
Abstract
Coronavirus disease 2019 (COVID-19) is a consequence of infection of the upper and lower respiratory tract with severe acute respiratory syndrome coronavirus 2 but often becomes a systemic disease, with important involvement of other organs. A bidirectional relationship exists between COVID-19 and cardiovascular disease. On the one hand, preexisting comorbidities, in particular high prevalence of cardiovascular risk factors such as hypertension and diabetes and chronic cardiovascular conditions predispose to severe disease. On the other hand, biomarkers of myocardial injury are frequently raised in patients with COVID-19, along with arrhythmia and heart failure. Localized thrombosis is a common finding in the lungs but can also increase the occurrence of thrombotic events systemically. Thrombosis is consequent to different pathogenic mechanisms, which include endothelial dysfunction and immunothrombosis. Thrombocytopenia is common in patients with COVID-19 and alterations in platelet function participate in the pro-thrombotic phenotype. Involvement of the cardiovascular system in COVID-19 has important consequences during recovery from infection and the development of long COVID.
Similar content being viewed by others


Main
Two years into the pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), it is now clear that novel COVID-19 is mild or even asymptomatic in the majority of infected individuals. However, 4–5% of infected individuals develop severe disease that requires hospitalization. It was evident from the very beginning of the pandemic that preexisting comorbidities are highly prevalent among people hospitalized with COVID-19. In particular, admitted patients have a high prevalence of cardiovascular risk factors such as hypertension and diabetes mellitus as well as chronic cardiovascular conditions such as ischemic heart disease and heart failure1,2,3,4,5. Severe COVID-19 is a multisystem disease; beyond pneumonitis and hypoxia, other features may include systemic coagulopathy and thrombosis, cardiac injury, renal failure, hepatic dysfunction, gastrointestinal abnormalities and neurocognitive dysfunction. Moreover, incident cardiovascular complications may be life-threatening during severe COVID-19. As such, the bidirectional relationship between cardiovascular disease and COVID-19 is highly pertinent to the pathophysiology and outcome of this complex disease (Fig. 1).

Severe COVID-19 is a multisystem disease, in which the lung–heart axis has an essential role. Lung infection causes systemic inflammation, predisposition to microthrombosis and hypoxia, which can damage the myocardium. This in turn can worsen lung function, leading to right heart failure. The presence of predisposing conditions is fundamental to worsen this vicious cycle. AF, atrial fibrillation; CVD, cardiovascular disease (including ischemic heart disease, heart failure and stroke).
Here, we review the available evidence on the involvement of the cardiovascular system in COVID-19.
Epidemiology of COVID-19 and cardiovascular disease
The early descriptions of clinical characteristics among patient cohorts hospitalized with COVID-19 in China, Europe and the United States consistently noted a high prevalence of individuals with cardiovascular disease and diabetes3,5,6. Subsequent large cohort studies and population-based analyses have confirmed the association between preexisting cardiometabolic comorbidities and severe COVID-19; these studies have also identified age >60 years, male sex, other comorbidities, obesity, a non-white ethnic background, socioeconomic deprivation and occupational exposure as additional factors associated with severe COVID-19 disease4,7. Age is by far the strongest risk factor; individuals over the age of 80 have a more than 20-fold higher risk of mortality than 50–59 year olds, after adjusting for other risk factors4. Hypertension, however, does not appear to be associated with an increased risk of death even though it is highly prevalent among those admitted with COVID-19 disease4.
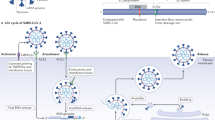
What is the underlying basis for the association between preexisting cardiovascular disease and COVID-19? The first possibility to consider is whether these individuals have a higher susceptibility to become infected upon exposure to SARS-CoV-2. Obtaining good direct evidence to support this idea is challenging because exposure to the virus is uncertain or unknown in most studies. However, systematic longitudinal community-based household surveys such as the UK Office of National Statistics Coronavirus Infection Survey in approximately 150,000 people have not provided much evidence to support the idea8. An early hypothesis was that treatment with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers, which are prescribed to many individuals with cardiometabolic conditions, may increase infection with SARS-CoV-2. The rationale was that these drugs increase the expression of the ACE2 viral receptor9 and thus viral internalization, a process that can occur directly at the plasma membrane or after endocytosis (Fig. 2). However, numerous studies have now convincingly demonstrated a lack of detrimental impact of treatment with these drugs10,11,12,13.

The spike protein on the envelope of SARS-CoV-2 virions (orange) binds the ACE2 receptor and then needs activation by a cellular protease. This priming step, which is essential for membrane fusion, can be carried out by furin or TMPRSS2 at the plasma membrane level (left) or after endocytosis in the acidic environment of late endosomes by endosomal low pH-activated proteases such as cathepsin B and cathepsin L (right and bottom). Proteases are in red. In infected cells, a fraction of the spike protein is present on the plasma membrane. When this surface-expressed spike is activated by the plasma membrane proteases, infected cells can fuse to neighboring cells expressing the ACE2 receptor, thus mimicking the event occurring during fusion of the virion envelope with the cell plasma membrane.
An alternative possibility is that individuals with cardiometabolic comorbidities may be more prone to develop severe COVID-19 and life-threatening complications. As mentioned earlier, this possibility is strongly supported by numerous large population-based and cohort studies4,7. It is possible that the higher propensity to develop severe disease may not be specific to COVID-19 as it is well established that people with cardiometabolic comorbidities develop more severe illness with other infective disorders14,15. This may be related to the fact that these individuals already have a variety of subclinical pathophysiological abnormalities, such as endothelial dysfunction and activation, platelet hyperreactivity and propensity to coagulopathy and dysfunctional immune responses, such that they are more likely to develop systemic inflammation and thromboembolic complications to infection and/or increased cardiovascular complications16. Moreover, they might also be more likely to develop cardiac dysfunction in the face of severe lung injury (see below).
A specific additional aspect worth mentioning is the intersection between ethnicity and cardiometabolic morbidities. In the multiethnic populations of the United Kingdom and the United States, it was evident from the very beginning of the pandemic that individuals of non-white minority background are disproportionately affected by severe COVID-19. This is especially the case for Black and south Asian people in the United Kingdom and for African Americans and Hispanics in the United States4,17,18,19,20. While the relationship between ethnicity and COVID-19 is complex and multifactorial, it is clear that an important component of the higher mortality rate observed in minority groups may be attributed to socioeconomic factors (such as living conditions, household size, type of occupation, adherence/ability to socially distance and access to healthcare) that may affect exposure to the virus and ability to take mitigating measures21,22. In addition, a high prevalence of cardiometabolic comorbidities may be a contributory factor to higher mortality in some ethnic groups, for example, south Asians20,23. Indeed, south Asians in the general population as well as those admitted with COVID-19 tend to have a substantially higher prevalence of diabetes and cardiovascular disease than other ethnic groups, both in the United States and the United Kingdom24,25. Moreover, in the pre-COVID-19 era, the rate of cardiovascular complications such as myocardial infarction was disproportionately high in UK south Asians after controlling for the prevalence of comorbidities25,26, suggesting an enhanced susceptibility to complications.
Together, it appears that the association between cardiometabolic morbidities and COVID-19 may in large part be related to an increased likelihood of more severe disease due to the preexisting clinical and subclinical abnormalities in these individuals, which are exacerbated by the additional insult of SARS-CoV-2 infection.
Cardiovascular complications in COVID-19
Patients hospitalized with severe COVID-19 have a significantly higher rate of incident clinical cardiovascular complications27,28. These include acute heart failure, arrhythmias, venous thromboembolism, cardiogenic shock, arterial thrombosis, myocardial ischemia or infarction, acute stroke and myocarditis. The rates of individual complications are highly variable among studies, which may reflect selection bias impacting publications, variations in the investigations that are performed and/or improvements in the prevention and management of COVID-19 during the pandemic. A retrospective study early in the pandemic on 1,000 patients in New York indicated that the commonest clinical complications are arrhythmias (up to 20%), thromboembolism (>25%) and heart failure27. The rates of thromboembolism may be even higher as it may be occult in a large proportion of patients. A retrospective cohort study on 13,217 hospitalized patients in the Netherlands reported that the risk of thrombotic complications, in particular venous thromboembolism, is markedly higher in COVID-19 (25%) than influenza (11%)29. Acute heart failure is a frequent complication among patients in intensive care units (up to 30%), and a substantial component of this may relate to right ventricular (RV) failure secondary to severe lung injury and pulmonary thromboembolism. Overt myocardial infarction or acute coronary syndromes are relatively infrequent in most studies that reported on hospitalized patients (<10%)30. A recent population-based study from Sweden is one of the first to carefully quantify the increased risk of acute myocardial infarction or stroke among individuals with COVID-19 as compared to controls without COVID-19 but matched for comorbidities and other risk factors31. These authors reported an odds ratio of 6.61 for acute myocardial infarction and 6.74 for acute ischemic stroke in 86,742 patients with acute COVID-19 as compared to the matched cohort. Contrary to speculation in the early stages of the COVID-19 pandemic, clinical acute myocarditis appears to be rare (<1%) in this group.
Cardiac injury and dysfunction
A substantial proportion of patients hospitalized with COVID-19 have elevated biomarkers of myocardial injury (as assessed by increased levels of high-sensitivity (hs) cardiac troponin) but often in the absence of overt cardiac symptoms or impairment. The prevalence of elevated hs-troponin is 30% or higher in many studies and is strongly associated with a worse outcome28,32,33. Lala et al.28 analyzed a US cohort of 2,736 hospitalized patients and found that, after adjustment for disease severity and clinical factors, the hazard ratio for death was 1.75 with even a small increase in hs-troponin I ( > 0.03 to 0.09 ng ml−1), while greater elevation (>0.09 ng dl−1) was associated with an adjusted hazard ratio of 3.03. Patients with raised hs-troponin levels also have more frequent malignant arrhythmias34. In a multivariable logistic regression analysis in 671 hospitalized patients in Wuhan (China), older age, comorbidities and a high level of C-reactive protein (indicative of inflammation) were predictors of myocardial injury32.
An important question has been whether myocardial injury is a major causative factor contributing to poor outcomes in COVID-19 or whether it is the consequence of critical systemic infective illness per se. This is because it is well established that myocardial injury as evidenced by increases in hs-troponin frequently occurs in critically ill patients (especially those with sepsis) and is a predictor of poor outcomes35,36. A recent small study comparing 76 patients admitted with COVID-19 with those admitted with other pneumonias found that myocardial injury biomarkers were no worse in patients with COVID-19, but that these patients did have higher rates of venous thromboembolism37. This raises several important questions, including what is the mechanistic basis of the myocardial injury; whether it is related to myocardial ischemia (macro or microvascular), infection of the myocardium with SARS-CoV-2 (that is, viral myocarditis) or inflammation; what is the relationship of myocardial damage with preexisting chronic cardiovascular disease; and to what extent cardiac damage might have long-term consequences. The answers to some of these questions are emerging from clinical analyses, cardiac imaging studies and autopsy data.
Analysis of cardiac function by echocardiography
Assessment of cardiac function by transthoracic echocardiography has been undertaken in numerous studies. Dweck et al.38 reported an analysis involving 1,216 patients across multiple continents who underwent echocardiography during hospital admission. These authors found that 55% of patients had an abnormal echocardiogram with equal numbers showing left ventricular or right ventricular abnormalities. Findings suggestive of new myocardial infarction or myocarditis were, however, observed only in a small minority (3% each). The hs-troponin level was an independent predictor of left ventricular dysfunction, whereas COVID-19 severity was an independent predictor of right ventricular dysfunction in this study. Another international collaborative study reported left ventricular dysfunction in 20% and right ventricular dysfunction in 30% of hospitalized patients and found that both left and right ventricular dysfunction were independently associated with mortality in a multivariable analysis39. Szekely et al.40 reported a high proportion of right ventricular dysfunction in 100 consecutive patients who underwent echocardiography. Given the high prevalence of established cardiovascular disease among those admitted with COVID-19, a key question is whether such observed left ventricular dysfunction is new or long-standing. A direct answer is problematic, firstly because of the lack of pre-illness imaging data and secondly because those that undergo echocardiography represent a selected group even among hospitalized patients. A recent study assessing first-phase ejection fraction (EF1), an index associated with preclinical heart failure41, in patients admitted with COVID-19 and a control group with similar comorbidities42 offers a potential answer to the question. This study found that impaired EF1 was strongly associated with mortality independent of other risk factors but that it was similar between patients and controls, suggesting that it reflects preexisting cardiovascular disease. Along similar lines, a cohort study in 1,721 multiethnic UK patients found that most cardiovascular complications or hs-troponin elevation occurred in patients with preexisting cardiovascular disease43. This is likely to represent an enhanced susceptibility of individuals with preexisting cardiovascular abnormalities (for example, latent cardiac dysfunction, an arrhythmogenic substrate due to cardiac fibrosis and endothelial dysfunction) to the pathophysiological stresses imposed by COVID-19, such as severe lung injury and systemic inflammation.
Cardiac assessment by magnetic resonance imaging
Cardiac magnetic resonance (CMR) imaging may provide more mechanistic insight into the etiology of cardiac dysfunction with COVID-19. However, there are important logistic issues in performing CMR in the acute phase of severe COVID-19 and published data are even more prone to selection bias than for echocardiography. As such, there are only a few case series available during the acute phase44. Chen et al.45 reported a series of 25 patients who underwent CMR imaging within 10 days of an acute COVID-19 diagnosis and found evidence of minor left ventricular dysfunction (reduced longitudinal strain) and increased extracellular volume fraction (suggesting myocardial edema) as compared to healthy controls. The major limitations are that the control group was not matched for preexisting comorbidities and the study design does not demonstrate whether such findings might also be found in other critical illness conditions. An interesting analysis in the UK Biobank took advantage of the routine CMR phenotyping in this large prospective population-based study. Raisi-Estabragh et al.46 found that preexisting abnormal CMR phenotypes were associated with greater odds of COVID-19 positivity independent of classical cardiovascular risk factors, consistent with the suggestion that a notable component of left ventricular dysfunction in patients with COVID-19 might reflect preexisting disease.
Several studies have reported on CMR imaging performed in the convalescent phase 2 months or longer after COVID-19. A large German study in 100 patients reported a very high prevalence (78%) of abnormal findings including slightly impaired left ventricular function and raised native T1 and T2 values (suggesting possible diffuse fibrosis or edema and inflammation, respectively)47. A major limitation of this study is, again, the use of healthy controls as the comparator group and the absence of a convalescent group with other serious illnesses. In a multicenter series of 148 patients, 25% had evidence of ischemic heart disease (which may have been preexistent), and some had limited features consistent with myocarditis-like injury but with minimal functional impact48. In a very well-designed study in healthcare workers, Joy et al.49 showed that CMR findings were essentially similar in 74 seropositive individuals 6 months after mild COVID-19 and 75 matched seronegative individuals. Raman et al.50 compared 58 convalescent patients who had suffered moderate-to-severe COVID-19 and 30 age-, sex- and comorbidity-matched controls and found similar left and right ventricular function in the two groups but modest differences in native T1 and late gadolinium enhancement (suggestive of fibrosis).
Together, these CMR studies have not sufficiently illuminated the possible cardiac pathology in patients with acute severe COVID-19 but are consistent with at least a component of detected abnormalities being related to preexisting cardiovascular disease.
Heart failure during acute COVID-19
New left heart failure appears to be relatively uncommon in severe COVID-19 with a rate of <3% among 1,000 consecutive patients in a large US study27. Rates of overt left ventricular systolic dysfunction are also reported to be only around 10% among consecutive patients undergoing comprehensive echocardiography40. By contrast, right heart failure is a common feature in critically ill patients with COVID-19 and may be related to a combination of lung injury, pulmonary thromboembolism and pulmonary hypertension51. The prevalence of pulmonary hypertension in severely ill COVID-19 patients in intensive care units as assessed by echocardiography was estimated to be between 40% and 58% and of right ventricular dysfunction was over 35%52,53. Although patients with lung injury and acute respiratory distress syndrome due to other causes develop increased pulmonary microvascular resistance, interestingly, it has been reported that patients with COVID-19 disproportionately exhibit pulmonary venous (post-capillary) hypertension54, which is due to left heart dysfunction. This is probably related to the higher prevalence of preexisting cardiovascular comorbidities in patients with COVID-19, with associated structural and functional left ventricular dysfunction. Indeed, as described earlier, these patients tend to have abnormal values of EF1 (ref. 42), an index that sensitively detects subclinical left ventricular dysfunction and relates to a high likelihood of diastolic (that is, left ventricular filling) dysfunction41,55. This combination of post-capillary hypertension and severe lung injury with right ventricular dysfunction can readily be envisaged to lead to a vicious cycle of heart and lung dysfunction in severe COVID-19 (Fig. 1) and may be another important mechanism contributing to the higher mortality in those with preexisting cardiovascular disease.
Autopsy data in acute COVID-19
Several autopsy series have examined the hearts of patients who died of COVID-19. Halushka and Vander Heide56 reviewed 277 autopsy reports across 22 studies and concluded that histological evidence of myocarditis was present in <2% of reports. Findings that were noted in nearly half of the autopsies included macrovascular or microvascular thrombi, inflammation or intraluminal megakaryocytes, all of which may also occur with other critical illnesses. The low prevalence of myocarditis is also in line with our own study on a series of patients deceased with severe SARS-CoV-2 infection during the first wave of the pandemic in Italy57. A report on 39 consecutive autopsies identified viral RNA in the heart by PCR and in situ hybridization but found no evidence of virus in cardiomyocytes and no histological evidence of myocarditis58. Another recent study undertook detailed histological and transmission electron microscopic examination of 15 COVID-19 autopsies and 12 control hearts (including patients who died from other infections)59. These authors also found no evidence of cardiomyocyte infection nor any viral particles in myocardial endothelial cells but did note a high prevalence of nonocclusive fibrin microthrombi without ischemic injury in the COVID-19 cohort. Pellegrini et al.60 similarly found a high prevalence of cardiac thrombi in small vessels and capillaries among 40 autopsies along with some evidence of left ventricular cardiomyocyte necrosis. A small study of seven hearts from deceased COVID-19 patients found no evidence of endothelial activation but, interestingly, noted extensive neutrophil–platelet aggregates and neutrophil-rich clusters within macrothrombi as well as evidence of neutrophil extracellular trap (NET) formation61.
Together, these data suggest that viral infection of cardiomyocytes is very rare or absent in COVID-19. Histological evidence of myocarditis also appears to be uncommon. The most prominent histopathological feature is microvascular thrombosis and associated cardiomyocyte necrosis, which is a strong candidate etiological mechanism to explain the biomarker evidence of myocardial injury in severe COVID-19. The totality of evidence so far suggests that cardiac injury is likely to be a consequence of the thromboembolic abnormalities associated with COVID-19, especially in individuals with preexisting cardiovascular disease.
The finding that cardiomyocytes are not a frequent target of SARS-CoV-2 in vivo contrasts with the detectable expression of ACE2 in these cells62 and the possibility of infecting cardiomyocytes in the laboratory, either in two-dimensional cultures or within tissue organoids62,63,64,65. Other barriers to productive infection, besides receptor interaction, evidently exist in cardiomyocytes in vivo compared to tissue culture conditions.
COVID-19 and thrombosis
Early studies in 2020 already reported a high incidence of thrombosis (31%)66 and pulmonary embolism (20.6%)67 in hospitalized patients with severe disease. Subsequent meta-analyses of clinical studies have indicated that thrombosis is present clinically in over 20% of patients with COVID-19 and in at least half of patients requiring intensive care68,69,70,71. Compared to non-thrombotic patients, patients with thrombosis have a notably increased risk of death.
While a thrombotic response is common in other respiratory infections and other forms of acute respiratory distress syndrome, the magnitude of this response is increased in COVID-19. For example, a recent postmortem study reported that severe vascular injury, including alveolar microthrombi, was nine times more prevalent in the lungs of patients with COVID-19 than in patients with influenza72. Postmortem investigation indicates that overt thrombosis is largely confined to the lung, resulting in diffuse alveolar damage and the formation of pulmonary microthrombi affecting both ventilation and perfusion. Our postmortem analysis of 41 patients with COVID-19 revealed macroscopic appearances of thrombosis of large pulmonary vessels in four patients and the presence of microthrombosis in 83% of patients requiring intensive care and 75% of the other patients at microscopic examination73. These features resonate with those of several other pathological investigations over the last year72,74,75,76,77. The presence of macrovascular or microvascular thrombosis was less frequently detected in other organs in most of these studies.
Additional support for the local origin of thrombosis comes from the observation that fibrin deposition and thrombi in COVID-19 lungs show signs of heterochronicity, with several fresh thrombi infiltrated by inflammatory cells’ neighboring thrombi in an advanced, fibrotic stage of organization. The high levels of fibrin degradation D-dimers and fibrinogen in the blood are the consequence of this state of pulmonary intravascular coagulopathy78, which thus appears distinct from disseminated intravascular coagulation, a rare observation in coronavirus-infected patients79. This conclusion is also consistent with the observation that thrombocytopenia is usually mild, and most patients do not exhibit an increase in prothrombin time or a decrease in antithrombin levels, arguing against a consumptive coagulopathy. This increased pro-thrombotic state, however, is likely to be responsible for the substantially increased risk of developing venous thromboembolism (deep vein thrombosis and pulmonary embolism), which is observed in 8–69% of critically ill patients with COVID-19 despite thromboprophylaxis (reviewed in ref. 80) and with the microthrombotic events observed in the heart, as discussed earlier.
Pathogenic mechanisms of thrombosis
Although the local nature of the thrombotic status appears evident from these considerations, the pathogenic mechanisms connecting SARS-CoV-2 infection to the development of thrombosis remain poorly explained and probably involve all the main players of the hemostasis process, including the endothelium, platelets, inflammatory cells, coagulation system and complement (Fig. 3).

The main involved events include endothelial dysfunction, which can be caused by inflammation or direct infection of endothelial cells by SARS-CoV-2; immunothrombosis, in which NETs are released by activated neutrophils; platelet direct stimulation by the SARS-CoV-2 spike protein, when either on the virion envelopes or on the plasma membrane of infected cells; or the direct activation of complement.
The role of the endothelium
The lung vascular endothelium plays a fundamental role in the pathogenesis of COVID-19 thrombosis. Vessels display features of endothelialitis, characterized by hyperplastic endothelium, intimal proliferation and adherence of macrophages and lymphocytes to the endothelium and perivascular space72,81 and markers of endothelial dysfunction are detectable in the blood82. Lung endothelial cells express markers of activation (such as VCAM-1 and E-selectin) and dysfunction (tissue factor)73. Similar microangiopathy and thrombosis are also characteristically detectable in rhesus macaques infected with SARS-CoV-2 (ref. 83). Endothelial dysfunction can be the consequence of the hyper-inflammatory state that accompanies COVID-19, including highly elevated pro-inflammatory cytokines (in particular, interleukin (IL)-6, tumor necrosis factor (TNF) and IL-1β)84,85,86,87,88. These are part of a hyperactive immune reactivity, characterized by the release of interferons, interleukins, chemokines and other mediators that belong to a conserved innate response to clear infectious agents. A specific connection between inflammation and thrombosis is immunothrombosis, a mechanism by which neutrophils and monocytes activate the coagulation cascade as a host immune defense against infection89,90,91. In the lungs of patients with COVID-19, NETs released by neutrophils may contribute to vascular occlusion and formation of thrombi92,93. This process might be enhanced by the presence, in several of these patients, of antiphospholipid autoantibodies94. By contrast, anti-platelet factor 4 autoantibodies, which have been causally related to vaccine-induced immune thrombotic thrombocytopenia following SARS-CoV-2 vaccination95,96, do not appear different in patients with natural COVID-19 nor do they participate in the thrombotic predisposition of these patients97.
In addition to being the target of the exaggerated production of inflammatory cytokines, the vascular endothelium itself can be directly infected by SARS-CoV-2. This conclusion is supported by the detection of viral RNA and the expression of viral antigens by endothelial cells of patients with COVID-19 (refs. 73,81) and evidence for viral inclusion bodies by electron microscopy in these cells in both the lung72 and other body districts—even if interpretation of electron microscopy data have recently been questioned (reviewed in ref. 98). While the levels of ACE2 are relatively low in the normal vasculature, inflammation-induced activation of endothelial cells increases receptor expression, which is consistent with the augmented ACE2 levels observed in the lungs of patients with COVID-19 (ref. 72). Increased ACE2 can exert a protective role in limiting excessive vascular dysfunction99, but, at the same time, would render endothelial cells more susceptible to direct viral infection.
A central activator of the COVID-19 coagulopathy is tissue factor, the expression of which can be activated both directly by SARS-CoV-2 infection and by inflammatory cytokines80,100,101. Recent work also indicates that patients with COVID-19 have increased levels of circulating extracellular vesicles containing tissue factor, which could contribute to the pro-thrombotic state102.
The role of platelets
Several observations point to a specific involvement of platelets in the pathogenesis of COVID-19 thrombosis. A specific characteristic that accompanies the pro-thrombotic state in patients is thrombocytopenia, which is observed in the absence of overt disseminated intravascular coagulation80,103,104,105. Lungs from patients with COVID-19 show increased numbers of pulmonary megakaryocytes106. SARS-CoV-2 infection is associated with platelet hyperreactivity, including increased P-selectin expression, increased activation and spreading on both fibrinogen and collagen, increased levels of circulating platelet–neutrophil, platelet–monocyte and platelet–T cell aggregates, and increased platelet degranulation and extracellular vesicle-mediated inflammation107,108,109. Increased levels of von Willebrand factor, a key mediator of platelet adhesion and aggregation110, are detectable in most patients with severe disease82. Finally, a recent study based on massive single-cell image-based profiling in 110 patients with COVID-19 has revealed the anomalous presence of excessive platelet aggregates in nearly 90% of all analyzed patients111.
The specific mechanisms linking SARS-CoV-2 infection to platelet hyperreactivity remain elusive. We have recently identified a new mechanism that regulates spike protein-induced cell–cell fusion, which involves cellular factors of lung epithelial cells that may also be relevant for platelet function. We discovered that spike, when expressed on the plasma membrane, is primed by furin and triggers fusion of the SARS-CoV-2-infected cells with neighboring cells expressing ACE2 (Fig. 2). Formation of these syncytial cells depends on the activation, by spike, of the calcium-dependent chloride channel and scramblase transmembrane protein 16F (TMEM16F; also known as anoctamin 6), with consequent externalization of phosphatidylserine onto the outer leaflet of the plasma membrane112 (Fig. 4). Of note, the TMEM16F channel is also expressed in platelets, where it is essential for the activation of platelet procoagulant activity113,114,115. Our recent data indicate that spike, either on the surface of SARS-CoV-2-infected cells or on the envelope of virions, is a potent activator of platelet adhesion and aggregation, eventually inducing a procoagulant phenotype characterized by increased calcium levels, phosphatidylserine externalization and thrombin generation116. A drug that we identified as an inhibitor of spike-induced epithelial syncytia formation, the antihelminthic agent niclosamide112, acts as a TMEM16F inhibitor and also blocks platelet activation.

Current data indicate that spike can lead to the activation of the TMEM16F protein, which in platelets acts as the main calcium-dependent scramblase that promotes externalization of phosphatidylserine onto the outer leaflet of the plasma membrane. This is an essential step in the activation of the procoagulant activity of platelets.
The finding that spike activates TMEM16F, which in turn stimulates phosphatidylserine externalization in platelets, could also explain the thrombocytopenia commonly observed in individuals with COVID-19, as platelets with increased phosphatidylserine on their membranes are cleared by splenic reticuloendothelial cells and macrophages117. This is known to occur in other conditions of thrombocytopenia in animal models, for example, in knock-out mice for the Bcl2l1 antiapoptotic gene117 or in mice deficient in sphingomyelin synthase 1, a platelet membrane enzyme that normally blunts TMEM16F activity117.
The role of complement
Finally, patients with severe COVID-19 show elevated plasma levels of the C5b-C9 components of complement as compared with patients with influenza and other causes of acute respiratory distress syndrome118,119, and deposits of terminal complement components have been reported in the lung microvasculature of patients with COVID-19 disease120. As the complement system represents a first-line innate response to SARS-CoV-2, it is tempting to speculate that its hyperactivation might play a role in both inflammation and endothelial cell dysfunction, including the COVID-19 pro-thrombotic state. In this respect, both the spike121 and N122 proteins of SARS-CoV-2 can activate the alternative and the lectin pathways of complement, respectively. To what extent complement activation might be the main trigger of thrombosis or, more likely, a potentiator of the thrombotic state remains to be understood.
Cardiovascular disease and COVID-19 in children
The vast majority of children who are infected with SARS-CoV-2 may be asymptomatic or develop very mild disease123. There is also some evidence from meta-analyses that the susceptibility to infection upon exposure to virus may be lower in children, and especially those below the ages of 10–14 years, than in adults. However, a very small proportion of children develop multisystem inflammatory syndrome in children (MIS-C). This condition occurs a few weeks after acute COVID-19 and may have a predilection for those of Black and Hispanic background124,125,126,127. It is thought that MIS-C may represent the consequences of a dysregulated inflammatory response to SARS-CoV-2 infection. The commonest features of MIS-C are persistent pyrexia and gastrointestinal disturbance, but cardiac manifestations are also prominent. These include ventricular impairment, arrhythmia, conduction abnormalities, coronary artery dilation, coronary aneurysms and cardiogenic shock. In contrast to acute COVID-19 in adults, myocarditis may be a prominent feature of MIS-C. The acute outcome is reported to be good in the vast majority of children presenting with MIS-C, who typically recover within days or a couple of weeks. However, the longer-term consequences of the condition are still unclear126.
The cardiovascular system and COVID-19 vaccination
Although no cases of myocarditis or pericarditis were observed in the original phase 3 clinical trials of the BNT162b2 (Pfizer–BioNTech)128, mRNA-1273 (Moderna)129 or ChAdOx1 nCoV-19 (AstraZeneca)130 vaccines, a few cases later started to be reported during immunization of the population. The largest epidemiological data derive from studies in Israel, where approximately 5.12 million residents had received two doses of the BNT162b2 mRNA vaccine by the end of May 2021. A survey of over 880,000 vaccinated individuals initially revealed an excess risk of myocarditis (2.7 events per 100,000 persons) after vaccination, as compared with after natural SARS-CoV-2 infection (11.0 events per 100,000 persons)131. Two further studies on the same population later confirmed these findings, reporting 136 cases of myocarditis in over 5 million people in one study132 and 54 cases in 2.5 million people in the other133. The highest incidence (10 cases per 100,000 persons) was reported in male patients between the ages of 16 and 29 years133, with the first week after the second vaccine dose being the main risk window. Female patients had a probability of myocarditis of less than 1 in 100,000 persons. Most of the observed cases were mild or moderate in severity, with resolution of symptoms and improvement in imaging markers with or without treatment.
The pathogenic mechanisms underlying these rare myocarditis and pericarditis events are still unclear. Acute onset of chest pain, commonly 3–5 days after vaccine administration, as is reported in several cases of myocarditis, suggests an immune-mediated mechanism134. As discussed earlier, SARS-CoV-2 infection itself can lead to CMR findings suggestive of myocarditis, and some of the postvaccination cases of myocarditis were in individuals with prior documented SARS-CoV-2 infection. These observations are again consistent with an autoimmune mechanism triggered by the viral spike protein135. In two cases in which a cardiac biopsy was performed, there was no histological evidence of myocardial infiltrate in one136 or even signs of myocarditis in the other137. Myocarditis and pericarditis can be the consequence of an exaggerated inflammatory response fueled by excessive activation of innate immunity pathways135. The mRNA-based spike vaccines consist of a modified mRNA molecule encoding the spike protein encased in a lipid nanoparticle formed by four different lipids according to the stable nucleic acid lipid particle technology128,129. The inflammatory response is probably a consequence of SARS-CoV-2 spike protein as the four lipid components of the vaccine nanoparticle are not reported to elicit immune or inflammatory responses. Chemical modifications of the mRNA include incorporation of modified nucleosides138, which are known to reduce innate immunogenicity. However, the modified mRNA could still evoke a strong response in some predisposed individuals139.
Alternatively, myocarditis could be a consequence of molecular mimicry between spike and an unknown cardiac protein. As far as this possibility is concerned, the presence of autoantibodies has long been postulated as a pathogenic mechanism for myocarditis, both as a consequence of viral infection or in the apparent absence of it140. In support of this possibility, autoantibodies against spike have been reported experimentally to cross-react with cardiac antigens141. The clinical significance of this observation remains to be confirmed.
Cardiovascular complications in long COVID
The term ‘long COVID’ has come to be used for symptoms that persist or develop beyond 4–12 weeks after an initial episode of acute COVID-19 infection and cannot be explained by an alternative diagnosis. The syndrome is highly heterogeneous with a wide variety of symptoms that are potentially attributable to multiple organ systems, including myalgia, fatigue, breathlessness, impaired concentration and sleep quality, joint pain or swelling, short-term memory loss, chest pain and palpitations142. Rarer cardiovascular problems include late venous and arterial thromboembolism, stroke, acute coronary syndromes and pericarditis. There are also reports of postural orthostatic tachycardia syndrome (POTS) after acute COVID-19 (ref. 143). The specific condition of MIS-C was discussed earlier.
In the UK population-based Office for National Statistics Coronavirus Infection Survey, around 1 in 50 people in the United Kingdom who tested positive for COVID-19 (including very mild disease) were experiencing self-reported long COVID based on self-reported symptoms (https://www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/articles/coronaviruscovid19latestinsights/infections/; last accessed 31 January 2022). An analysis of people enrolled in the COVID Symptom Study app144 indicated that 2.3% of individuals with a positive SARS-CoV-2 test (predominantly nonhospitalized individuals) reported symptoms lasting for ≥12 weeks, which was more common in women145. These authors also found that experiencing more than five symptoms during the first week of illness was associated with long COVID. Looking at patients discharged from hospital after acute COVID-19, 1,077 patients were assessed at 2–7 months in the PHOSP-COVID UK study146. Only 29% had fully recovered and 20% had developed a new disability. Common cardiovascular symptoms included chest tightness (27%), chest pain (21%) and palpitations (20%). Female sex, middle age (40–59 years), the presence of ≥2 comorbidities (most commonly cardiovascular, respiratory and type 2 diabetes) and more severe acute illness were associated with persistence of symptoms. Other studies of patients who had been hospitalized also suggest a higher prevalence of long COVID in women147.
The pathogenesis of cardiovascular symptoms and complications in long COVID remains unclear. In patients who suffered severe acute COVID-19, it seems likely that myocardial injury and vascular thrombotic complications superimposed on preexisting cardiovascular and metabolic comorbidity are the primary drivers. A combination of lung injury and cardiac dysfunction also exacerbates symptoms as discussed earlier. In the PHOSP-COVID study, symptom persistence and severity were associated with a continued elevation of C-reactive protein, suggesting that persistent inflammation may be an additional etiological mechanism. The pathogenesis of long COVID in previously healthy individuals who suffered only mild acute illness is less clear and may have overlap with other post-viral syndromes. There have been a number of studies in which competitive athletes have undergone CMR imaging after an episode of mild COVID-19. An early study suggested a high incidence of abnormalities but was flawed in that there was no control group148, but the athletic heart is well known to undergo remodeling. A more recent analysis compared athletes who had recovered from acute COVID-19 with a COVID-negative athletic control group and found a much lower prevalence of abnormalities by CMR imaging149. However, they did identify features suggestive of myocarditis in 2 out of 59 athletes, who were both asymptomatic. There are many ongoing large prospective studies of patients who have recovered from acute COVID-19 of varying severity, which should in time shed light on the important question of long-term damage and symptom persistence.
Conclusion
Cardiovascular disease has a bidirectional relationship with COVID-19. On the one hand, preexisting chronic cardiovascular disease is a major risk factor for severe COVID-19 while, on the other, cardiovascular complications have an important role in its pathophysiology. While there is limited evidence that the heart itself is directly affected by SARS-CoV-2, mild-to-moderate myocardial injury is a common and adverse prognostic feature of severe COVID-19. The causes of such myocardial injury are multifactorial and include systemic inflammation, microvascular thrombosis, hypoxia and a higher susceptibility in those with preexisting disease. Importantly, cardiac impairment may aggravate pulmonary dysfunction and lead to a downward spiral in patients. More still needs to be learnt about the pathogenesis of cardiovascular problems in patients who recover from initial acute illness.
Unfortunately, a major limitation of studies in this area is a lack of suitable animal models. Although some animal coronaviruses can recapitulate a few characteristics of human disease and SARS-CoV-2 can infect other animal species or humanized rodents150,151, none of these fully recapitulate all the clinical features of human disease.
A specific area that deserves deeper investigation is thrombosis, as this is a major clinical aspect of severe COVID-19, with several features that are more pronounced than in other common severe pulmonary infections. Recent findings raise the possibility that SARS-CoV-2-specific mechanisms may be involved in platelet activation, potentially opening up specific therapeutic avenues.
References
Yang, J. et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: a systematic review and meta-analysis. Int. J. Infect. Dis. 94, 91–95 (2020).
Docherty, A. B. et al. Features of 20,133 UK patients in hospital with COVID-19 using the ISARIC WHO Clinical Characterisation Protocol: prospective observational cohort study. BMJ 369, m1985 (2020).
Richardson, S. et al. Presenting characteristics, comorbidities and outcomes among 5,700 patients hospitalized with COVID-19 in the New York City area. JAMA 323, 2052–2059 (2020).
Williamson, E. J. et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 584, 430–436 (2020). Retrospective analysis of over 17,000 primary care records, of which almost 11,000 linked to COVID-19-related deaths from the OpenSAFELY database generated on behalf of NHS England, to examine factors associated with COVID-19-related deaths.
Wu, Z. & McGoogan, J. M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72,314 cases from the Chinese Center for Disease Control and Prevention. JAMA 323, 1239–1242 (2020).
Inciardi, R. M. et al. Characteristics and outcomes of patients hospitalized for COVID-19 and cardiac disease in Northern Italy. Eur. Heart J. 41, 1821–1829 (2020).
Holman, N. et al. Risk factors for COVID-19-related mortality in people with type 1 and type 2 diabetes in England: a population-based cohort study. Lancet Diabetes Endocrinol. 8, 823–833 (2020).
Pouwels, K. B. et al. Community prevalence of SARS-CoV-2 in England from April to November, 2020: results from the ONS Coronavirus Infection Survey. Lancet Public Health 6, e30–e38 (2021).
Vuille-dit-Bille, R. N. et al. Human intestine luminal ACE2 and amino acid transporter expression increased by ACE inhibitors. Amino Acids 47, 693–705 (2015).
Bean, D. M. et al. Angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers are not associated with severe COVID-19 infection in a multisite UK acute hospital trust. Eur. J. Heart Fail. 22, 967–974 (2020).
Hippisley-Cox, J. et al. Risk of severe COVID-19 disease with ACE inhibitors and angiotensin receptor blockers: cohort study including 8.3 million people. Heart 106, 1503–1511 (2020).
Mancia, G., Rea, F., Ludergnani, M., Apolone, G. & Corrao, G. Renin–angiotensin–aldosterone system blockers and the risk of COVID-19. N. Engl. J. Med. 382, 2431–2440 (2020).
Morales, D. R. et al. Renin–angiotensin system blockers and susceptibility to COVID-19: an international, open science, cohort analysis. Lancet Digit. Health 3, e98–e114 (2021).
Koivula, I., Sten, M. & Makela, P. H. Risk factors for pneumonia in the elderly. Am. J. Med. 96, 313–320 (1994).
Arias-Fernandez, L., Gil-Prieto, R. & Gil-de-Miguel, A. Incidence, mortality and lethality of hospitalizations for community-acquired pneumonia with comorbid cardiovascular disease in Spain (1997–2015). BMC Infect. Dis. 20, 477 (2020).
Gu, S. X. et al. Thrombocytopathy and endotheliopathy: crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 18, 194–209 (2021).
Webb Hooper, M., Napoles, A. M. & Perez-Stable, E. J. COVID-19 and racial/ethnic disparities. JAMA 323, 2466–2467 (2020).
Karaca-Mandic, P., Georgiou, A. & Sen, S. Assessment of COVID-19 hospitalizations by race/ethnicity in 12 states. JAMA Intern. Med. 181, 131–134 (2021).
Bennett, H. Q. et al. The contribution of multiple long-term conditions to widening inequalities in disability-free life expectancy over two decades: longitudinal analysis of two cohorts using the Cognitive Function and Ageing Studies. EClinicalMedicine 39, 101041 (2021).
Nafilyan, V. et al. Ethnic differences in COVID-19 mortality during the first two waves of the coronavirus pandemic: a nationwide cohort study of 29 million adults in England. Eur. J. Epidemiol. 36, 605–617 (2021).
Feldman, J. M. & Bassett, M. T. Variation in COVID-19 mortality in the US by race and ethnicity and educational attainment. JAMA Netw. Open 4, e2135967 (2021).
Mathur, R. et al. Ethnic differences in SARS-CoV-2 infection and COVID-19-related hospitalisation, intensive care unit admission, and death in 17 million adults in England: an observational cohort study using the OpenSAFELY platform. Lancet 397, 1711–1724 (2021).
Zakeri, R. et al. A case–control and cohort study to determine the relationship between ethnic background and severe COVID-19. EClinicalMedicine 28, 100574 (2020).
Brancati, F. L., Kao, W. H., Folsom, A. R., Watson, R. L. & Szklo, M. Incident type 2 diabetes mellitus in African American and white adults: the Atherosclerosis Risk in Communities Study. JAMA 283, 2253–2259 (2000).
Tillin, T., Forouhi, N. G., McKeigue, P. M., Chaturvedi, N. & Group, S. S. Southall And Brent REvisited: cohort profile of SABRE, a UK population-based comparison of cardiovascular disease and diabetes in people of European, Indian Asian and African Caribbean origins. Int. J. Epidemiol. 41, 33–42 (2012).
Patel, A. P., Wang, M., Kartoun, U., Ng, K. & Khera, A. V. Quantifying and understanding the higher risk of atherosclerotic cardiovascular disease among South Asian individuals: results from the UK Biobank Prospective Cohort Study. Circulation 144, 410–422 (2021).
Argenziano, M. G. et al. Characterization and clinical course of 1,000 patients with coronavirus disease 2019 in New York: retrospective case series. BMJ 369, m1996 (2020).
Lala, A. et al. Prevalence and impact of myocardial injury in patients hospitalized with COVID-19 infection. J. Am. Coll. Cardiol. 76, 533–546 (2020).
Stals, M. et al. Risk of thrombotic complications in influenza versus COVID-19-hospitalized patients. Res. Pract. Thromb. Haemost. https://doi.org/10.1002/rth2.12496 (2021).
Bangalore, S. et al. ST-segment elevation in patients with COVID-19—a case series. N. Engl. J. Med. 382, 2478–2480 (2020).
Katsoularis, I., Fonseca-Rodriguez, O., Farrington, P., Lindmark, K. & Fors Connolly, A. M. Risk of acute myocardial infarction and ischaemic stroke following COVID-19 in Sweden: a self-controlled case series and matched cohort study. Lancet 398, 599–607 (2021). Population-based epidemiological study in over 86,000 patients with COVID-19 in Sweden showing a significantly increased risk for acute myocardial infarction and ischemic stroke.
Shi, S. et al. Characteristics and clinical significance of myocardial injury in patients with severe coronavirus disease 2019. Eur. Heart J. 41, 2070–2079 (2020).
Shi, S. et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. 5, 802–810 (2020).
Guo, T. et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 5, 811–818 (2020).
Ammann, P. et al. Troponin as a risk factor for mortality in critically ill patients without acute coronary syndromes. J. Am. Coll. Cardiol. 41, 2004–2009 (2003).
Bajwa, E. K. et al. Biomarker evidence of myocardial cell injury is associated with mortality in acute respiratory distress syndrome. Crit. Care Med. 35, 2484–2490 (2007).
Jirak, P. et al. Myocardial injury in severe COVID-19 is similar to pneumonias of other origin: results from a multicentre study. ESC Heart Fail. 8, 37–46 (2021).
Dweck, M. R. et al. Global evaluation of echocardiography in patients with COVID-19. Eur. Heart J. Cardiovasc. Imaging 21, 949–958 (2020).
Karagodin, I. et al. Echocardiographic correlates of in-hospital death in patients with acute COVID-19 infection: The World Alliance Societies of Echocardiography (WASE-COVID) Study. J. Am. Soc. Echocardiogr. 34, 819–830 (2021).
Szekely, Y. et al. Spectrum of cardiac manifestations in COVID-19: a systematic echocardiographic study. Circulation 142, 342–353 (2020).
Gu, H. et al. Reduced first-phase ejection fraction and sustained myocardial wall stress in hypertensive patients with diastolic dysfunction: a manifestation of impaired shortening deactivation that links systolic to diastolic dysfunction and preserves systolic ejection fraction. Hypertension 69, 633–640 (2017).
Gu, H. et al. First-phase ejection fraction, a measure of preclinical heart failure, is strongly associated with increased mortality in patients with COVID-19. Hypertension 77, 2014–2022 (2021).
O'Gallagher, K. et al. preexisting cardiovascular disease rather than cardiovascular risk factors drives mortality in COVID-19. BMC Cardiovasc. Disord. 21, 327 (2021). Results from a cohort study of more than 1,700 consecutive adults hospitalized for severe COVID-19 in the United Kingdom showing that preexisting established cardiovascular disease is a more important contributor to mortality than cardiovascular risk factors in the absence of cardiovascular disease.
Petersen, S. E. et al. Cardiovascular magnetic resonance for patients with COVID-19. JACC Cardiovasc. Imaging https://doi.org/10.1016/j.jcmg.2021.08.021 (2021).
Chen, B. H. et al. Early cardiac involvement in patients with acute COVID-19 infection identified by multiparametric cardiovascular magnetic resonance imaging. Eur. Heart J. Cardiovasc. Imaging 22, 844–851 (2021).
Raisi-Estabragh, Z. et al. Adverse cardiovascular magnetic resonance phenotypes are associated with greater likelihood of incident coronavirus disease 2019: findings from the UK Biobank. Aging Clin. Exp. Res. 33, 1133–1144 (2021).
Puntmann, V. O. et al. Outcomes of cardiovascular magnetic resonance imaging in patients recently recovered from coronavirus disease 2019 (COVID-19). JAMA Cardiol. 5, 1265–1273 (2020).
Kotecha, T. et al. Patterns of myocardial injury in recovered troponin-positive COVID-19 patients assessed by cardiovascular magnetic resonance. Eur. Heart J. 42, 1866–1878 (2021).
Joy, G. et al. Prospective case–control study of cardiovascular abnormalities 6 months following mild COVID-19 in healthcare workers. JACC Cardiovasc. Imaging https://doi.org/10.1016/j.jcmg.2021.04.011 (2021).
Raman, B. et al. Medium-term effects of SARS-CoV-2 infection on multiple vital organs, exercise capacity, cognition, quality of life and mental health, post-hospital discharge. EClinicalMedicine 31, 100683 (2021).
Brault, C. et al. COVID-19- versus non-COVID-19-related acute respiratory distress syndrome: differences and similarities. Am. J. Respir. Crit. Care Med. 202, 1301–1304 (2020).
Norderfeldt, J. et al. Acute pulmonary hypertension and short-term outcomes in severe COVID-19 patients needing intensive care. Acta Anaesthesiol. Scand. 65, 761–769 (2021).
Manzur-Sandoval, D. et al. Right ventricular dysfunction and right ventricular-arterial uncoupling at admission increase the in-hospital mortality in patients with COVID-19 disease. Echocardiography 38, 1345–1351 (2021).
Caravita, S. et al. Haemodynamic characteristics of COVID-19 patients with acute respiratory distress syndrome requiring mechanical ventilation. An invasive assessment using right heart catheterization. Eur. J. Heart Fail. 22, 2228–2237 (2020).
Gu, H. et al. First-phase ejection fraction is a powerful predictor of adverse events in asymptomatic patients with aortic stenosis and preserved total ejection fraction. JACC Cardiovasc. Imaging 12, 52–63 (2019).
Halushka, M. K. & Vander Heide, R. S. Myocarditis is rare in COVID-19 autopsies: cardiovascular findings across 277 postmortem examinations. Cardiovasc. Pathol. 50, 107300 (2021). Meta-analysis of 22 different postmortem studies showing that secondary or preexisting myocardial abnormalities are common in patients with COVID-19, whereas myocarditis is rare.
Dal Ferro, M. et al. SARS-CoV-2, myocardial injury and inflammation: insights from a large clinical and autopsy study. Clin. Res. Cardiol. 110, 1822–1831 (2021).
Lindner, D. et al. Association of cardiac infection with SARS-CoV-2 in confirmed COVID-19 autopsy cases. JAMA Cardiol. 5, 1281–1285 (2020).
Bois, M. C. et al. COVID-19-associated nonocclusive fibrin microthrombi in the heart. Circulation 143, 230–243 (2021).
Pellegrini, D. et al. Microthrombi as a major cause of cardiac injury in COVID-19: a pathologic study. Circulation 143, 1031–1042 (2021).
Johnson, J. E. et al. COVID coronary vascular thrombosis: correlation with neutrophil but not endothelial activation. Am. J. Pathol. 192, 112–120 (2021).
Nicin, L. et al. Cell-type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur. Heart J. 41, 1804–1806 (2020).
Navaratnarajah, C. K. et al. Highly efficient SARS-CoV-2 infection of human cardiomyocytes: spike protein-mediated cell fusion and its inhibition. J. Virol. 95, e0136821 (2021).
Sharma, A. et al. Human iPSC-derived cardiomyocytes are susceptible to SARS-CoV-2 infection. Cell Rep. Med. 1, 100052 (2020).
Bailey Adam, L. et al. SARS-CoV-2 infects human engineered heart tissues and models COVID-19 myocarditis. JACC: Basic Transl. Sci. 6, 331–345 (2021).
Klok, F. A. et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 191, 145–147 (2020).
Poissy, J. et al. Pulmonary embolism in patients with COVID-19: awareness of an increased prevalence. Circulation 142, 184–186 (2020).
Xiong, X., Chi, J. & Gao, Q. Prevalence and risk factors of thrombotic events on patients with COVID-19: a systematic review and meta-analysis. Thromb. J. 19, 32 (2021).
Jenner, W. J. & Gorog, D. A. Incidence of thrombotic complications in COVID-19: on behalf of ICODE: The International COVID-19 Thrombosis Biomarkers Colloquium. J. Thromb. Thrombolysis 52, 999–1006 (2021).
Zhang, L. et al. D-dimer levels on admission to predict in-hospital mortality in patients with COVID-19. J. Thromb. Haemost. 18, 1324–1329 (2020).
Lippi, G. & Favaloro, E. J. D-dimer is associated with severity of coronavirus disease 2019: a pooled analysis. Thromb. Haemost. 120, 876–878 (2020).
Ackermann, M. et al. Pulmonary vascular endothelialitis, thrombosis and angiogenesis in COVID-19. N. Engl. J. Med. 383, 120–128 (2020). Postmortem analysis of lungs from patients who died of COVID-19 or of influenza A (H1N1), showing diffuse alveolar damage with perivascular T cell infiltration in both cases, but distinctive features in COVID-19 only, consisting of severe endothelial injury, widespread thrombosis and microangiopathy.
Bussani, R. et al. Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID-19 pathology. Lancet EBioMedicine 61, 103104 (2020). Postmortem pathological analysis in 41 consecutive patients who died of COVID-19 in Italy, showing COVID-19-specific features in the lungs, such as thrombosis of the microvasculature and macrovasculature and several dysmorphic pneumocytes; no virus-related alterations were found in other organs.
Carsana, L. et al. Pulmonary postmortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect. Dis. 20, 1135–1140 (2020).
Fox, S. E. et al. Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respir. Med. 8, 681–686 (2020).
Buja, L. M. et al. The emerging spectrum of cardiopulmonary pathology of the coronavirus disease 2019 (COVID-19): report of 3 autopsies from Houston, Texas, and review of autopsy findings from other United States cities. Cardiovasc. Pathol. 48, 107233 (2020).
Wichmann, D. et al. Autopsy findings and venous thromboembolism in patients with COVID-19. Ann. Intern. Med. 173, 268–277 (2020).
McGonagle, D., O’Donnell, J. S., Sharif, K., Emery, P. & Bridgewood, C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2, e437–e445 (2020).
Giannis, D., Ziogas, I. A. & Gianni, P. Coagulation disorders in coronavirus-infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. J. Clin. Virol. 127, 104362 (2020).
Mackman, N., Antoniak, S., Wolberg, A. S., Kasthuri, R. & Key, N. S. Coagulation abnormalities and thrombosis in patients infected with SARS-CoV-2 and other pandemic viruses. Arterioscler. Thromb. Vasc. Biol. 40, 2033–2044 (2020).
Varga, Z. et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 395, 1417–1418 (2020).
Goshua, G. et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 7, e575–e582 (2020).
Aid, M. et al. Vascular disease and thrombosis in SARS-CoV-2-infected rhesus macaques. Cell 183, 1354–1366 (2020).
McKechnie, J. L. & Blish, C. A. The innate immune system: fighting on the front lines or fanning the flames of COVID-19? Cell Host Microbe 27, 863–869 (2020).
Zhou, Z. et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 27, 883–890 (2020). Results from a meta-transcriptomic profiling study from the bronchoalveolar lavage fluid of eight patients with COVID-19, showing marked upregulation of pro-inflammatory cytokines and robust activation of numerous interferon stimulated genes.
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Blanco-Melo, D. et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1036–1045 (2020).
Chen, G. et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 130, 2620–2629 (2020).
Engelmann, B. & Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 13, 34–45 (2013).
Bonaventura, A. et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 21, 319–329 (2021).
Stark, K. & Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 18, 666–682 (2021).
Zuo, Y. et al. Neutrophil extracellular traps in COVID-19. JCI Insight https://doi.org/10.1172/jci.insight.138999 (2020).
Barnes, B. J. et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J. Exp. Med. 217, 20200652 (2020).
Zuo, Y. et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 12, eabd3876 (2020).
Greinacher, A. et al. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N. Engl. J. Med. 384, 2092–2101 (2021).
Schultz, N. H. et al. Thrombosis and thrombocytopenia after ChAdOx1 nCoV-19 vaccination. N. Engl. J. Med. 384, 2124–2130 (2021).
Arepally, G. M. & Ortel, T. L. Vaccine-induced immune thrombotic thrombocytopenia: what we know and do not know. Blood 138, 293–298 (2021).
Bernard, I., Limonta, D., Mahal, L. K. & Hobman, T. C. Endothelium infection and dysregulation by SARS-CoV-2: evidence and caveats in COVID-19. Viruses https://doi.org/10.3390/v13010029 (2020).
Lovren, F. et al. Angiotensin-converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 295, H1377–H1384 (2008).
Bautista-Vargas, M., Bonilla-Abadia, F. & Canas, C. A. Potential role for tissue factor in the pathogenesis of hypercoagulability associated with in COVID-19. J. Thromb. Thrombolysis 50, 479–483 (2020).
Canzano, P. et al. Platelet and endothelial activation as potential mechanisms behind the thrombotic complications of COVID-19 patients. JACC Basic Transl. Sci. 6, 202–218 (2021).
Rosell, A. et al. Patients with COVID-19 have elevated levels of circulating extracellular vesicle tissue factor activity that is associated with severity and mortality-brief report. Arterioscler. Thromb. Vasc. Biol. 41, 878–882 (2021).
Lippi, G., Plebani, M. & Henry, B. M. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin. Chim. Acta 506, 145–148 (2020).
Bikdeli, B. et al. COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy and follow-up. J. Am. Coll. Cardiol. 75, 2950–2973 (2020).
Chaudhary, R. et al. Thromboinflammatory biomarkers in COVID-19: systematic review and meta-analysis of 17,052 patients. Mayo Clin. Proc. Innov. Qual. Outcomes 5, 388–402 (2021).
Valdivia-Mazeyra, M. F. et al. Increased number of pulmonary megakaryocytes in COVID-19 patients with diffuse alveolar damage: an autopsy study with clinical correlation and review of the literature. Virchows Arch. 478, 487–496 (2021).
Zaid, Y. et al. Platelets can associate with SARS-CoV-2 RNA and are hyperactivated in COVID-19. Circ. Res. 127, 1404–1418 (2020).
Manne, B. K. et al. Platelet gene expression and function in patients with COVID-19. Blood 136, 1317–1329 (2020).
Bongiovanni, D. et al. SARS-CoV-2 infection is associated with a pro-thrombotic platelet phenotype. Cell Death Dis. 12, 50 (2021).
Gragnano, F. et al. The role of von Willebrand factor in vascular inflammation: from pathogenesis to targeted therapy. Mediators Inflamm. 2017, 5620314 (2017).
Nishikawa, M. et al. Massive image-based single-cell profiling reveals high levels of circulating platelet aggregates in patients with COVID-19. Nat. Commun. 12, 7135 (2021).
Braga, L. et al. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature 594, 88–93 (2021). The study reports high-throughput screening of over 3,000 clinically approved drugs for small molecules that inhibit cell–cell fusion induced by the SARS-CoV-2 spike protein; a mechanism for cell fusion based on the activation of the cellular scramblase TMEM16F and a drug (niclosamide) that blocks this effect are identified.
Yang, H. et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 151, 111–122 (2012).
Baig, A. A. et al. TMEM16F-mediated platelet membrane phospholipid scrambling is critical for hemostasis and thrombosis but not thromboinflammation in mice. Arterioscler. Thromb. Vasc. Biol. 36, 2152–2157 (2016).
Fujii, T., Sakata, A., Nishimura, S., Eto, K. & Nagata, S. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc. Natl Acad. Sci. USA 112, 12800–12805 (2015).
Cappelletto, A. et al. SARS-CoV-2 spike protein activates TMEM16F-mediated platelet procoagulant activity. Preprint at bioRxiv https://doi.org/10.1101/2021.1112.1114.472668 (2021).
Wagner, K. U. et al. Conditional deletion of the Bcl-x gene from erythroid cells results in hemolytic anemia and profound splenomegaly. Development 127, 4949–4958 (2000).
Holter, J. C. et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl Acad. Sci. USA 117, 25018–25025 (2020).
Ma, L. et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. Sci. Immunol. https://doi.org/10.1126/sciimmunol.abh2259 (2021).
Magro, C. et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl. Res. 220, 1–13 (2020).
Yu, J. et al. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood 136, 2080–2089 (2020).
Gao, T. et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. Preprint at medRxiv https://doi.org/10.1101/2020.1103.1129.20041962 (2020).
Viner, R. M. et al. Susceptibility to SARS-CoV-2 infection among children and adolescents compared with adults: a systematic review and meta-analysis. JAMA Pediatr. 175, 143–156 (2021).
Riphagen, S., Gomez, X., Gonzalez-Martinez, C., Wilkinson, N. & Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 395, 1607–1608 (2020).
Whittaker, E. et al. Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA 324, 259–269 (2020).
Alsaied, T. et al. Review of cardiac involvement in multisystem inflammatory syndrome in children. Circulation 143, 78–88 (2021).
Carter, M. J., Shankar-Hari, M. & Tibby, S. M. Paediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2 infection: an overview. Intensive Care Med. 47, 90–93 (2021).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2021).
Voysey, M. et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 397, 99–111 (2021).
Barda, N. Safety of the BNT162b2 mRNA COVID-19 vaccine in a nationwide setting. N. Engl. J. Med. 385, 1078–1090 (2021). Data analysis from a vaccination programme in Israel (over 880,000 individuals) with the BNT162b2 mRNA vaccine shows that vaccination was associated with an excess risk of myocarditis (2.7 events per 100,000 persons), which was significantly lower than after natural SARS-CoV-2 infection.
Mevorach, D. et al. Myocarditis after BNT162b2 mRNA vaccine against COVID-19 in Israel. N. Engl. J. Med. 385, 2140–2149 (2021).
Witberg, G. et al. Myocarditis after COVID-19 vaccination in a large healthcare organization. N. Engl. J. Med. 385, 2132–2139 (2021).
Shay, D. K., Shimabukuro, T. T. & DeStefano, F. Myocarditis occurring after immunization with mRNA-based COVID-19 vaccines. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2021.2821 (2021).
Bozkurt, B., Kamat, I. & Hotez, P. J. Myocarditis with COVID-19 mRNA vaccines. Circulation 144, 471–484 (2021).
Larson, K. F. et al. Myocarditis after BNT162b2 and mRNA-1273 vaccination. Circulation 144, 506–508 (2021).
Rosner, C. M. et al. Myocarditis temporally associated with COVID-19 vaccination. Circulation 144, 502–505 (2021).
Kariko, K., Buckstein, M., Ni, H. & Weissman, D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005).
Caso, F. et al. Could SARS-coronavirus-2 trigger autoimmune and/or autoinflammatory mechanisms in genetically predisposed subjects? Autoimmun. Rev. 19, 102524 (2020).
Caforio, A. L., Mahon, N. J., Tona, F. & McKenna, W. J. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: pathogenetic and clinical significance. Eur. J. Heart Fail. 4, 411–417 (2002).
Vojdani, A. & Kharrazian, D. Potential antigenic cross-reactivity between SARS-CoV-2 and human tissue with a possible link to an increase in autoimmune diseases. Clin. Immunol. 217, 108480 (2020).
Crook, H., Raza, S., Nowell, J., Young, M. & Edison, P. Long COVID mechanisms, risk factors and management. BMJ 374, n1648 (2021).
Johansson, M. et al. Long-haul post-COVID-19 symptoms presenting as a variant of postural orthostatic tachycardia syndrome: the Swedish experience. JACC Case Rep. 3, 573–580 (2021).
Menni, C. et al. Real-time tracking of self-reported symptoms to predict potential COVID-19. Nat. Med. 26, 1037–1040 (2020).
Sudre, C. H. et al. Attributes and predictors of long COVID. Nat. Med. 27, 626–631 (2021). Analysis of data from self-reported symptoms in the COVID Symptom Study app to understand symptoms associated with the development of long COVID. Experiencing more than five symptoms during the first week of illness was associated with long COVID. Long COVID was also characterized by fatigue, headache, dyspnea and anosmia and was more likely with increasing age and body mass index and female sex.
Evans, R. A. et al. Physical, cognitive and mental health impacts of COVID-19 after hospitalisation (PHOSP-COVID): a UK multicentre, prospective cohort study. Lancet Respir. Med. https://doi.org/10.1016/S2213-2600(21)00383-0 (2021).
Sigfrid, L. et al. Long COVID in adults discharged from UK hospitals after COVID-19: a prospective, multicentre cohort study using the ISARIC WHO Clinical Characterisation Protocol. Lancet Reg. Health Eur. 8, 100186 (2021).
Rajpal, S. et al. Cardiovascular magnetic resonance findings in competitive athletes recovering from COVID-19 infection. JAMA Cardiol. 6, 116–118 (2021).
Clark, D. E. et al. COVID-19 myocardial pathology evaluation in athletes with cardiac magnetic resonance (COMPETE CMR). Circulation 143, 609–612 (2021).
Shou, S. et al. Animal models for COVID-19: hamsters, mouse, ferret, mink, tree shrew and non-human primates. Front. Microbiol. 12, 626553 (2021).
Lee, C. Y. & Lowen, A. C. Animal models for SARS-CoV-2. Curr. Opin. Virol. 48, 73–81 (2021).
Acknowledgements
M.G. is supported by the European Research Council (Advanced Grant 787971), the British Heart Foundation (RG/19/11/34633) and the European Commission Horizon 2020 grants 825670 and 874764. A.M.S. is supported by the British Heart Foundation (CH/1999001/11735 and RE/18/2/34213) and the Medical Research Council (MR/V040162/1). The authors acknowledge support from the National Institute for Health Research Biomedical Research Centres at Guy’s & St Thomas’ NHS Foundation Trust with King’s College London (IS-BRC-1215-20006).
Author information
Authors and Affiliations
Contributions
M.G. and A.M.S. jointly wrote the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cardiovascular Research thanks John Hwa, Eduardo Marban and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Giacca, M., Shah, A.M. The pathological maelstrom of COVID-19 and cardiovascular disease. Nat Cardiovasc Res 1, 200–210 (2022). https://doi.org/10.1038/s44161-022-00029-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44161-022-00029-5
This article is cited by
-
Three-dimensional morphologic and molecular atlases of nasal vasculature
Nature Cardiovascular Research (2023)
-
SARS-CoV-2 infection boosts inflammation in atherosclerotic plaques
Nature Cardiovascular Research (2023)
