles venom23. Although the L. intermedia venom was analyzed using Multi-Dimensional Protein Identification Technology (MudPIT)24, only 39 proteins were identified. Of these proteins, only 14 were described as toxins generally found in animal venoms23. Thus, this proteomic study seems to have severely underestimated the great toxin diversity of the Loxosceles venom, particularly in comparison to the many publications that already described distinct molecular clones from venoms of different Loxosceles species25–32. Transcriptome analyses of the L. laeta and L. intermedia venoms revealed a huge complexity of brown-spider venoms19,20. Specifically, the analysis of the L. intermedia venom described three classes of toxins comprising most toxin-encoding transcripts, such as peptides of low molecular mass (55.9%), astacin-like proteases (22.6%), and PLDs (20.2%). Also, transcripts similar to hyaluronidases, serine proteases, serine protease inhibitors, venom allergens, and members of the translationally controlled tumor protein (TCTP) family presented low levels of expression20. Although considerable information is now available on venom gland transcripts of L. intermedia, the total protein content of this venom has remained unclear. A previous study from our group applied two-dimensional immunoblots and zymograms on the venom of L. intermedia, L. laeta, and L. gaucho, and revealed several spots with differential volume containing proteins having gelatinolytic activity corresponding to astacin-like proteases33. These results corroborate that venoms from these species present a broad astacin-like family with many isoforms22,33,34.
The lack of genomic data from this arachnid prevents employing the PSM approach in full, so most of the weightlifting must be accomplished through de novo sequencing. Mainstream de novo sequencing, however, cannot efficiently handle unanticipated post-translational modifications, being far more prone to generating sequencing errors. This is because various molecules fail to provide enough mass spectral peaks during fragmentation to enable the sequencing of full peptides. To overcome these limitations, our dataset was acquired with multiple dissociation strategies applied to the same precursor (e.g., collision-induced dissociation (CID), higher-energy collisional dissociation (HCD), and electron-transfer dissociation (ETD)), thereby enabling the use of state-of-the-art de novo sequencing algorithms. These capitalize on complementary dissociation information and thus achieve unprecedented sequencing accuracy35,36. The use of different proteolytic enzymes on the venom aliquots unlocks the application of another very powerful paradigm, that of spectral networks37,38. These ‘specnets’ align spectra against one another, ultimately allowing the detection of unanticipated post-translational modifications. Moreover, they can assemble consensus mass spectra from overlapping peptides yielded by different proteolytic digests. A consensus spectrum thus obtained presents a better signal-to-noise ratio and allows for the de novo sequencing of amino-acid stretches far longer than those handled by the conventional approach. Once high-confidence de novo data are available, it becomes possible to employ tools, such as PepExplorer39 or Meta-SPS37, that apply pattern recognition approaches to the mapping of de novo sequencing data against sequences from homologous organisms, thereby facilitating biological interpretation.
By themselves, the meta-contig assemblies provided by spectral networks are not enough for one to conclude whether a biomolecule obtained 100% coverage. To pave the way in this direction, top-down proteomic data in combination with MS3 (i.e., product ion(s) selected from an MS/MS spectrum further fragmented and producing another tandem mass spectrum) and ETD were also acquired for a partition of the venom molecules into two sets (<~10 kDa and >~10 kDa). The top-down strategy consists of injecting intact proteins into the mass spectrometer, thus doing away with the inference limitations of the peptide-centric approach40. This provides complementary information to that of the networks and helps in the discovery of how much is required for obtaining full coverage. We anticipate that these data will be fundamental in the development of next-generation algorithms capable of bridging the gap between bottom-up, middle-down, and top-down proteomics.
Here, we present the first multi-protease, multi-dissociation, bottom-up-to-top-down proteomic dataset of the venom of L. intermedia, the ‘urban’ spider species commonly found in the city of Curitiba, Brazil41, along with an analysis using state-of-the-art tools. The approach stems from the motivation that multiple enzyme digestion increases protein coverage42, besides relying on different activation and acquisition methods.
Methods
Sample preparation
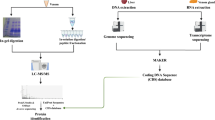
Adult L. intermedia specimens (both male and female) were collected in the wild in accordance with the Brazilian Federal System for Authorization and Information on Biodiversity (SISBIO-ICMBIO, license number 29801-1). Venom from 200 spiders was extracted through the electrostimulation method43 and immediately diluted in ammonium bicarbonate buffer 0.4 M/urea 8 M. Protein concentration was determined through the Coomassie blue method, using bovine serum albumin (BSA) as standard curve44. First, the venom was separated into two fractions using an ultra-filter unit (MW cutoff 10 kDa) (Millipore), one fraction containing venom proteins above ~10 kDa (400 μg) and the other containing venom proteins and peptides bellow ~10 kDa (90 μg). All procedures described next were performed equally for each fraction, after further dividing it into four aliquots, each of which was reduced with dithiothreitol (DTT) to a final concentration of 25 mM for 3 h at room temperature. Afterwards, the samples were alkylated with iodacetamide (IAA) to a final concentration of 80 mM for 15 min at room temperature in the dark. Each aliquot was digested with one of the follow enzymes: trypsin (Trypsin Gold, Mass Spectrometry Grade, Promega Corporation, Madison, cat. No. V5280, WI, USA), chymotrypsin (Promega, cat. No. V1062), and pepsin (Promega, cat. No. V1959) at the ratio of 1:50 (E:S). We note that an additional aliquot was stored and not digested. Three aliquots were incubated individually with each enzyme for 18 h, at 25 °C for chymotrypsin and 37 °C for trypsin and pepsin. The other aliquot was incubated for only 4 h with trypsin at 37 °C. Each digested fraction was divided into three aliquots and desalted with ultra-micro C-18 spin columns according to the manufacturer’s instructions (Harvard Apparatus). One of these three aliquots was stored for future use, another had its peptides desalted and directly submitted to reverse phase chromatography coupled online with an Orbitrap XL mass spectrometer. The third aliquot of the desalted peptides was eluted with 70% acetonitrile (ACN) and 0.1% formic acid, then dried in a speed vacuum concentrator, suspending buffer C (i.e., 10 mM of K2HPO4, 25%ACN, pH=3.0). Afterwards, the sample was passed through a micro strong cation exchanged spin column (SCX) according to the manufacturer’s instructions (Harvard Apparatus). Briefly, the column was equilibrated with buffer C, centrifuged for 1 min at 100×g, and the sample was eluted from the SCX spin column with increasing concentration of KCl, i.e., 100, 170, 290, and 400 mM. Finally, each fraction was desalted once more with ultra-micro C-18 spin columns according to the manufacturer’s instructions (Harvard Apparatus). All columns were then washed ten times with 0.1% formic acid and the peptides were eluted with buffer B (i.e., 70% acetonitrile, 0.1% formic acid) to proceed to next step.
Mass spectrometry analysis
Each fraction of peptides, including the non-fractionated as well as those from the SCX fractionation, was previously desalted and subjected to an LC-MS/MS analysis on a nano-LC 1D plus System (Eksigent, Dublin, CA), an ultra-high performance liquid chromatography (UHPLC) system coupled with an LTQ-Orbitrap XL ETD (Thermo, San Jose, CA) mass spectrometer, at the Mass Spectrometry Facility RPT02H of the Carlos Chagas Institute (Fiocruz, Brazil). In these analyses, the peptide mixtures were loaded onto a column (75 mm i.d., 15 cm long), packed in-house with a 3.2 μm ReproSil-Pur C18-AQ resin (Dr Maisch) with a flow of 500 nl/min and subsequently eluted with a flow of 250 nl/min from 5 to 40% ACN in 0.5% formic acid in a 120 min gradient. The mass spectrometer was set to data-dependent mode to automatically switch between MS and MS/MS acquisition. Full-scan MS spectra (m/z 350–1,800) were acquired in the Orbitrap analyzer with resolution R=60,000 at m/z 400 (after accumulation to a target value of 1,000,000 in the linear trap) using survey mode. The three most intense ions were sequentially isolated and fragmented using CID, HCD, and ETD for the same precursor. Previous target ions selected for MS/MS were dynamically excluded for 60 s. The total cycle time was approximately 5 s. The general mass spectrometric conditions were: spray voltage, 2.4 kV; no sheath or auxiliary gas flow; ion transfer tube temperature, 100 °C; collision gas pressure, 1.3 mTorr; normalized collision energy using wide-band activation mode; 35% for MS/MS. Ion selection thresholds were of 5,000 counts for MS/MS. The parameters for each fragmentation type in MS/MS acquisitions were as follows. For CID: isolation width, m/z 2.5; normalized collision energy, 35; activation, q=0.25; activation time, 30 ms. For HCD: isolation width, m/z 2.5; normalized collision energy, 35; activation time, 30 ms; full width at half maximum resolution, 15,000. For ETD: isolation width, m/z 2.5; activation time, 100 ms.
Bioinformatics analysis
The de novo sequencing approach employed in this work utilized multiple MS/MS spectra from overlapping peptides, generated from multiple proteases and of precursors analyzed with CID, HCD, and ETD spectrum triples. Each was then converted into prefix residue mass (PRM) spectra. In this conversion, MS/MS peak masses were converted into putative cumulative precursor fragment masses, with intensity scores determined from likelihood models specific to each fragmentation mode. Triples of PRM spectra from the same precursor were then merged into a single PRM spectrum per precursor by adding scores for matching peak masses. Spectral-network algorithms, implemented in the ProteoSAFe web platform that is freely accessible at http://proteomics.ucsd.edu/ProteoSAFe/, were then used to align merged PRM spectra from peptides with overlapping sequences. Moreover, A-Bruijn algorithms were used to integrate these alignments into assembled contigs.
Each contig was then used to construct a consensus contig spectrum, or meta-contig, capitalizing on the corroborating evidence from all of its assembled spectra to yield a high-quality consensus de novo sequence36. Subsequently, the Meta-SPS algorithm was used to align the meta-contigs against a FASTA sequence database37. This database contained all Loxosceles sequences from UniProt, all from the transcriptome of the L. intermedia venom gland20, and an internal database with common mass spectrometry contaminants and proteases.
A summary of this methodology is found in Fig. 1.

Summary of the sequence of procedures that constitute the methodology employed, from venom extraction to the meta-contig assembling that enabled the identifications of venom proteins.
Data Records
Our bioinformatics analysis disclosed a list of 190 proteins (Table 1). As far as we know, this is the most complete comprehensive proteomic profiling of the L. intermedia venom. All mass spectrometry data are available from both the ProteomeXchange Consortium via the PRIDE45 partner repository, with dataset identifier PXD005523 (Data Citation 1), and our servers (http://proteomics.fiocruz.br/pcarvalho/lintermedia/venom/). A full list of the proteins, meta-contigs, and homologous sequences is made available in Table 1.
All Meta-SPS results for >~10 kDa and <~10 kDa, together with the parameter files used for running the software, are available as separate material (MetaSPS_Results.xlsx, Data Citation 2). The results are presented in six tabs, viz., for >~10 kDa grouped by contig, >~10 kDa grouped by spectrum, >~10 kDa parameter file, <~10 kDa grouped by contig, <~10 kDa grouped by spectrum, and <~10 kDa parameter file.
Technical Validation
The lack of any previous comprehensive proteomic analysis of the Loxosceles venom demonstrates that studying this venom in detail has been a challenge, one that stems from the organism being highly non-canonical and from the fact that protein sequences for it have remained scarce in databases. The present work circumvented these obstacles by using a combination of shotgun proteomic experiments and different tools to generate and analyze large proteomic datasets and de novo sequencing results.
Our results revealed 190 protein identifications, including all classes of toxins described in previous transcriptome analyses19,20 (Table 2 (available online only)). Our approach identified both high- and low-abundance toxins of the L. intermedia venom, as well as homolog sequences from distinct Loxosceles species (astacin-like proteases, PLDs, peptides, TCTPs, hyaluronidases, allergens, serine proteases, serine protease inhibitors, and housekeeping proteins) (Table 2 (available online only)). These data reinforce the holocrine nature of the Loxosceles venom gland23 and demonstrate that its venom is composed of toxins and housekeeping proteins originating from epithelial-cell content, such as the angiotensin converting enzyme, the 60S ribosomal protein, the Na-Pi co-transporter, and the myosin heavy chain (Table 2 (available online only)). Our results, therefore, validate the method used for analyzing the proteome of an organism with non-sequenced genome.
Taken together, the identified toxins in the L. intermedia venom include representatives from all toxin groups, even if in low abundances (as in the case of, e.g., hyaluronidases and serine proteases). We also find it noteworthy that we obtained significant coverage of the three major families present in the venom, viz., PLDs, astacin-like metalloproteases, and ICK peptides. These families are of great importance for studies of the brown-spider envenomation features and of biotechnological and medical applications.
Many of the aligned contigs mapped to distinct PLD isoforms from a variety of Loxosceles species. In fact, these toxins are the most studied and well-characterized components of the Loxosceles venom5,20,26,31,46–48. PLDs are able to reproduce the deleterious effects observed in loxoscelism and represent a great target for drug discovery against brown-spider envenomation2,5.
As for the astacin-like metalloproteases identified, we note that astacins were first described as an animal-venom component in 2007 (ref. 28) and only later recognized as a family of toxins present in the Loxosceles venom33. These toxins present proteolytic activity on distinct extracellular matrix proteins and are related to the hemostatic effects in loxoscelism43,49.
ICK peptides, the major components of the L. intermedia venom-gland transcriptome (54,9% of the expressed sequence tags), were identified with correspondence to all four different ICK peptides described for L. intermedia (LiTx1, LiTx2, LiTx3, and LiTx4)50,51. These ICK peptides, also called knottins, are characterized by the neurotoxic properties they exhibit on ion channels and receptors expressed in the nervous systems of insects and mammals52. The high expression of LiTx transcripts, which correlates with the proteomic results found herein, are consistent with the venom’s effects of paralyzing and killing both preys and predators1,20,51.
Additional Information
How to cite this article: Trevisan-Silva, D. et al. A multi-protease, multi-dissociation, bottom-up-to-top-down proteomic view of the Loxosceles intermedia venom. Sci. Data 4:170090 doi: 10.1038/sdata.2017.90 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Escoubas, P. Molecular diversification in spider venoms: a web of combinatorial peptide libraries. Mol. Divers. 10, 545–554 (2006).
Gremski, L. H. et al. Recent advances in the understanding of brown spider venoms: From the biology of spiders to the molecular mechanisms of toxins. Toxicon Off. J. Int. Soc. Toxinology 83, 91–120 (2014).
You, K. E. et al. The effective control of a bleeding injury using a medical adhesive containing batroxobin. Biomed. Mater. Bristol Engl 9, 025002 (2014).
Byron, M., Zochert, S., Hellwig, T., Gavozdea-Barna, M. & Gulseth, M. P. Successful use of laboratory monitoring to facilitate an invasive procedure for a patient treated with dabigatran. Am. J. Health-Syst. Pharm. AJHP Off. J. Am. Soc. Health-Syst. Pharm. 74, 461–465 (2017).
Chaim, O. M. et al. Brown spider (Loxosceles genus) venom toxins: tools for biological purposes. Toxins 3, 309–344 (2011).
Neves-Ferreira, A. G. C. et al. Structural and functional analyses of DM43, a snake venom metalloproteinase inhibitor from Didelphis marsupialis serum. J. Biol. Chem. 277, 13129–13137 (2002).
Rocha, S. L. G. et al. Functional analysis of DM64, an antimyotoxic protein with immunoglobulin-like structure from Didelphis marsupialis serum. Eur. J. Biochem. 269, 6052–6062 (2002).
Mendes, T. M. et al. Generation and characterization of a recombinant chimeric protein (rCpLi) consisting of B-cell epitopes of a dermonecrotic protein from Loxosceles intermedia spider venom. Vaccine 31, 2749–2755 (2013).
Gui, J. et al. A tarantula-venom peptide antagonizes the TRPA1 nociceptor ion channel by binding to the S1-S4 gating domain. Curr. Biol. CB 24, 473–483 (2014).
Jung, A. R. et al. The effect of PnTx2-6 protein from Phoneutria nigriventer spider toxin on improvement of erectile dysfunction in a rat model of cavernous nerve injury. Urology 84, 730.e9–17 (2014).
Liu, Z. et al. A novel spider peptide toxin suppresses tumor growth through dual signaling pathways. Curr. Mol. Med. 12, 1350–1360 (2012).
Zobel-Thropp, P. A., Bodner, M. R. & Binford, G. J. Comparative analyses of venoms from American and African Sicarius spiders that differ in sphingomyelinase D activity. Toxicon Off. J. Int. Soc. Toxinology 55, 1274–1282 (2010).
King, G. F. & Hardy, M. C. Spider-venom peptides: structure, pharmacology, and potential for control of insect pests. Annu. Rev. Entomol. 58, 475–496 (2013).
Junqueira, M. & Carvalho, P. C. Tools and challenges for diversity-driven proteomics in Brazil. Proteomics 12, 2601–2606 (2012).
Eng, J. K., McCormack, A. L. & Yates, J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989 (1994).
Perkins, D. N., Pappin, D. J., Creasy, D. M. & Cottrell, J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 (1999).
Seidler, J., Zinn, N., Boehm, M. E. & Lehmann, W. D. De novo sequencing of peptides by MS/MS. Proteomics 10, 634–649 (2010).
Futrell, J. M. Loxoscelism. Am. J. Med. Sci. 304, 261–267 (1992).
Fernandes-Pedrosa, M. de F. et al. Transcriptome analysis of Loxosceles laeta (Araneae, Sicariidae) spider venomous gland using expressed sequence tags. BMC Genomics 9, 279 (2008).
Gremski, L. H. et al. A novel expression profile of the Loxosceles intermedia spider venomous gland revealed by transcriptome analysis. Mol. Biosyst. 6, 2403–2416 (2010).
Binford, G. J. & Wells, M. A. The phylogenetic distribution of sphingomyelinase D activity in venoms of Haplogyne spiders. Comp. Biochem. Physiol. B Biochem. Mol. Biol 135, 25–33 (2003).
Machado, L. F. et al. Proteome analysis of brown spider venom: identification of loxnecrogin isoforms in Loxosceles gaucho venom. Proteomics 5, 2167–2176 (2005).
dos Santos, L. D., Dias, N. B., Roberto, J., Pinto, A. S. & Palma, M. S. Brown recluse spider venom: proteomic analysis and proposal of a putative mechanism of action. Protein Pept. Lett 16, 933–943 (2009).
Washburn, M. P., Wolters, D. & Yates, J. R. 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 (2001).
Binford, G. J., Cordes, M. H. J. & Wells, M. A. Sphingomyelinase D from venoms of Loxosceles spiders: evolutionary insights from cDNA sequences and gene structure. Toxicon Off. J. Int. Soc. Toxinology 45, 547–560 (2005).
Chaim, O. M. et al. Brown spider dermonecrotic toxin directly induces nephrotoxicity. Toxicol. Appl. Pharmacol. 211, 64–77 (2006).
da Silveira, R. B. et al. Molecular cloning and functional characterization of two isoforms of dermonecrotic toxin from Loxosceles intermedia (brown spider) venom gland. Biochimie 88, 1241–1253 (2006).
da Silveira, R. B. et al. Identification, cloning, expression and functional characterization of an astacin-like metalloprotease toxin from Loxosceles intermedia (brown spider) venom. Biochem. J. 406, 355–363 (2007).
da Silveira, R. B. et al. Hyaluronidases in Loxosceles intermedia (Brown spider) venom are endo-beta-N-acetyl-d-hexosaminidases hydrolases. Toxicon Off. J. Int. Soc. Toxinology 49, 758–768 (2007).
da Silveira, R. B. et al. Two novel dermonecrotic toxins LiRecDT4 and LiRecDT5 from brown spider (Loxosceles intermedia) venom: from cloning to functional characterization. Biochimie 89, 289–300 (2007).
Appel, M. H. et al. Identification, cloning and functional characterization of a novel dermonecrotic toxin (phospholipase D) from brown spider (Loxosceles intermedia) venom. Biochim. Biophys. Acta 1780, 167–178 (2008).
de Santi Ferrara, G. I. et al. SMase II, a new sphingomyelinase D from Loxosceles laeta venom gland: molecular cloning, expression, function and structural analysis. Toxicon Off. J. Int. Soc. Toxinology 53, 743–753 (2009).
Trevisan-Silva, D. et al. Differential metalloprotease content and activity of three Loxosceles spider venoms revealed using two-dimensional electrophoresis approaches. Toxicon Off. J. Int. Soc. Toxinology 76, 11–22 (2013).
Trevisan-Silva, D. et al. Astacin-like metalloproteases are a gene family of toxins present in the venom of different species of the brown spider (genus Loxosceles). Biochimie 92, 21–32 (2010).
Jeong, K., Kim, S. & Pevzner, P. A. UniNovo: a universal tool for de novo peptide sequencing. Bioinformatics 29, 1953–1962 (2013).
Guthals, A., Clauser, K. R., Frank, A. M. & Bandeira, N. Sequencing-Grade De novo Analysis of MS/MS Triplets (CID/HCD/ETD) From Overlapping Peptides. J. Proteome Res. 12, 2846–2857 (2013).
Guthals, A., Clauser, K. R. & Bandeira, N. Shotgun protein sequencing with meta-contig assembly. Mol. Cell. Proteomics MCP 11, 1084–1096 (2012).
Bandeira, N. Spectral networks: a new approach to de novo discovery of protein sequences and posttranslational modifications. BioTechniques, 691 passim 42, 687 (2007).
Leprevost, F. V. et al. PepExplorer: A Similarity-driven Tool for Analyzing de Novo Sequencing Results. Mol. Cell. Proteomics MCP 13, 2480–2489 (2014).
Catherman, A. D., Skinner, O. S. & Kelleher, N. L. Top Down proteomics: Facts and perspectives. Biochem. Biophys. Res. Commun. 445, 683–693 (2014).
Fischer, M. L. & Vasconcellos-Neto, J. Microhabitats occupied by Loxosceles intermedia and Loxosceles laeta (Araneae: Sicariidae) in Curitiba, Paraná, Brazil. J. Med. Entomol. 42, 756–765 (2005).
Choudhary, G., Wu, S.-L., Shieh, P. & Hancock, W. S. Multiple enzymatic digestion for enhanced sequence coverage of proteins in complex proteomic mixtures using capillary LC with ion trap MS/MS. J. Proteome Res. 2, 59–67 (2003).
Feitosa, L. et al. Detection and characterization of metalloproteinases with gelatinolytic, fibronectinolytic and fibrinogenolytic activities in brown spider (Loxosceles intermedia) venom. Toxicon Off. J. Int. Soc. Toxinology 36, 1039–1051 (1998).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 (1976).
Vizcaíno, J. A. et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res 41, D1063–D1069 (2013).
Kalapothakis, E. et al. Molecular cloning, expression and immunological properties of LiD1, a protein from the dermonecrotic family of Loxosceles intermedia spider venom. Toxicon Off. J. Int. Soc. Toxinology 40, 1691–1699 (2002).
Ribeiro, R. O. S. et al. Biological and structural comparison of recombinant phospholipase D toxins from Loxosceles intermedia (brown spider) venom. Toxicon Off. J. Int. Soc. Toxinology 50, 1162–1174 (2007).
Vuitika, L. et al. Brown spider phospholipase-D containing a conservative mutation (D233E) in the catalytic site: identification and functional characterization. J. Cell. Biochem. 114, 2479–2492 (2013).
da Silveira, R. B. et al. Identification of proteases in the extract of venom glands from brown spiders. Toxicon Off. J. Int. Soc. Toxinology 40, 815–822 (2002).
de Castro, C. S. et al. Identification and molecular cloning of insecticidal toxins from the venom of the brown spider Loxosceles intermedia. Toxicon Off. J. Int. Soc. Toxinology 44, 273–280 (2004).
Matsubara, F. H. et al. A novel ICK peptide from the Loxosceles intermedia (brown spider) venom gland: cloning, heterologous expression and immunological cross-reactivity approaches. Toxicon Off. J. Int. Soc. Toxinology 71, 147–158 (2013).
Dutertre, S. & Lewis, R. J. Use of venom peptides to probe ion channel structure and function. J. Biol. Chem. 285, 13315–13320 (2010).
Zhang, B., Chambers, M. C. & Tabb, D. L. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 6, 3549–3557 (2007).
Data Citations
Trevisan-Silva, D. PRIDE PXD005523 (2016)
Trevisan-Silva, D. Figshare https://doi.org/10.6084/m9.figshare.c.3709168 (2017)
Acknowledgements
The authors thank CNPq and CAPES for financial support. They also thank Fiocruz for use of Mass Spectrometry Facility RPT02H at the Carlos Chagas Institute, as well as Dr Michel Batista for aiding in the mass spectrometry procedures. V.C.B. acknowledges support from a FAPERJ BBP grant. N.B. is an Alfred P. Sloan Research Fellow and was partially supported by the US National Institutes of Health Grant 2 P41 GM103484-06A1 from the National Institute of General Medical Sciences. The authors thank Wagner Nagib from Carlos Chagas Instituto, Fiocruz—Paraná for generating the final cover art.
Author information
Authors and Affiliations
Contributions
P.C.C., S.S.V., A.S.-R., J.S.G.F., and D.T.-S. conceived and designed the work. D.T.-S. and A.V.B. collected venom samples. D.T.-S. and A.V.B. performed sample preparation. P.C.C. designed the protocol for data acquisition by mass spectrometry and F.K.M. ran the samples; together with V.C.B., they discussed alternatives for generating and analyzing the data. N.B. and A.G. developed and applied the method for data analysis. P.C.C., D.T.-S., F.V.L., V.C.B., and A.S.-R. analyzed the results and wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
N.B. has an equity interest in Digital Proteomics, LLC, a company that may potentially benefit from the research results; Digital Proteomics, LLC was not involved in any aspects of this research. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. The remaining authors declares no competing financial interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Trevisan-Silva, D., Bednaski, A., Fischer, J. et al. A multi-protease, multi-dissociation, bottom-up-to-top-down proteomic view of the Loxosceles intermedia venom. Sci Data 4, 170090 (2017). https://doi.org/10.1038/sdata.2017.90
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2017.90
