
Abstract
Streptococcus pneumoniae is common nasopharyngeal commensal bacterium and important human pathogen. Vaccines against a subset of pneumococcal antigenic diversity have reduced rates of disease, without changing the frequency of asymptomatic carriage, through altering the bacterial population structure. These changes can be studied in detail through using genome sequencing to characterise systematically-sampled collections of carried S. pneumoniae. This dataset consists of 616 annotated draft genomes of isolates collected from children during routine visits to primary care physicians in Massachusetts between 2001, shortly after the seven valent polysaccharide conjugate vaccine was introduced, and 2007. Also made available are a core genome alignment and phylogeny describing the overall population structure, clusters of orthologous protein sequences, software for inferring serotype from Illumina reads, and whole genome alignments for the analysis of closely-related sets of pneumococci. These data can be used to study both bacterial evolution and the epidemiology of a pathogen population under selection from vaccine-induced immunity.
Design Type(s) | time series design • Whole Genome Sequencing • strain comparison design |
Measurement Type(s) | Genome Assembly Sequence |
Technology Type(s) | DNA sequencer |
Factor Type(s) | year • population • Serotype |
Sample Characteristic(s) | Streptococcus pneumoniae |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
Streptococcus pneumoniae (the pneumococcus) is a genetically diverse bacterial species commonly asymptomatically carried in the nasopharynx of infants, an age group not capable of generating a strong adaptive immune response to the polysaccharide capsule that envelopes most pneumococcal cells1. This capsule inhibits immune clearance by both complement- and neutrophil-mediated pathways2, and is a critical factor in allowing S. pneumoniae to invade other anatomical sites and cause disease such as pneumonia, bacteraemia and meningitis, particularly in both the young and elderly. Consequently, polysaccharide conjugate vaccines (PCVs) based on the pneumococcal capsule have been developed to induce anti-pneumococcal immunity in children3. These vaccine formulations are limited in the number of antigens they contain: the first licensed formulation (PCV7) contained seven serotypes. However, over 90 immunologically distinct capsule polysaccharides (serotypes) have been identified in S. pneumoniae4, which is the consequence of extensive genetic variation at the capsule polysaccharide synthesis (cps) locus. Hence at the point of vaccine introduction the pneumococcal population consists of a mix of ‘vaccine serotypes’, susceptible to artificially-induced host immunity, and ‘non-vaccine serotypes’. Nevertheless, the widespread use of PCVs has caused a substantial fall in the incidence of invasive pneumococcal disease5.
This is the consequence of the PCV7 vaccine having at least 50% efficacy in preventing nasopharyngeal carriage of vaccine serotypes6,7, and around 98% (ref. 8) efficacy against invasive disease caused by the same types. However, the overall levels of pneumococcal carriage have not changed post-vaccination owing to serotype replacement9,10: the increase in frequency of non-vaccine serotypes. The reduced incidence of pneumococcal disease has therefore been attributed to the lower rate at which these non-vaccine serotypes cause symptomatic infections relative to vaccine serotypes11. Additionally, as many multidrug-resistant pneumococci were of PCV7 serotypes prior to the vaccine’s introduction, it was anticipated that PCV7 would decrease the levels of S. pneumoniae antimicrobial resistance; however, any such benefit in this regard observed shortly after vaccine introduction12 was not sustained over the longer term13. Hence understanding how the carried pneumococcal population structure changes following the implementation of partial coverage PCVs is important for relating the intervention to its subsequent clinical outcomes.
To address this question, the carried population of pneumococci in Massachusetts has been followed by the Streptococcus pneumoniae Antimicrobial Resistance in Children (SPARC) project. The instigation of this study coincided with the introduction of PCV7 in the USA in 2000. Samples were obtained through swabbing the nasopharynx of children seven years of age or under during routine visits to primary care physicians14. In the spring of 2001, winter and spring of 2004, and winter and spring of 2006–2007, 742, 994 and 972 individuals were sampled, respectively13–15. The detected level of pneumococcal colonisation remained stable over these winters, with 190 (26% prevalence), 232 (23% prevalence) and 294 (30% prevalence) S. pneumoniae isolates recovered in the three successive sampling periods. This collection has been used to analyse the changing antigenic profile of the population in response to vaccine-induced immunity through serological typing13–15. The same collection was also genotyped by multilocus sequence typing16 (MLST) to relate these changes to the elimination, emergence and diversification of individual bacterial lineages17,18. Subsequently, whole genome sequencing was applied to the collection to investigate the population dynamics in greater detail19. This dataset consists of 616 annotated draft S. pneumoniae genomes representing the evolutionary epidemiology of the species as the population structure changed in response to vaccine-induced selection pressures. To aid analysis of the overall set of isolates, the species-wide core genome alignment and phylogeny are also made available, as are the predicted protein sequences, a method for inferring serotype from Illumina reads, and whole genome alignments for fifteen sets of closely-related isolates.
Methods
Culturing and phenotyping of strains
Following retrieval from storage, all bacterial samples were colony purified, then grown on 5% sheep’s blood agar overnight at 37 °C in the presence of 5% CO2. Samples were serologically typed using latex agglutination (Statens Serum Institut, Copenhagen) as a check on sample handling. Discrepant results with previous typing were verified using the Quellung reaction (Statens Serum Institut, Copenhagen; Table 1 (available online only)).
Overnight plate growth was harvested through resuspension in phosphate buffered saline, and genomic DNA was extracted using DNeasy columns (Qiagen) following manufacturer’s instructions. The concentration of genomic DNA was quantified using the Qubit system (Life Technologies); all samples yielded at least 3 μg of DNA. The integrity of the genomic DNA was checked using agarose gel electrophoresis relative to a λHindIII ladder (New England Biolabs).
Generation of sequence data
Illumina sequencing libraries were constructed as described previously20,21. Briefly, genomic DNA was first fragmented using Adaptive Focused Acoustics technology (Covaris). The resulting fragments were then repaired to ensure they had blunt ends, phosphorylated at their 5′ end, A-tailed at the 3′ end, and ligated to adapter molecules. This library of fragments was then separated by agarose gel electrophoresis. DNA constructs of the appropriate size range (generating an insert size of approximately 150–300 bp) were then extracted from the gel and amplified by a polymerase chain reaction using Kapa HiFi polymerase (Kapa Biosystems) that added one of the 96 index tags used in this project. Libraries were then quantified using qPCR, and combined into an equimolar pool of 96 samples prior to denaturation, cluster generation and sequencing in a single flow cell lane with an Illumina HiSeq machine. Isolates from 2001 and 2003–2004 were sequenced as paired end libraries generating 75 nt reads; isolates from 2006–2007 were sequenced as paired end libraries generating 100 nt reads.
Assembly and annotation of sequence data
Sequences were assembled de novo using Velvet22 version 1.2 with parameters selected to be optimal for individual datasets as described previously23. Both Glimmer3 (ref. 24) and Prodigal25 were trained on the reference sequence of S. pneumoniae ATCC 700669 (ref. 26; Data Citation 1), then applied to the complete draft assembly with an 11 nt sequence encoding stop codons in each reading frame added to each end, to facilitate the identification of partial coding sequences (CDSs) broken by the assembly. Putative CDSs were then trimmed at the 3′ end to stop them spanning contig breaks within the assembly. Final CDS predictions were identified as the consensus of Glimmer3 and Prodigal outputs, as described previously19 (Table 1 (available online only)). Protein sequences were then translated, aligned using BLAT27 suite 0.34, and ‘clusters of orthologous genes’ (COGs) identified using COGsoft28. Pairs of orthologous sequences were then manually identified as described previously19.
To generate functional annotations of genome sequences, all CDSs were labelled with a unique identifier (of the form, ‘ERSX_Y’, where ‘ERSX’ is the sample accession code in the European Nucleotide Archive listed in Table 1 (available online only) and Y is an incrementing index) and their COG (of the form, ‘SPARC1_CLSZ’ or ‘SPARC1_CLSTZ’, where Z is a number). COGs relating to antibiotic resistance and the newly-characterised variable restriction-modification system loci were annotated as described previously29; the 590 COGs found to be specific to prophage, the three COGs found to be specific to a particular prophage remnant, the 142 COGs found to be specific to phage-related chromosomal islands, and the 355 COGs found to be specific to integrative and conjugative elements were also appropriately identified in these datasets29. All COGs not belonging to one of these categories were annotated using a database of pneumococcal CDS information. This was constructed by extracting the protein sequences and annotated functions from publicly-available complete genomes and the annotation of 90 capsule polysaccharide synthesis loci30. Where a CDS in one of the draft genomes had a putative protein sequence identical to the translated sequence of a previously annotated locus, the annotation was directly transferred; otherwise, the annotation was transferred on the basis of orthology, if another putative protein in the same COG was identical to the translation of a CDS in an annotated genome sequence. In cases where no such information could be obtained, CDSs were labelled as producing ‘hypothetical proteins’. Pneumococcal small interspersed repeats were annotated as described previously31, and tRNA and rRNA loci were annotated with tRNAscan-SE32 version 1.3.1 and rnammer33 version 1.2, respectively (Table 1 (available online only)).
Generation of core genome alignment and overall phylogeny
As described previously19, the 1,194 COGs found to have a single representative in each of the 616 genomes were individually aligned at the protein level using MUSCLE34, prior to backtranslation to generate a 1.14 Mb codon alignment. The 106,196 polymorphic sites were extracted and used to generate a phylogeny using RAxML35 version 7.0.4 with the general time reversible substitution model and a four category gamma distribution to account for rate heterogeneity. This tree was midpoint rooted on the longest branch, which separated sequence cluster 12 from the rest of the population. This is consistent with a wider phylogenetic analysis of multiple species that suggested sequence cluster 12 was the earliest lineage to diverge from the other isolates19. The same alignment was analysed with BAPS36 version 5 to identify the sequence clusters. Both the core genome alignment and tree are made available as part of Data Citation 2.
Generation of whole genome alignments
For each of the fifteen monophyletic sequence clusters identified using BAPS and RAxML, a single reference draft assembly was selected for manual curation. The Illumina read data were reassembled with SGA37 version 0.9, and these contigs merged with those from Velvet using Zorro38 version 2.2. These were arranged into scaffolds using SSPACE39 version 2, then manually curated and ordered using ABACAS40 and ACT41. These fifteen assemblies are made available as part of Data Citation 2. Illumina read pairs from isolates of the same sequence cluster were then mapped against this reference using SMALT42 version 0.5.8. The resulting read alignment was processed with Samtools43, VCFtools44 and Biopython45 to generate a consensus sequence. Bases were called at positions spanned by at least two reads in each direction, where at least a 75% consensus on the allele was evident; additionally, the base quality at the site had to be at least 50, and the mapping quality had to be at least 30, on the Phred scale46,47. These consensus sequences from each representative of the sequence cluster were then combined to generate a reference-based multiple genome alignment, each of which was analysed as described previously using an earlier version of the Gubbins48 software. These fifteen whole genome alignments, which do not include the reference assemblies themselves, are also made available as part of Data Citation 2.
Code Availability
The algorithm used to predict recombination events has been developed into a software package, named Gubbins48, which can be installed on Linux and Max OSX operating systems. It can also be run on Windows operating systems using a virtual machine environment, and is freely available from http://sanger-pathogens.github.io/gubbins/. Code and reference sequences for the serological typing of S. pneumoniae using sequence reads is made available as part of Data Citation 2.
Data Records
The raw sequence data (FASTQ format) and annotated draft genome sequences (EMBL format) for the 616 isolates in the dataset have been deposited in the European Nucleotide Archive (http://www.ebi.ac.uk/ena/) with the project accession code in Data Citation 3. Individual accession codes for both raw read data and annotated draft genome assemblies are listed in Table 1 (available online only) and in the machine-readable ISA-tab metadata files associated with this article.
Further files are made available through the Dryad repository (https://datadryad.org) with the digital object identifier in Data Citation 2. The core genome codon alignment (FASTA format) and maximum likelihood phylogeny (Newick format) describe the overall population structure. For the study of individual lineages, a whole genome alignment (FASTA format) and draft reference assembly (FASTA format) are included for each of the 15 monophyletic sequence clusters19. These encompass 491 of the isolates, excluding those in the diverse polyphyletic sequence cluster 16. In addition, the full set of protein coding sequences (FASTA format), and the translated proteins (FASTA format), can be used to study the diversity of individual COGs. To minimise the manipulation of the protein sequences, the initial amino acid of each protein is directly translated, rather than being converted to a methionine.
The epidemiological and phylogenetic data can also be interactively visualised and analysed online using the Microreact website (http://microreact.org/) with the URL in Data Citation 4.
Technical Validation
Integrity of sample handling and quality control
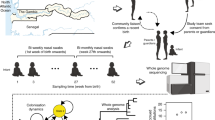
An overview of the processing pipeline, including the technical validation steps, is shown in Fig. 1. Of the 716 samples collected as part of the surveillance project between 2001 and 2007, 631 could be revived and cultured. All these isolates were subjected to serological typing using latex agglutination to ensure consistency of sample handling relative to previous studies (Fig. 1 and Table 1 (available online only)). As multiple colonisation is often observed in children49 it was not necessarily expected that the original strain would be recovered in all cases; in the earlier studies, only a single isolate per individual was analysed in order to maximise the size of the host population sample, as detecting strains carried at low frequencies is inefficient using standard microbiological techniques50. The serology differed from that previously recorded for 12 of the 631 samples. These new serotype inferences were all independently verified as being correct using a second culture of the same isolate, and subsequently confirmed in all seven cases where the samples were retyped in a separate laboratory using the Quellung reaction51. One sample yielded two clearly morphologically distinct strains that proved to be of different serotypes, increasing the number of isolates in the study to 632. All isolates found to be non-typeable serologically were tested for optochin susceptibility using ‘P discs’; this identified one isolate as likely to represent a non-pneumococcal streptococcus, the exclusion of which reduced the overall collection size to 631.

The flow chart shows the steps taken to generate the draft genome dataset; boxes in orange indicate steps at which typing was performed, allowing the integrity of sample handling to be checked, and boxes in blue indicate steps at which checks were performed to allow for the identification and elimination of low quality samples.
Sufficient high-quality genomic DNA for analysis was extracted from all isolates. After sequencing, nine samples failed either automated quality control checks implemented by the Sanger Institute, or manual investigation of dataset properties, such as adapter content, insert size and GC content; one of these failures was successfully resequenced. After assembly of the remaining 623 samples, seven further samples were rejected as generating low quality draft genomes. These represented cases where the N50 was below 15 kb and the total assembly length was greater than 2.4 Mb, likely as a consequence of sequence from more than one isolate being mixed in the raw Illumina reads. This resulted in the final dataset of 616 draft genomes.
Integrity of data handling
Serotypes and multilocus sequence types (STs) were inferred from Illumina read data as described previously23. Excluding isolates determined as being ‘non typeable’ by either microbiological or bioinformatic serotyping, across the final set of 616 samples the capsule polysaccharide synthesis (cps) locus was congruent with the serogroup (a set of antigenically similar serotypes) identified through immunological tests in all but two cases. Of the 594 isolates for which an ST had been previously established, 553 (93%) were identical with those inferred from Illumina reads (Fig. 2a). These included both cases where the genome’s cps locus did not match the experimentally ascertained serogroup, indicating these discrepancies were not likely to result from sampling handling issues. Of the 41 cases in which the original ST was discrepant with that inferred from the reads, 29 differed at only one of the seven loci (Fig. 2b). All remaining inconsistencies are likely to reflect instances of multiple colonisation, resulting in different strains being originally genotyped before storage and subsequently retrieved for sequencing. This is consistent with the cps locus from the sequence reads in these cases matching the serology of the revived isolates from which the genomic DNA was extracted.

(a) Venn diagram showing the overlap between the sequence types from the original genotyping of the collection, those inferred from Illumina sequence read mapping, and those inferred from the genome assemblies. Only data for the 594 isolates for which all three datatypes were available are represented here. (b) Venn diagram showing the overlap between sequence types inferred by different methodologies, in this case treating results as being consistent if only one of the seven loci differed between results. In this case, the sequence types inferred from read mapping and de novo assembly are identical, and differ from the original genotyping in only twelve cases. (c) Histogram showing the number of CDSs in publicly available annotated complete, or high quality draft, S. pneumoniae genomes. (d) Histogram showing the number of CDSs in the 616 draft genomes from Massachusetts. This distribution shows that the count of putative CDSs in each draft genome is within the range of CDSs identified in manually annotated genomes, consistent with the draft genomes being near-complete, and the CDS predictions being accurate.
Quality of genome assemblies
The 616 samples in the dataset each yielded between 267 and 1,865 Mb of sequence data (median of 652 Mb). Assuming a typical 2 Mb S. pneumoniae genome26, this meant each isolate had a sequencing depth of over 100 fold coverage. Based on a random sample of 10,000 reads aligned to a set of prokaryotic and eukaryotic reference genomes using BWA52, a substantial majority of reads matched to the S. pneumoniae representative (strain ATCC 700669) in each dataset, confirming these data were primarily derived from the submitted genomic DNA sample.
All draft assemblies had an N50 greater than 15 kb and a total length between 1.98 and 2.19 Mb, similar to complete S. pneumoniae genomes. Additionally, the number of CDSs in the de novo assemblies was within the range of CDSs found within annotated complete or high-quality draft S. pneumoniae genomes (Fig. 2c,d). Each isolate’s annotation included at least 101 of the 102 protein functional domains recently suggested to be essential and ubiquitous across cellular genomes53; the only discrepancy was the short ribosomal protein coding sequence rpsN, which was not consistently identified by the automated gene annotation software even when present within assemblies. Assembly quality was also judged on the basis of non-coding RNA content: using previously-defined criteria53, the majority of isolates had a full-length representative of each of the ribosomal RNAs. Similarly, quantifying tRNA content found the majority of isolates had at least one tRNA for each standard amino acid.
To ascertain the accuracy of the de novo assemblies relative to the original epidemiological data and Illumina sequence reads, the STs were extracted from the contigs through identification of the relevant loci using BLAST54 (Fig. 2a,b). All seven loci could be recovered from each assembly. Across the 616 samples, the ST extracted from the assembly and Illumina reads was identical in 602 cases (98% accuracy). In four cases, the ST inferred from the assemblies differed from the consensus of the original genotyping and the ST extracted from the reads at a single locus (Fig. 2a). In six cases, the ST inferred from the Illumina reads differed from the consensus of the original genotyping and the assembly at a single locus; the assemblies indicated these all corresponded to a single ST, suggesting a rare systematic error. In four cases, the STs inferred from the assembly and Illumina reads differed at a single locus, and the original genotyping data were missing or inconsistent with both.
The wider ‘core’ genome was defined as a set of 1,194 COGs, a single representative of which was found in each genome19. Independent re-analysis of this dataset with a different method for defining COGs found 1,206 ‘core’ COGs, of which 1,027 were identical to the 1,194 originally identified55. Concatenated codon alignments of the ‘core’ COGs were subject to three independent analyses with BAPS version 5, which converged on identical membership of the sixteen sequence clusters, in two cases, with additional isolates included within SC7 in the third. The fifteen sequence clusters containing similar isolates were correspondingly monophyletic in the core genome phylogeny, confirming them as being closely related sets of bacteria (Fig. 3).

The maximum likelihood phylogeny generated from the core genome alignment is presented, as displayed in the Microreact system (Data Citation 4), with each leaf node coloured according to its sequence cluster (SC).
Validation of recombination analyses
Recombination detection was only attempted within the 15 monophyletic sequence clusters, as they consisted of groups of isolates with detectable similarity that was likely to reflect recent common ancestry. Simulations indicated that the type of approach that was used to identify recombinations in whole genome alignments is most accurate when applied to sets of closely-related sequences48. In the analyses presented in this work, all alignments in which at least ten recombination events were detected formed an exponential recombination length distribution with a rate constant consistent with other genomic data23,56–58 and experimental work47. The positions of recombination ‘hotspots’ relative to the reference genome annotations were also consistent with these independent analyses. In the cases where evidence of a molecular clock could be detected, the substitution rate was also found to be consistent with the analyses of other S. pneumoniae datasets23,56–58. Additionally, the consistency of the final phylogenies with the epidemiological data allowed a phylogeographic signal to be detected19.
Further analysis of the isolates’ inferred serology (Table 1 (available online only)) identified cases where closely-related isolates differed at their cps loci, suggesting ‘serotype switching’ had occurred. In all cases where the pattern of switching could be robustly established, the change at the cps locus could be attributed to an inferred recombination affecting the relevant genes4.
Usage Notes
Sequence data may be downloaded from the European Nucleotide Archive using the project accession codes ERP000809 or PRJEB2632. All accession codes for raw sequence data and annotated individual assemblies are listed in Table 1 (available online only). Associated epidemiological data was published previously19. Sequences and functional annotation can be displayed using Artemis41. Whole genome alignments can be viewed and analysed using standard software. Gubbins48 can be applied to them for the inference of recombined sequence. The software for serological typing can be run on Linux or Mac OSX as described in the accompanying README file.
Additional Information
Tables 1 is only available in the online version of this paper.
How to cite this article: Croucher, N. J. et al. Population genomic datasets describing the post-vaccine evolutionary epidemiology of Streptococcus pneumoniae. Sci. Data 2:150058 doi: 10.1038/sdata.2015.58 (2015).
References
References
Weintraub, A. Immunology of bacterial polysaccharide antigens. Carbohydrate Research 338, 2539–2547 (2003).
Hyams, C., Camberlein, E., Cohen, J. M., Bax, K. & Brown, J. S. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect Immun. 78, 704–715 (2010).
Austrian, R. Pneumococcal otitis media and pneumococcal vaccines, a historical perspective. Vaccine 19 (Suppl 1): S71–S77 (2000).
Croucher, N. J. et al. Selective and Genetic Constraints on Pneumococcal Serotype Switching. PLoS Genet 11, e1005095 (2015).
Whitney, C. G. et al. Effectiveness of seven-valent pneumococcal conjugate vaccine against invasive pneumococcal disease: a matched case-control study. Lancet 368, 1495–1502 (2006).
Ghaffar, F. et al. Effect of the 7-valent pneumococcal conjugate vaccine on nasopharyngeal colonization by Streptococcus pneumoniae in the first 2 years of life. Clin. Infect. Dis. 39, 930–938 (2004).
Rinta-Kokko, H., Dagan, R., Givon-Lavi, N. & Auranen, K. Estimation of vaccine efficacy against acquisition of pneumococcal carriage. Vaccine 27, 3831–3837 (2009).
Black, S. et al. Clinical effectiveness of seven-valent pneumococcal conjugate vaccine (Prevenar) against invasive pneumococcal diseases: prospects for children in France. Arch. Pediatr 11, 843–853 (2004).
Weinberger, D. M., Malley, R. & Lipsitch, M. Serotype replacement in disease after pneumococcal vaccination. The Lancet 378, 1962–1973 (2011).
Spratt, B. G. & Greenwood, B. M. Prevention of pneumococcal disease by vaccination: does serotype replacement matter? Lancet 356, 1210–1211 (2000).
Gladstone, R. A., Jefferies, J. M., Faust, S. N. & Clarke, S. C. Continued control of pneumococcal disease in the UK - the impact of vaccination. J Med. Microbiol. 60, 1–8 (2011).
Kyaw, M. H. et al. Effect of introduction of the pneumococcal conjugate vaccine on drug-resistant Streptococcus pneumoniae . N Engl. J Med. 354, 1455–1463 (2006).
Huang, S. S. et al. Continued impact of pneumococcal conjugate vaccine on carriage in young children. Pediatrics 124, e1–e11 (2009).
Finkelstein, J. A. et al. Antibiotic-resistant Streptococcus pneumoniae in the heptavalent pneumococcal conjugate vaccine era: predictors of carriage in a multicommunity sample. Pediatrics 112, 862–869 (2003).
Huang, S. S. et al. Post-PCV7 changes in colonizing pneumococcal serotypes in 16 Massachusetts communities, 2001 and 2004. Pediatrics 116, e408–e413 (2005).
Enright, M. C. & Spratt, B. G. A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiology 144, 3049–3060 (1998).
Hanage, W. P. et al. Diversity and antibiotic resistance among nonvaccine serotypes of Streptococcus pneumoniae carriage isolates in the post-heptavalent conjugate vaccine era. J Infect Dis. 195, 347–352 (2007).
Hanage, W. P. et al. Clonal replacement among 19 A Streptococcus pneumoniae in Massachusetts, prior to 13 valent conjugate vaccination. Vaccine 29, 8877–8881 (2011).
Croucher, N. J. et al. Population genomics of post-vaccine changes in pneumococcal epidemiology. Nat. Genet. 45, 656–663 (2013).
Quail, M. A. et al. A large genome center’s improvements to the Illumina sequencing system. Nat. Methods 5, 1005–1010 (2008).
Quail, M. A. et al. Optimal enzymes for amplifying sequencing libraries. Nat. Methods 9, 10–11 (2012).
Zerbino, D. R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Croucher, N. J. et al. Rapid pneumococcal evolution in response to clinical interventions. Science 331, 430–434 (2011).
Delcher, A. L., Bratke, K. A., Powers, E. C. & Salzberg, S. L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679 (2007).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010).
Croucher, N. J. et al. Role of conjugative elements in the evolution of the multidrug-resistant pandemic clone Streptococcus pneumoniaeSpain23F ST81. J Bacteriol. 191, 1480–1489 (2009).
Kent, W. J. BLAT--the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Kristensen, D. M. et al. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 26, 1481–1487 (2010).
Croucher, N. J. et al. Diversification of bacterial genome content through distinct mechanisms over different timescales. Nat. Commun. 5, 5471 (2014).
Bentley, S. D. et al. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet. 2, e31 (2006).
Croucher, N. J., Vernikos, G. S., Parkhill, J. & Bentley, S. D. Identification, variation and transcription of pneumococcal repeat sequences. BMC Genomics 12, 120 (2011).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Lagesen, K. et al. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Stamatakis, A. et al. RAxML-Light: a tool for computing terabyte phylogenies. Bioinformatics 28, 2064–2066 (2012).
Tang, J., Hanage, W. P., Fraser, C. & Corander, J. Identifying currents in the gene pool for bacterial populations using an integrative approach. PLoS Comput. Biol. 5, e1000455 (2009).
Simpson, J. T. & Durbin, R. Efficient construction of an assembly string graph using the FM-index. Bioinformatics 26, i367–i373 (2010).
Costa, G. G., Vidal, R. O. & Carazzolle, M. F. Zorro. Available at <http://www.lge.ibi.unicamp.br/zorro/> (2011).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Assefa, S., Keane, T. M., Otto, T. D., Newbold, C. & Berriman, M. ABACAS: Algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25, 1968–1969 (2009).
Carver, T. et al. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 24, 2672–2676 (2008).
Postingl, H. SMALT. Available at <http://www.sanger.ac.uk/resources/software/smalt/> (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Cock, P. J. A. et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423 (2009).
Harris, S. R. et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science (80-. ). 327, 469–474 (2010).
Croucher, N. J., Harris, S. R., Barquist, L., Parkhill, J. & Bentley, S. D. A high-resolution view of genome-wide pneumococcal transformation. PLoS Pathog 8, e1002745 (2012).
Croucher, N. J. et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15 (2015).
Turner, P. et al. Improved detection of nasopharyngeal cocolonization by multiple pneumococcal serotypes by use of latex agglutination or molecular serotyping by microarray. J. Clin. Microbiol. 49, 1784–1789 (2011).
Huebner, R. E., Dagan, R., Porath, N., Wasas, A. D. & Klugman, K. P. Lack of utility of serotyping multiple colonies for detection of simultaneous nasopharyngeal carriage of different pneumococcal serotypes. Pediatr. Infect. Dis. J. 19, 1017–1020 (2000).
Habib, M., Porter, B. D. & Satzke, C. Capsular serotyping of Streptococcus pneumoniae using the Quellung reaction. J. Vis. Exp. 84, e51208 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Land, M. L. et al. Quality scores for 32,000 genomes. Stand. Genomic. Sci. 9, 20 (2014).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Van Tonder, A. J. et al. Defining the Estimated Core Genome of Bacterial Populations Using a Bayesian Decision Model. PLoS Comput. Biol. 10, e1003788 (2014).
Croucher, N. J. et al. Variable recombination dynamics during the emergence, transmission and ‘disarming’ of a multidrug-resistant pneumococcal clone. BMC Biol. 12, 49 (2014).
Croucher, N. J. et al. Evidence for Soft Selective Sweeps in the Evolution of Pneumococcal Multidrug Resistance and Vaccine Escape. Genome Biol. Evol. 6, 1589–1602 (2014).
Chewapreecha, C. et al. Dense genomic sampling identifies highways of pneumococcal recombination. Nat. Genet. 46, 305–309 (2014).
Data Citations
Croucher, N.J. International Nucleotide Sequence Database FM211187 (2009)
Croucher, N.J. Dryad http://dx.doi.org/10.5061/dryad.t55gq (2015)
Croucher, N.J. International Nucleotide Sequence Database PRJEB2632 (2013)
Croucher, N.J. Microreact http://microreact.org/project/NJwviE7F (2015)
Acknowledgements
N.J.C. is funded by a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and Royal Society (Grant Number 104169/Z/14/Z). Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the US National Institutes of Health (NIH) under award R01AI066304 and by Wellcome Trust grant 098051. We are grateful for the support of the Sanger Institute core sequencing and informatics teams.
Author information
Authors and Affiliations
Contributions
N.J.C. assembled, analysed and annotated sequences. J.A.F. designed the project and managed the collection of samples. S.I.P. managed the phenotyping of samples. S.D.B. managed the generation and deposition of sequence data. J.P. managed the generation and deposition of sequence data. M.L. designed the project, managed the phenotyping of samples and generation of genomic DNA. W.P.H. designed the project, managed the genotyping of samples and generation of genomic DNA. All authors contributed to, and approved, the final manuscript.
Corresponding author
Ethics declarations
Competing interests
S.I.P. has investigator-initiated grants from Merck and Pfizer and has consulted for GlaxoSmithKline, Merck, Pfizer and Novartis. W.P.H. has consulted for GlaxoSmithKline. M.L. has consulted for Pfizer and Novartis.
ISA-Tab metadata
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0 Metadata associated with this Data Descriptor is available at http://www.nature.com/sdata/ and is released under the CC0 waiver to maximize reuse.
About this article

Cite this article
Croucher, N., Finkelstein, J., Pelton, S. et al. Population genomic datasets describing the post-vaccine evolutionary epidemiology of Streptococcus pneumoniae. Sci Data 2, 150058 (2015). https://doi.org/10.1038/sdata.2015.58
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2015.58
