
Abstract
The early-matured japonica (Geng) rice variety, Suijing18 (SJ18), carries multiple elite traits including durable blast resistance, good grain quality, and high yield. Using PacBio SMRT technology, we produced over 25 Gb of long-read sequencing raw data from SJ18 with a coverage of 62×. Using Illumina paired-end whole-genome shotgun sequencing technology, we generated 59 Gb of short-read sequencing data from SJ18 (23.6 Gb from a 200 bp library with a coverage of 59× and 35.4 Gb from an 800 bp library with a coverage of 88×). With these data, we assembled a single SJ18 genome and then generated a set of annotation data. These data sets can be used to test new programs for variation deep mining, and will provide new insights into the genome structure, function, and evolution of SJ18, and will provide essential support for biological research in general.
Design Type(s) | sequence assembly objective • whole genome sequencing |
Measurement Type(s) | genome assembly |
Technology Type(s) | DNA sequencing |
Factor Type(s) | protocol |
Sample Characteristic(s) | Oryza sativa Japonica Group |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
As the leading staple food resource for humans, rice has been adopted as an important model organism for biological research, especially for monocots. Asian cultivated rice (Oryza sativa L.) comprises two subspecies: O. sativa subsp. japonica (also known as Keng1 with the corresponding Pinyin, Geng) and subsp. indica (also known as Hsien1 with the corresponding Pinyin, Xian). Currently, japonica/Geng, especially the early-matured type, is becoming more and more important in rice production. In 2016, the cultivation area of early-mature japonica/Geng was more than 4 million ha in Northeast China.
It has become clear that one single genome is not enough to represent the huge amount of variation in rice genomes. Recently, in addition to the previously published de novo assemblies, including 93–11 (indica/Xian, two-line hybrid restorer), PA64S (admixture type with roughly 55% indica/Xian, 25% japonica/Geng, and 20% javanica, a two-line hybrid sterile line), IR64 (indica/Xian), DJ123 (aus type indica/Xian)2, HR-12 (indica/Xian)3, and Swarna (indica/Xian)4, three new sets of indica/Xian genomes have been released with the aid of third-generation sequencing technology for variation deep mining, including MH63RS1 (indica/Xian, three-line hybrid restorer), ZS97RS1 (indica/Xian, three-line hybrid maintainer)5, and R498 (indica/Xian, three-line hybrid restorer)6. These data sets have enriched our knowledge of the genomic variations of indica/Xian rice. Nevertheless, the genome of japonica/Geng is quite different from that of indica/Xian. Since the release of the gold standard genome of Nipponbare7,8, a medium-matured japonica/Geng variety with photosensitivity, the public availability of japonica/Geng genomes, especially for the early-mature type, remains largely blank.
According to our breeder’s experiences, early-matured japonica/Geng is a relatively unique type compared with the medium-matured japonica/Geng. In addition, common variations, such as single nucleotide polymorphisms (SNPs) within early-matured japonica/Geng group are relatively sparse. To improve the efficiency of molecular breeding in early-matured japonica/Geng, deep mining of further genome variations is urgently required. Short-read sequencing (SRS) technologies, such as Illumina HiSeq, have offered us an opportunity to access huge amounts of variations, including SNPs and short InDels, instantly from large sets of genomes9; however, to perform deeper mining of complex but critical variations, such as repeat sequence variations, long InDels, and structure variations (SVs), the technical bottleneck of the short sequencing read length remains a challenge. Currently, long-read sequencing (LRS) data are available with the aid of new technology, such as PacBio. However, the cost and error rate still remain relatively high. Thus, a scheme comprising LRS amended by SRS would represent a balanced choice for deep mining of genome variations6,10.
Early-mature japonica/Geng cultivar Suijing18 (SJ18) was newly developed by our joint project and was licensed for release in Northern China in 2014. It is a representative early-matured japonica/Geng cultivar harboring multiple elite traits (such as durable blast resistance, good grain quality, and high yield) and now represents more than 10% of the planting area of early-matured japonica/Geng in China. Therefore, we initiated a collaborative project to generate one high quality genome assembly for SJ18 to be used as a fundamental tool to help us investigate underlying genome variations in early-matured japonica/Geng. In this study, we report the resources and data sets that were generated and used for the deep mining of SJ18 genome variations: (1) raw PacBio LRS data, (2) Illumina whole-genome shotgun (WGS) SRS data, (3) the amended assembly of SJ18, (4) the annotation data based on the amended assembly of SJ18, and (5) the functional analysis results based on this annotation.
With the resources and data generated in this study, not only were we able assemble de novo a good quality genome sequence for early-matured japonica/Geng, but also were able to provide the scientific community with data to advance biological research at the genomic level, especially for the deep mining of genetic variations, and provided more information for genome-based molecular breeding of crops.
Methods
Plant material and library construction
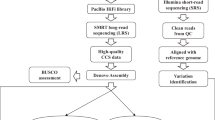
The early-matured japonica/Geng cultivar SJ18, which was developed by our own group, was licensed for release in 2014 and is now widely planted (more than 0.8 million hectare) in Heilongjiang province in Northeast China. High-molecular-weight genomic DNA was extracted from 10-day-old leaves of SJ18 (multiple seeds) using the modified CTAB method11, followed by 0.5× bead purification twice. The quality of the DNA sample was assessed using 0.75% agarose gel assays and Nanodrop (Nanodrop Technologies, Wilmington, DE, US), and was quantified using Qubit system (Thermo Fisher Scientific, Waltham, MA). The sample that met the quantity and quality standards was split into two parts, which were used to construct PacBio Sequel and Illumina libraries for LRS and SRS, respectively (Fig. 1).

Outline of the workflow used to generate and analyze the genome data for Suijing18 (SJ18).
The Sequel 20 K libraries were prepared using the standard protocol from PacBio and sequenced in the wet laboratory department of the Beijing Computing Center (http://www.bcc.ac.cn/) using a PacBio LRS instrument, model Sequel. The 200 bp- and 800 bp-libraries, with peak insert sizes of ~200 bp and ~800 bp, respectively, were prepared using an Illumina Truseq DNA library protocol (Illumina Kit FC-121-4001; Illumina Inc., San Diego, CA, USA). The qualities of libraries were checked using a standard protocol involving an Agilent 2,100 Bioanalyzer High Sensitivity Kit. After library profile analysis, the libraries were sequenced using 150 bp pair-end strategies with the Illumina HiSeq X10 platform (Illumina Inc.).
The amount of raw data from LRS was no less than 25 Gb, with a coverage of 62×. Using SRS, 59 Gb of raw data was generated, including 23.6 and 35.4 Gb of data from the 200 and 800 bp libraries, respectively. The total coverage of SRS was about 147×.
Data analysis
The LRS data was screened and adjusted by the procedures embed in CANU12. Data that met the threshold of Q20 (corresponding to a 1% error rate) were adopted. De novo assembly was carried out for the LRS data using the CANU pipeline with default parameters, except for errorRate=0.045 and genomeSize=350 m. The SRS data were then aligned to the preliminary assembly using BWA13. In addition, the pilon package14 was adopted for the amendment process. The amended assembly represented the submitted version of the SJ18 sequence.
Based on the amended version of the SJ18 assembly, genome annotation was carried out using the following steps with default parameters, except for those indicated:
-
1)
Tandem repeats were recognized by the TRF package15 with the following parameter settings: Match=2, Mismatch=7, Delta=7, PM=80, PI=10, Minscore=50, MaxPeriod=2,000. Other types of repeat sequences were recognized by RepeatModeler (http://www.repeatmasker.org/RepeatModeler/) with default settings. The database adopted for RepeatModeler analysis was an integrated library comprising Repbase16 (updated in January 2017), Dfam2 (ref. 17), and publicly available libraries containing de novo information for rice.
-
2)
Annotation for non-coding RNA (ncRNA) was carried out by using cmsearch in Infernal18, searching the Rfam database V12.2 (http://rfam.xfam.org/) with a parameter setting of ‘—cyk -T10’ for microRNAs (miRNAs), small nuclear ribonucleic acid (snRNA), and small RNA (sRNA). The transfer RNA (tRNA) annotation was carried out using tRNAscan-SE19 with default settings.
-
3)
We masked the repeats using RepeatMask with the parameter setting of ‘-nolow -no_is -norna’ and then annotated the SJ18 genome using multiple tools, including GENEID20 with parameter settings for rice, GeneMark21 with a setting of —ES —cores 24 —min_contig 100, SNAP22 with default setting, and AUGUSTUS23 with -species=rice. We compared the coding sequences (CDSs) and protein sequences from other rice genomes using PASA24 with default settings and GeneWise25 with a setting of ‘splice_gtag -sum -gff -quiet’. All the annotation results were integrated and screened using EVidenceModeler (EVM)26.
-
4)
The predicted coding genes from SJ18 were translated into protein sequences and aligned to the proteins from plant species in the Uniprot database (http://www.uniprot.org/) and Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/), respectively, using BLASTP. The threshold was set to e-value<1e-8, and the best hits were submitted for further analysis.
-
5)
Gene ontology (GO) analysis was carried out based on the above functional annotation results by using topGO27. The biological process (BP), cellular component (CC), and molecular function (MF) matches were listed. The secondary binding point was chosen in the analysis. The annotated proteins from SJ18 were submitted for pathway analysis using KEGG.
Data Records
Raw PacBio long-read sequencing (LRS) data are available through the NCBI SRA with the accession number SRR5877285 (Data Citation 1). All Illumina short-read sequencing (SRS) data for SJ18 can be found at the NCBI SRA with accession numbers SRR5880534 (Data Citation 2) and SRR5880533 (Data Citation 3). The assembled SJ18 genome version 1 is available at the NCBI with the accession number PDFQ00000000 (Data Citation 4). All these raw data are also available at figshare (Data Citation 5). The analyzed data are available at figshare (Data Citation 5) or through the URLs offered by figshare and the Rice Functional Genomics and Breeding (RFGB) database28 (Table 1).
Technical Validation
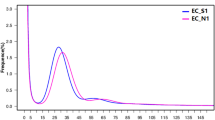
The LRS data were screened and amended with the SRS data using the CANU package with default settings. Possible sequencing errors were further minimized by removing reads that aligned with high scores to the downloaded sequences from bacteria, fungi, or human genomes from GenBank using BWA. Finally, a total of 648,237 high-quality LRS reads that passed this quality check step were submitted for assembly. The distribution of these reads is shown in Fig. 2.

Distribution of high quality reads from PacBio long-read sequencing (LRS) for Suijing18 (SJ18).
The raw SRS data was screened using the Trimmomatic package29, which removed the adaptors and the reads with a quality value lower than 20 (corresponding to a 1% error rate).
We also compared the parameters of SJ18 with other assemblies. The statistics of the assembled contigs are shown in Table 2. The statistics of repeat sequences are shown in Table 3 in comparison with Nipponbare (medium-matured japonica/Geng) and R498 (indica/Xian, the most recently available rice assembly).
Additional information
How to cite this article: Nie, S.-J. et al. Assembly of an early-matured japonica (Geng) rice genome, Suijing18, based on PacBio and Illumina sequencing. Sci. Data 4:170195 doi: 10.1038/sdata.2017.195 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Morishima, H. & Oka, H.-I. Phylogenetic differentiation of cultivated rice, XXII. Numerical evaluation of the indica-japonica differentiation. Jpn J Breeding 31, 402–413 (1981).
Schatz, M. C. et al. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol 15, 506 (2014).
Mahesh, H. B. et al. Indica rice genome assembly, annotation and mining of blast disease resistance genes. BMC Genomics 17, 242 (2016).
Rathinasabapathi, P., Purushothaman, N., Ramprasad, V. L. & Parani, M. Whole genome sequencing and analysis of Swarna, a widely cultivated indica rice variety with low glycemic index. Sci Rep 5, 11303 (2015).
Zhang, J. et al. Building two indica rice reference genomes with PacBio long-read and Illumina paired-end sequencing data. Scientific Data 3, 160076 (2016).
Du, H. et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat Commun 8, 15324 (2017).
Kawahara, Y. et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6, 1–10 (2013).
Sequencing ProjectInternational Rice, G. The map-based sequence of the rice genome. Nature 436, 793–800 (2005).
3K-RGP. The 3,000 rice genomes project. GigaScience 3, 7 (2014).
Zhang, J. et al. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc. Natl. Acad. Sci. USA 113, E5163–E5171 (2016).
Clarke, J. D. Cetyltrimethyl Ammonium Bromide (CTAB) DNA Miniprep for Plant DNA Isolation. CSH Protocols 2009, pdb.prot5177 (2009).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Walker, B. J. et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLOS ONE 9, e112963 (2014).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110, 462–467 (2005).
Wheeler, T. J. et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res 41, D70–D82 (2013).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964 (1997).
Blanco, E., Parra, G. & Guigó, R. in Current Protocols in Bioinformatics (John Wiley & Sons, Inc., 2002).
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O. & Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res 33, 6494–6506 (2005).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res 33, W465–W467 (2005).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res 31, 5654–5666 (2003).
Birney, E. & Durbin, R. Using GeneWise in the Drosophila Annotation Experiment. Genome Res 10, 547–548 (2000).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Alexa, A., Rahnenführer, J. & Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607 (2006).
Zheng, T. Q. et al. Rice functional genomics and breeding database (RFGB)-3K-rice SNP and InDel sub-database. Chinese Sci Bull 60, 367–371 (2015).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Data Citations
NCBI Sequence Read Archive SRP113746 (2017)
NCBI Sequence Read Archive SRP113817 (2017)
NCBI Sequence Read Archive SRP113816 (2017)
NCBI Assembly GCA_002573525 (2017)
Zheng, T. Q. Figshare https://doi.org/10.6084/m9.figshare.c.3835939 (2017)
Acknowledgements
We appreciate support from a subprogram of the National Key R&D Program of China (2016YFD0101801) from the Chinese Ministry of Science & Technology to T.-Q.Z. and also the support of the Shenzhen Peacock Plan to Z.-K.L. and J.-L.X.
Author information
Authors and Affiliations
Contributions
T.-Q.Z., S.-J.N. J.-L.X., and Z.-K.L. designed and conceived research; Y.-Q.L., S.-W.G., T.-T.X., Q.L., and H.-L.C. prepared the samples for sequencing; C.-C.W., and P.-C.Y. performed the data collection and analysis; Y.-B.C., and W.P. contributed new reagents and analytical tools; T.-Q.Z., S.-J.N., and J.-L.X. wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Nie, SJ., Liu, YQ., Wang, CC. et al. Assembly of an early-matured japonica (Geng) rice genome, Suijing18, based on PacBio and Illumina sequencing. Sci Data 4, 170195 (2017). https://doi.org/10.1038/sdata.2017.195
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2017.195
This article is cited by
-
Pedigree genome data of an early-matured Geng/japonica glutinous rice mega variety Longgeng 57
Scientific Data (2024)
-
Chromosomal-level genome assembly of the high-quality Xian/Indica rice (Oryza sativa L.) Xiangyaxiangzhan
BMC Plant Biology (2023)
-
LongStitch: high-quality genome assembly correction and scaffolding using long reads
BMC Bioinformatics (2021)
-
Hecaton: reliably detecting copy number variation in plant genomes using short read sequencing data
BMC Genomics (2019)
