
Abstract
We present bacterial 16S rRNA gene datasets derived from stool samples of 44 patients with diarrhea indicative of a Clostridioides difficile infection. For 20 of these patients, C. difficile infection was confirmed by clinical evidence. Stool samples from patients originating from Germany, Ghana, and Indonesia were taken and subjected to DNA isolation. DNA isolations of stool samples from 35 asymptomatic control individuals were performed. The bacterial community structure was assessed by 16S rRNA gene analysis (V3-V4 region). Metadata from patients and control individuals include gender, age, country, presence of diarrhea, concomitant diseases, and results of microbiological tests to diagnose C. difficile presence. We provide initial data analysis and a dataset overview. After processing of paired-end sequencing data, reads were merged, quality-filtered, primer sequences removed, reads truncated to 400 bp and dereplicated. Singletons were removed and sequences were sorted by cluster size, clustered at 97% sequence similarity and chimeric sequences were discarded. Taxonomy to each operational taxonomic unit was assigned by BLASTn searches against Silva database 123.1 and a table was constructed.
Design Type(s) | observation design • disease state design |
Measurement Type(s) | rRNA_16S |
Technology Type(s) | DNA sequencing |
Factor Type(s) | National Origin • Sign or Symptom |
Sample Characteristic(s) | Homo sapiens • feces |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others
Background & Summary
Infections with Clostridioides difficile (formerly Clostridium difficile, see Lawson et al.1) have significantly increased over the past decade2–5. The organism is a Gram-positive, obligate anaerobic spore-forming bacterium, which is frequently found as member of the gut microbiome in healthy individuals, but eventually can also act as human pathogen causing disease that ranges from severe diarrhea to life-threatening toxic megacolon6. It produces two potent exotoxins, toxin A (enterotoxin, tcdA) and toxin B (cytotoxin, tcdB)7. Some isolates also express a third, so-called binary toxin (C. difficile transferase, CDT)8. The risk to suffer from a C. difficile infection increases with prior broad-spectrum antibiotic treatment, which supports the assumption that an imbalanced gut microbiome increases the likelihood of a C. difficile infection9.
In this data report, we provide the bacterial community composition in stool samples of 79 human individuals including 44 patients with diarrhea indicative for infection with C. difficile and 35 asymptomatic control individuals from regions of Germany (Seesen, Lower Saxony), Ghana (Eikwe, Western Region), and Indonesia (Medan, Sumatra)., For 20 of the 44 patients, clinical evidence of a C. difficile infection was obtained. For the remaining patients, the presence of C. difficile was indicated by 16S rRNA gene data or MALDI-TOF mass spectrometry. In total, we provide 20,844,594 paired-end 16S rRNA gene reads sequenced with the v3 chemistry of Illumina and a MiSeq instrument. Correspondingly, this dataset represents a total of 10,422,297 bacterial 16S rRNA gene sequences. After all processing steps, which included read-merging, quality-filtering, primer sequence removal, dereplication, singleton removal, read-trimming, chimera removal, and removal of extrinsic domains (Archaea, chloroplasts) 7.204.189 (69.1%) high quality 16S rRNA gene sequences remained for analysis (see Table 1 (available online only) for 16S rRNA gene sequence processing statistics). Additionally, we supply metadata including gender, age, country, presence or absence of diarrhea, C. difficile ribotype, toxin PCR ribotype, toxin test from stool, concomitant diseases at time of sampling, and antiobiotic treatment within the last three months (Table 2 (available online only)).
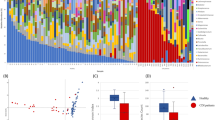
The dataset contributes to unveil the significance of the gut microbiome in diseased and asymptomatic patients. In a first analysis, we observed C. difficile as a rather low abundant (mainly <1%, with one exception) bacterial community member in stool samples (Fig. 1). The exception was patient_029 (male, age 91), who showed a high abundance of C. difficile (42.67%).

The bacterial community profiles are based on operational taxonomic unit (OTU, defined at 97% genetic identity) frequency in stool samples of 44 patients with diarrhea indicative of C. difficile infection and 35 asymptomatic control individuals (n=79). One stool sample per patient was used and amplicon PCRs were performed in triplicate for this analysis. Families, which exhibited an abundance of lower than 1% in the entire dataset, were summarized as rare taxa. Relative abundance of C. difficile (Peptoclostridium difficile in SILVA database 123.1) is displayed separately and exhibited highest similarity to Clostridioides difficile strain 630 delta erm (Accession number CP016318). Occurrence of diarrhea in patents is indicated by plus (patient exhibited diarrhea) and minus (no diarrhea), results from microbiological diagnosis of C. difficile infection (C. d. m. t.) are shown below (plus, positively tested for C. difficile; minus, negatively tested for C. difficile). Presence and absence of C. difficile in amplicon data (C. d. NGS) are indicated by plus (present) and minus (absent). Data processing and employed tools are described in detail in the methods section.
Whether the low abundance of C. difficile in most stool samples from diarrheic patients might indicate adhesion or invasion of C. difficile to the intestinal epithelium remains to be analyzed. However, a similar study also observed low abundances of C. difficile in CDI patients10. Furthermore, C. difficile is not the only potential pathogen of diseased patients. The stool samples of some patients contain other potentially pathogenic bacterial species belonging to different genera such as Escherichia/Shigella, Salmonella or Staphylococcus. In addition, some stool samples also contained facultative human-pathogenic Klebsiella and Pseudomonas species. These results support the hypothesis that the gut microbiome contributes to the pathogenic potential or at least can be used as an indicator of C. difficile infections. This is of special interest for C. difficile infections from Ghana, as most of the so far analyzed genomes of strains from this African country lack the toxin genes11. Furthermore, most German patients had a higher age than the patients from the other regions and showed a typical C. difficile infection profile, including treatment with antibiotics and presence of mainly toxin-positive strains. In contrast patients from Ghana and Indonesia were younger and had less antibiotic treatment than the German patients, and harboured predominantly toxin-negative strains (Table 2 (available online only)).
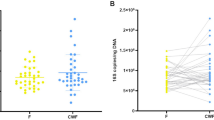
The Unifrac12 based bacterial community structure comparison shows variations in structure and diversity within potentially C. difficile-infected and reference patients (Fig. 2). We observed a low but significant correlation of the bacterial microbiome to patients who exhibited diarrhea (P=0.006, r2=0.0709) and diagnosed C. difficile positive by microbiological tests (P=0.017, r2=0.0628), respectively. In general, patients that have been diagnosed C. difficile positive harbour a less diverse bacterial microbiome (Fig. 2), which has also been observed recently13,14.

Non-metric multidimensional scaling (NMDS) based on weighted Unifrac12 was used to display the bacterial community structure in 79 stool samples at same sequencing effort (10.000 reads per sample). Samples from patients who exhibited diarrhea at time of sampling are encased by diamond. Samples from patients that were positively tested on C. difficile by microbiological test are marked by plus, samples of patients where C. difficile was detected in the amplicon dataset are marked by cross. Point size represents the phylogenetic diversity (PD, Faith's Phylogenetic Diversity26) of the microbiome, samples are encircled by PD ranges from 0–10, 10–15, 15–20, and 20–30. Data processing and employed tools are described in detail in the methods section. All alpha diversity metrics obtained by QIIME are listed in Table 3 (available online only).
Methods
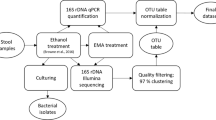
Stool sample preparation and processing
This study was approved by the Ethical Committee of the University Medical Center, Göttingen, Germany (2011-03-29). Diarrhea was defined as the passage of ≥three loose or liquid defecations per day. Upon informed consent, randomly selected patients with diarrhea and non-diarrheal volunteers agreed to submit a stool sample using stool containers and complete a standardised questionnaire about their lifestyle and medical history. Within two hours after providing the stool samples, they were cultured on Clostridium difficile agar base used with selective supplement (Oxoid, Basingstoke, Hampshire, UK) and 7% (v/v) defibrinated human blood for 48 h at 38 °C in anaerobic condition using gas packs (bioMérieux, Marcy-l’Ètoile, France). Stool samples were also tested for the presence of C. difficile glutamate dehydrogenase (GDH) antigen and toxins A and B by the C. DIFF QUIK CHEK COMPLETE test (Techlab, Blacksburg, USA). In addition, the stool sample that was used for C. difficile identification was also frozen immediately after taken from the patients, stored at −20 °C for a maximum of 11 months (based on duration of local sampling period) and transported within 24 h to Göttingen (Germany), where identification of C. difficile was confirmed by recultivation and MALDI-TOF mass spectrometry using Biotyper (Bruker Daltonics, Bremen, Germany) with score values of ≥2,000. All C. difficile strains were further characterized by toxin determination using the RealStar Clostridium difficile PCR Kit 1.0 (Altona Diagnostics, Hamburg, Germany). Ribotyping and toxinotyping was kindly performed by L. von Müller (Homburg, Germany) and M. Rupnik (Maribor, Slovenia) as previously be reported11. In addition, the Luminex xTag GPP test was used for all Ghanaian stool samples according to the manufacturer’s instructions (Luminex, Hertogenbosch, The Netherlands) in order to identify C. difficile and other potential intestinal pathogens11. The stool sample was also used for DNA isolation in order to determine bacterial community composition.
Nucleic acid extraction and amplification of 16S rRNA genes
DNA was extracted from all stool samples using the MagNA Pure LC 2.0 Instrument with the MagNA Pure LC Total Nucleic Acid Isolation kit following the instructions of the manufacturer (Roche, Mannheim, Germany). Bacterial 16S rRNA gene amplicons were generated using fusion primers TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-CCTACGGGNGGCWGCAG (MiSeq_overhang-D-Bact-0341-b-S-17) and GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-GACTACHVGGGTATCTAATCC (MiSeq_overhang-S-D-Bact-0785-a-A-21) including bacteria targeting primers from Klindworth et al.15. The PCR reaction mixture with a total volume 50 μl contained 1 U Phusion high fidelity DNA polymerase (Biozym Scientific, Oldendorf, Germany), 5% DMSO, 0.2 mM of each primer, 200 μM dNTP, 0.2 μl of 50 mM MgCl2, and 25 ng of isolated DNA. Thermal cycling scheme for bacterial amplicons was as follows: initial denaturation for 1 min at 98 °C, 25 cycles at 98 °C for 45 s, 45 s at 60 °C, and 30 s at 72 °C, and a final extension at 72 °C for 5 min. The resulting PCR products were checked by agarose gel electrophoresis for appropriate size and purified using the magnetic bead capture kit NucleoMag PCR (Macherey-Nagel, Düren, Germany) as recommended by the manufacturer. Quantification of the PCR products was performed using the Quant-iT dsDNA HS assay kit and a Qubit fluorometer (Invitrogen GmbH, Karlsruhe, Germany) following the manufacturer’s instructions. PCR products were used to attach indices and Illumina sequencing adapters using the Nextera XT Index kit (Illumina, San Diego). Index PCR was performed using 5 μl of template PCR product, 2.5 μl of each index primer, 12.5 μl of 2x KAPA HiFi HotStart ReadyMix and 2.5 μl PCR grade water. Thermal cycling scheme was as follows: 95 °C for 3 min, 8 cycles of 30 s at 95 °C, 30 s at 55 °C and 30 s at 72 °C and a final extension at 72 °C for 5 min. Bacterial 16S rRNA genes were sequenced using the dual index paired-end (v3, 2×300 bp) approach for the Illumina MiSeq platform as recommended by the manufacturer.
16S rRNA gene sequence processing and analyses
Demultiplexing and clipping of sequence adapters from raw sequences were performed by employing CASAVA data analysis software (Illumina). Paired-end sequences were merged using PEAR v0.9.1016 with default parameters. Subsequently, sequences with an average quality score lower than 20 and containing unresolved bases were removed with the split_libraries_fastq.py script from QIIME 1.9.117. We additionally removed non-clipped reverse and forward primer sequences by employing cutadapt 1.1018 with default settings. For operational taxonomic unit (OTU) clustering, we used USEARCH version 8.1.186119 with the UPARSE20 algorithm to truncate reads to 400 bp (-fastx_truncate), dereplicate (-derep_fulllength), sort by cluster size and remove singletons (-sortbysize). Subsequently, OTUs were clustered at 97% sequence identity using USEARCH (-cluster_otus), which includes de novo chimera removal. Additionally, chimeric sequences were removed using UCHIME21 included in software package USEARCH with reference mode (-uchime_ref) against RDPs trainset15_092015.fasta22. All quality-filtered sequences were mapped to chimera-free OTUs and an OTU table was created using USEARCH (-usearch_global). Taxonomic classification of the picked reference sequences (OTUs) was performed with parallel_assign_taxonomy_blast.py against SILVA SSU database release 123.123. Extrinsic domain OTUs, chloroplasts, and unclassified OTUs were removed from the dataset by employing filter_otu_table.py. Sample comparisons were performed at the same surveying effort, utilizing the lowest number of sequences by random resampling (10.000 reads per sample). Species richness, alpha and beta diversity estimates were determined using the QIIME script alpha_rarefaction.py. Non-metric multidimensional scaling (NMDS) and statistical tests were performed with the vegan package24 in R25.
Data Records
The paired-end reads of the 16S rRNA gene sequencing were deposited in the National Center for Biotechnology Information (Data Citation 1). The dataset consists of 158 zipped FASTQ files that were processed by the CASAVA software (Illumina), which includes demultiplexing and removal of adapter sequences. The OTU table (otu_table_PRJNA353065.xlsx) used for all analyses and the corresponding representative OTU sequences clustered at 97% genetic identity (otu_sequences_PRJNA353065.fasta) are accessible at figshare.com (Data Citation 2).
Technical Validation
Success of 16S rRNA gene amplicon generation was controlled by reviewing the amplicon size (approximately 550 bp) and absence of contaminations on an agarose gel. Additionally, negative (PCR reaction without template) and positive controls (genomic DNA of E. coli DH5a) were performed to ensure purity of the employed reagents. To reduce possible PCR biases, all PCRs were performed in triplicate and after purification pooled equimolar.
Usage Notes
The OTU table (otu_table_PRJNA353065.xlsx) used for all analyses and the corresponding representative OTU sequences clustered at 97% genetic identity (otu_sequences_PRJNA353065.fasta) are accessible at figshare (Data Citation 2).
Additional Information
How to cite this article: Schneider, D. et al. Gut bacterial communities of diarrheic patients with indications of Clostridioides difficile infection. Sci. Data 4:170152 doi: 10.1038/sdata.2017.152 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Lawson, P. A., Citron, D. M., Tyrrell, K. L. & Finegold, S. M. Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O’Toole 1935) Prévot 1938. Anaerobe 40, 95–99 (2016).
Honda, H. & Dubberke, E. R. Clostridium difficile infection: a re-emerging threat. Mo. Med. 106, 287–291 (2009).
Rupnik, M., Wilcox, M. H. & Gerding, D. N. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 7, 526–536 (2009).
Lessa, F. C., Gould, C. V. & McDonald, L. C. Current status of Clostridium difficile infection epidemiology. Clin. Infect. Dis. 55 (Suppl 2): S65–S70 (2012).
Ghose, C. Clostridium difficile infection in the twenty-first century. Emerg. Microbes Infect. 2, e62 (2013).
Hensgens, M. P. M. et al. Clostridium difficile infection in the community: a zoonotic disease? Clin. Microbiol. Infect. 16, 635–645 (2012).
Hatheway, C. L. Toxigenic clostridia. Clin. Microbiol. Rev. 3, 66–98 (1990).
Hemmasi, S. et al. Interaction of the Clostridium difficile Binary Toxin CDT and Its Host Cell Receptor, Lipolysis-stimulated Lipoprotein Receptor (LSR). J. Biol. Chem. 290, 14031–14044 (2015).
Almeida, R., Gerbaba, T. & Petrof, E. O. Recurrent Clostridium difficile infection and the microbiome. J. Gastroenterol. 51, 1–10 (2016).
Schubert, A. M. et al. Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. MBio. 5, e01021–14 (2014).
Janssen, I. et al. High prevalence of nontoxigenic Clostridium difficile isolated from hospitalized and non-hospitalized individuals in rural Ghana. Int. J. Med. Microbiol. 306, 652–656 (2016).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235 (2005).
Seekatz, A. M., Rao, K., Santhosh, K. & Young, V. B. Dynamics of the fecal microbiome in patients with recurrent and nonrecurrent Clostridium difficile infection. Genome Med. 8, 47 (2016).
Longo, D. L., Leffler, D. A. & Lamont, J. T. Clostridium difficile Infection. N. Engl. J. Med. 372, 1539–1548 (2015).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1 (2013).
Zhang, J., Kobert, K., Flouri, T. & Stamatakis, A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620 (2014).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 17, 10–12 (2011).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998 (2013).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011).
Cole, J. R. et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642 (2014).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Oksanen, J. et al. vegan: Community Ecology Package (2016).
Team, R. C. R. A Language and Environment for Statistical Computing (2015).
Faith, D. P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 61, 1–10 (1992).
Data Citations
Schneider, D. NCBI Sequence Read Archive SRP093596 (2016)
Schneider, D. Figshare https://doi.org/10.6084/m9.figshare.c.3877591.v1 (2017)
Acknowledgements
This work was funded by the Federal State of Lower Saxony, Niedersächsisches Vorab (VWZN2889). We thank L. von Müller and M. Rupnik for kindly having performed ribotyping and toxinotyping. We thank the patients and healthy volunteers who provided their stool samples.
Author information
Authors and Affiliations
Contributions
R.D. and D.S. conceived the study and the experiments. R.L. extracted the DNA. A.T. and K.Go. performed sample preparation and sequencing. U.G. organized the sample collection. K.Gu. and U.G. contributed data. D.S. analyzed sequence data. D.S., U.G., A.T. and R.D. wrote the manuscript. All authors interpreted the results and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Schneider, D., Thürmer, A., Gollnow, K. et al. Gut bacterial communities of diarrheic patients with indications of Clostridioides difficile infection. Sci Data 4, 170152 (2017). https://doi.org/10.1038/sdata.2017.152
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2017.152
This article is cited by
-
Microbiome differential abundance methods produce different results across 38 datasets
Nature Communications (2022)
-
The microbial diversity following antibiotic treatment of Clostridioides difficile infection
BMC Gastroenterology (2021)
-
Characterization of Gut Microbiota in Hospitalized Patients with Clostridioides difficile Infection
Current Microbiology (2020)
-
Bacterial succession along a sediment porewater gradient at Lake Neusiedl in Austria
Scientific Data (2019)
