
Abstract
Conductive hydrogels that are highly elastic, fatigue resistant and environmentally adaptive are promising materials in the fields of wearable electronics, bioelectronics and soft robotics. However, these materials are challenging to develop, especially for use in harsh environments including organic solvents and extreme temperatures. Here we report a simple method for the fabrication of highly compressible and fatigue-resistant conductive hydrogels with reinforced-concrete-type constituents and high-tortuosity interconnected cellular architecture through a self-assembly and two-stage in situ polymerization process. The obtained composites exhibit excellent mechanical compressibility with negligible residual strain at 50% strain for >104 cyclic loadings both in air and water. Due to the structure-favoured anisotropic response to tensile deformations coupled with elastic recovery, the hydrogel is endowed with sensing dimensions which allow the direction and velocity of movement on the sensor surface to be distinguished. In addition, by interpenetrating with an oleophilic polymer network, highly elastic and adaptive organohydrogels are developed with outstanding sensing performance in a wide variety of organic solvents and cryogenic temperatures. These materials may therefore be suitable for use in flexible and wearable devices in harsh environments.

Similar content being viewed by others

Main
Conductive hydrogels capable of adhering to arbitrary and curved surfaces and able to transduce mechanical deformations into electrical signals are highly attractive in the fields of flexible and wearable electronics and soft robotics1,2,3. However, the mechanical properties of synthetic hydrogels, especially fatigue resistance under repetitive elastic deformation, are much lower than that of natural biotissues, which severely restrain their use in applications that involve heavy cyclic loadings. For example, skeletal human muscle can withstand >109 deforming cycles up to compression strains of >40% (ref. 4). Furthermore, the inevitable swelling-induced decrease in robustness greatly limits their use in solvent environments5.
Inspired by the extraordinary mechanical properties of natural cellular materials6, great efforts have been devoted to constructing cellular architectures with superelasticity and fatigue resistance7,8,9,10,11,12,13. Existing highly compressible and conductive materials are predominantly carbon-based aerogels, sponges and foams that are prepared using flexible and robust carbon nanotubes14,15,16,17,18, carbon nanofibres19,20 graphene nanosheets8,10,21,22,23,24,25, organic-derived carbon species9,26,27,28 and two carbon materials29,30,31 as building blocks. Although directional freeze-casting is a widely used and cost-efficient wet-shaping technique for the construction of cellular materials8,9,20,21,32, it remains challenging to precisely control hierarchically spatial geometries of the cellular architectures including the voids and solid sections, and to rationally design the mechanically resilient constituents to move the mechanical elasticity and fatigue resistance to their theoretical limits33,34. Hierarchical structuring across multiple length scales has been, and continues to be, the source of inspiration to realize revolutionary materials with extraordinary performance32. Until now, environmentally adaptive and conductive materials that can reversibly undergo large compressive strain in harsh environments have been rarely reported24,35. Furthermore, there remain serious issues of permanent decay in mechanical strength and residual strain upon cyclic compression.
Metal nanowires, especially silver nanowires (AgNWs), are potential candidates for fabricating mechanically compressible conductors because they offer high electrical conductivity and mechanical resilience, coupled with large aspect ratios, and can be prepared by large-scale solution synthesis36. Unfortunately, no compressible and fatigue-resistant conductors based on engineering metal nanowires have been reported, probably due to a lack of efficient fabrication strategies. AgNWs have confirmed their broad usefulness in the construction of mechanically stretchable conductors via structure-enabled and materials-based strategies37,38. Thus, we can envision that it should be possible to produce mechanically compressible AgNW-based conductors with strong fatigue resistance and serviceability in harsh environments via a combination of designing constituents and controlling elaborate cellular structures from the nano- to the microscale, and further to the macroscale.
Here we demonstrate a simple approach for the fabrication of highly compressible and fatigue-resistant conductive hydrogels with an anisotropic, high-tortuosity endoplasmic-reticulum-like structure and interconnected lamellar network in orthogonal directions by a two-stage in situ polymerization of the predesigned AgNW-composed matrix. Benefiting from a hierarchically cellular structure and reinforced-concrete-type constituents, the obtained composites exhibit outstanding mechanical compressibility, retaining 79% of maximum stress and 1.5% of permanent deformation at a large compressive strain of 50% for 3 × 104 cycles. It is worth noting that the composite delivers unprecedented elasticity with no residual strain when compressed for 5 × 104 cycles at 50% strain in water. As a result of its distinct response to stretching strains coupled with elastic recovery, the hydrogel possesses sensing dimensions that can detect the direction and velocity of movements. Due to its mechanical properties, the composite can function as an underwater pressure sensor to reliably distinguish human motions in water. Furthermore, by incorporating oleophilic polymer chains, the resulting elastic and conductive organohydrogel with an interpenetrating oleophilic–hydrophilic network offers great sensing serviceability in a wide variety of organic solvents and subzero temperatures. Therefore, these materials are promising for use in wearable devices in harsh environments, and offer simple and scalable fabrication and large-area integration.
Results and discussion
Fabrication of AgNW–polyacrylamide hydrogel
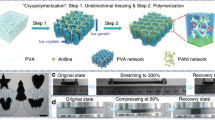
As shown in Fig. 1, a simple method combining directional-freezing-induced self-assembly and two-stage in situ polymerization was developed to fabricate the compressible and conductive AgNW–polyacrylamide (PAM) (CCAP) hydrogel. Importantly, the directional freezing assembly was carried out on the mixture at −120 °C with liquid nitrogen to obtain the polymerizable scaffold (Supplementary Fig. 1). Here, hydrogen bonding between acrylamide (AM) molecules limited the diffusion of water molecules and reduced the diffusion length of ice crystals, resulting in decreased rejection among AgNWs and the final finer porous structure39. With two-stage in situ polymerization of the above frozen monolith at −18 °C for 12 h and then at room temperature (r.t.) for 2 h, the CCAP hydrogel was fabricated with a typical endoplasmic-reticulum-like morphology and narrow open-cell network structure composed of PAM-wrapped, high-tortuosity thin walls (Fig. 2a–c). Parallel to the growth direction of ice crystals, a uniform 10-μm-spaced lamellar structure was observed where the neighbouring lamellae were interconnected and formed a secondary network (Fig. 2d–f). The homogeneous distributions confirmed successful incorporation of AgNWs with PAM in the hydrogel (Fig. 2g). The volume fraction of the AgNWs in the CCAP hydrogel was calculated to be ∼6.2% based on volume measurements of a bare AgNW framework produced by the directional-freezing-assembly method and CCAP gel.

a, Schematic illustration of the fabrication of the CCAP hydrogel by the combined directional freezing assembly and two-stage in situ polymerization process, that is, cryopolymerization at −18 °C for 12 h and subsequent polymerization at room temperature for 2 h. AM, MBAA and TEMED are used as the monomer, crosslinker and catalyst, respectively. b, Schematic of structural changes during the cryopolymerization process. In stage 1, the localized AgNW network in the compartmental wall was distorted under the uneven tension resulting from the phase transformation of the tightly and loosely bound water around the AgNW surfaces. Stabilized by the simultaneous in situ polymerization, a porous structure was produced in stage 2. c, Thawing of a large area of ice induced neighbouring lamellae to grow together in further polymerization at room temperature, forming an interconnected high-tortuosity network.

a–f, Scanning electron microscopy images with different magnifications of the CCAP hydrogel: top view (a–c); side view (d–f). g, Elemental mappings of the hydrogel. h, Cryo-Raman spectra showing the fitting peaks of ice (red and green lines) and bound water (blue and bright blue lines) in precursor solution for the fabrication of CCAP hydrogel recorded at −18 °C. i, Temperature-modulated DSC scan (black line) of the precursor solution for the fabrication of CCAP hydrogel at a heating rate of 5 K min−1. The red line shows the reversible part of the heat flow. The asterisks indicate the melting points of bound water (267.8 K) and ice (274.5 K).
We then tracked the structural evolution of the hydrogel during the cryopolymerization process. Typically, massive micropores were detected in the compartmental walls of the monolith during the first 2 h of cryopolymerization (Supplementary Fig. 2), arising from distortion of the AgNW network and interfacial polymerization during the phase transformation of water (Fig. 1b). Because of the presence of three water types existed in the AgNW-containing system40, the bound water around the AgNWs achieved appreciable mobility and gradually melted as the temperature increased from −120 to −18 °C (Supplementary Fig. 3)41,42. In the corresponding Raman spectra, the bands at 3,598 and 3,396 cm−1 were assigned to the antisymmetric O–H stretching vibration of asymmetrically bonded water molecules and bifurcated hydrogen bonds43, respectively, indicated the melting of the bound water (Fig. 2h). Furthermore, the appearance of two endotherms of water melting, including a band ranging from 252 to 269 K in the temperature-modulated differential scanning calorimetry (DSC) spectrum, proved the phase change of the bound water in the monolith (Fig. 2i). These changes indicate that the AgNW network was distorted by the action of the phase-transformation-induced unbalanced tension field. The in situ polymerization in the successively thawed water microregions triggered by hydrogen bonding between ‘intermediate water’ molecules and newly formed PAM chains44 in turn stabilized the distorted network and led to a porous structure. The obvious coating of the PAM layers was investigated over 12 h on the compartmental walls by examining the Ag–N bonds (Supplementary Figs. 4 and 5)45. Eventually, simultaneous r.t. polymerization and volume shrinkage caused by the thawing of a large area of ice resulted in a high-tortuosity endoplasmic-reticulum-like structure21. In addition, the greatly improved mass transfer and diffusion caused by ice thawing helped the neighbouring lamellae in the polymerization to grow together and form the interconnected network (Fig. 1c).
The directional freezing self-assembly made it possible to predesign the AgNW-containing cellular prototype similar to previously reported work9,20,26. However, imparting structural hierarchy at different length scales into the preformed cellular embryo was essential to the mechanical and sensing performances of the final products. This required sophisticated control over the AgNW network, cell shape and polymerization interfaces at multiscale levels, including phase transformation of water on a molecular scale, interfacial polymerization on the supramolecular scale, distortion of the AgNW network on the mesoscopic scale, and interconnection of neighbouring lamellae on the nano- and microscale. The interconnection of lamellae was controlled during the programmed two-stage in situ polymerization, which consisted of kinetically slow cryopolymerization for 12 h and subsequent r.t. polymerization for 2 h. The predesign–distortion–stabilization effects in the freezing assembly-assisted polymerization method result in the hydrogel exhibiting a hierarchical network comprising four levels: the AgNWs were crosslinked into compartmental walls at the nanoscale (first level), the film-shaped units were assembled into the cellular network (second level), and the metastable skeleton in the intermediate polymerization enabled a porous network in the compartmental wall (third level) and an interconnected network among the longitudinal lamellae (fourth level). This hierarchical network contributed to the appearance of a high-tortuosity endoplasmic reticulum in top view and a highly interconnected lamellar network in side view.
Mechanical properties
The mechanical properties of the hydrogel are shown in Fig. 3. In Fig. 3a, the loading stress–strain curve exhibits three characteristic deformation regions of open-cell foams: a nearly linear elastic regime for ε ≤ 7%, corresponding to bending of cell walls; a subsequent plateau regime for 7% < ε ≤ 50%, corresponding to buckling of cell walls; and a final densification regime with a sharply increased stress for ε > 50%. Combined with the AgNWs, the CCAP hydrogel delivered a high strength with the maximum stress of 18.2 kPa cm−3 normalized by volume at the strain of 70%. Furthermore, an obvious hysteresis loop was observed in the loading–unloading curves, suggesting a substantial capacity for energy dissipation. In the first cycle, the work performed in the compression and unloading processes was estimated to be 5.95 and 2.34 mJ cm−3, respectively, which yielded an energy dissipation of 3.61 mJ cm−3 and an energy loss coefficient of 0.61, much higher than those of carbon- and metal-based foams7,9,14,15. It was notable that the energy dissipation was low at σ = 15% and high-energy adsorption occurred in regions with large σ (Supplementary Fig. 6), guaranteeing a stable structure during the buckling of the cell walls and the final densification regimes.

a, Compressive stress–strain curves under cyclic compression. The photographs show the hydrogel before and after 500 compression cycles. b, Stress–strain curves at up to 50% strain for up to 3 × 104 cycles and at 15% strain for up to 105 cycles. c, Time-dependent swelling ratio of the CCAP hydrogel in terms of height and diameter. d, Photographs showing CCAP, DF-PAM and Ag/PAM hydrogels enduring a weight of 5 g after swelling in water for 10 days. e, Loading–unloading stress–strain curves at different compressive strains in water. f, Stress–strain curves at up to 50% strain for up to 5 × 104 cycles and at 15% strain for up to 5 × 105 cycles in water. g, Energy loss coefficient, maximum stress and residual strain over 5 × 104 cycles at 50% strain in water. h, Ashby chart showing residual strain versus compressive strain during long-term compression cycles of CCAP hydrogel compared with previously reported materials. The cycle numbers are marked next to the symbols with references in parentheses. The red, green, blue, pink and yellow hollow diamonds represent elastic materials using graphene14,15,17, carbon nanotubes8,23,25, carbon nanofibres19,20, two carbon materials29,30,31 and organic-derived species9,26 as building blocks, respectively. The purple solid diamond represents compression in water35. The red and green spheres in the blue area represent our CCAP hydrogel compressed in air and water, respectively. i, Schematic showing CCAP hydrogel with an interconnected lamellar network and a porous cell wall resistant to compression force (F). j, Schematic illustration of the energy-dissipation mechanism from three recoverable elements: (1) reversible elastic bending of the AgNW network at the nanoscale (F′1), (2) reversible radial buffer of porous structure at the microscale (F′2) and (3) global relaxation in the interconnected AgNW/PAM network at the macroscale (F′3).
For comparison, we also prepared two control hydrogels via similar two-stage polymerization without directional freezing and assembly, and two-stage polymerization without AgNWs, denoted as Ag/PAM hydrogel and DF-PAM hydrogel, respectively. The Ag/PAM hydrogel showed a closed-cell network structure with random pore distribution, whereas the DF-PAM hydrogel presented a disordered open-cell network resulting from the ice-template effect (Supplementary Fig. 7). In cyclic compression tests, the CCAP hydrogel displayed high mechanical stability with maximum stress retention of 80% and residual strain of 6% after 500 compression cycles at a large strain of 70% (Fig. 3a). In sharp contrast, two control hydrogels presented obviously irreversible shape deformations during compression (Supplementary Fig. 8). After 500 cycles, 40% and 50% stress reduction were investigated for the Ag/PAM and DF-PAM hydrogels, respectively (Supplementary Fig. 9a). In addition, 15% of residual strain was detected for DF-PAM hydrogel even in the first cycle (Supplementary Fig. 9b). After 500 cycles, severe mechanical attenuation took place with a large residual strain of 50%.
Long-term loading–unloading compression curves at different compression strains were further measured to estimate the fatigue resistance of the CCAP hydrogel. As recorded in Fig. 3b, negligible changes in maximum stress (7%) and residual strain (0.9%) were indicated at a strain of 15% even for up to 105 compression cycles (Supplementary Fig. 10a). Loaded with a large compressive strain of 50% for up to 3 × 104 cycles, the hydrogel still delivered well-shaped compression profiles with 79% of maximum stress maintained. Notably, the permanent deformation was only 1.5%, much lower than that of 10–30% for polymeric foams at ε ≈ 20% (ref. 33) and the energy loss coefficient was slightly improved from 0.38 to 0.43 after many cycles (Supplementary Fig. 11), revealing that a large portion of energy was absorbed and no serious accumulation of damage or even structural collapse occurred with compression cycles. These compression tests demonstrated the excellent fatigue tolerance of the CCAP hydrogel due to the combined advantages of hybrid composition and endoplasmic-reticulum-like architecture which offer highly effective dissipation of crack energy.
To evaluate its mechanical properties in water, the swelling performance of the CCAP hydrogel was investigated by monitoring the changes in its height and diameter over time (Fig. 3c). Swelling equilibrium was achieved rapidly, within 10 s, due to the capillary siphon effect, accompanied by a negligible change (~2%) in height and a 30% expansion of diameter. The weight of the gel remained essentially unchanged even after soaking in water for 10 days, and the gel could sustain a weight of 5 g (Fig. 3d). This unique restrained swelling performance was attributed to the strong equilibrium between osmotic and elastic energies in the CCAP network5, associated with the extremely stable polymer network fixed by the AgNW-assembled cellular scaffold46. In contrast, both of the comparative hydrogels were mechanically weak after swelling (Fig. 3d and Supplementary Fig. 12a). Due to this rapid and highly anisotropic swelling behaviour, the CCAP hydrogel may therefore be useful in complex liquid environments because of its anisotropically free channels and robust AgNW-assembled framework.
Figure 3e shows that the compressive profile at 70% strain of the CCAP hydrogel delivered characteristic linearly increasing, plateau and sharply rising stages, indicating the well-maintained open-cell network structure in water. The identical paths of the loading and unloading curves at strains from 15% to 70% confirmed the high mechanical resilience of the hydrogel. Cyclic compressions in water at a strain of 15% resulted in compressive curves that nearly overlapped and a 3% decrease in maximum stress after 5 × 105 loading–unloading fatigue cycles (Fig. 3f, inset, and Supplementary Fig. 10b). After 5 × 104 cycles at a strain up to 50%, no residual strain was observed and the maximum stress gradually decreased from 8.9 to 7.4 kPa (Fig. 3f,g). The energy loss coefficient stabilized at ∼0.3 for 5 × 104 compression cycles in water. Even after being compressed in water at a large strain of 70% for 1,000 cycles, the hydrogel still delivered constant compressive curves with 3.5% residual strain (Supplementary Fig. 13). These analyses suggested that the CCAP hydrogel is supercompressible in water in terms of residual strain, energy loss coefficient and fatigue resilience.
Understanding the supercompressible mechanism
The elastic performance of cellular materials is determined by the properties of the cell walls, including their composition, geometry and dimension14. The CCAP hydrogel showed a hierarchically cellular architecture with a high-tortuosity interconnected lamellar network (Fig. 3i). In this architecture, AgNWs with large length–diameter ratios combined with PAM chains are assembled into the film-like cell walls of the CCAP hydrogel, which have small wall-thickness-to-size ratios (Fig. 3j(1)). This structure, which features both elasticity and robustness at the nanoscale, favoured the switch of compressive deformations into elastic bending and buckling while tolerating mechanical failure7,12. At the microscale level, the micrometre-sized porous structure enclosed by the AgNW-PAM network was robust and elastic, and radially buffered external strain simply by shrinking the pore parallel to the compressive stress and elongating the pore in the vertical direction (Fig. 3j(2)). When the compression force was removed, the shape deformation of the pore recovered instantaneously, contributing to a larger dissipation of the crack energy. In sharp contrast to the isolated struts, the interconnected AgNW-PAM lamellar structure made the hydrogel network into an integrated and highly accommodating buffer capable of enduring a large elastic strain and relaxing the external loading throughout the entire flexible network (Fig. 3j(3)). In situ laser scanning images showed that the interconnected lamellar network was bent along the loading direction under a compressive strain of 20% (Supplementary Fig. 14a). The hydrogel maintained good structural integrity without fracture, however, under persistent strains of 30–50%. Meanwhile, it was observed that the bending deformation of the lamellae tended to be smooth, rather than heavy buckling, indicating that large compressive loading was spread out and shared over the entire interconnected network, and the out-of-plane deformation of the connecting branches prevented stress concentration in the lamellae. In this way, the compressive strain applied to the hydrogel (ε) was largely shared by the multilevel shape deformations of the AgNW network at the nanoscale, by the porous cell walls at the microscale, and by the interconnected lamellar network. These features enabled the AgNW-based units to suffer much less compression (ε′). When the compressive strain was released, the network structure could spring back to its original shape (Supplementary Fig. 14b). Furthermore, the hydrogel exhibited nearly complete structural recovery under repeated loading–unloading compression at 50% strain (Supplementary Fig. 15).
It is worth noting that the network of AgNWs and PAM not only afforded a robust concrete-type structure created by welding the AgNW network with PAM chains, but also perfectly inherited the flexibility of the polymeric gel. These advantages of a hierarchically designed cellular structure, reinforced with hard–soft constituents, endowed the CCAP hydrogel with supercompressive elasticity and fatigue resistance, which, combined with its superior mechanical properties in water, make the CCAP hydrogel among the top-performing reported elastic materials (Fig. 3h and Supplementary Table 1). Notably, the self-assembly–polymerization combined method presented here demonstrates a strong synthetic capability to integrate different types of assembly units into the hydrogels, including one-dimensional nanomaterials of copper nanowires (CuNWs), carbon nanotubes (CNTs) and SiO2 nanowires, two-dimensional graphene oxide nanosheets (GONSs) and conductive poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) networks (Supplementary Fig. 16). Based on the materials’ consistent structural characteristics and mechanical properties (Supplementary Figs. 17–19), we can conclude that this is a generic and relatively simple method to synthesize a diverse array of highly compressible and fatigue-resistant hydrogels without the need for complex facilities and tedious processing, provided that the necessary physical parameters are satisfied.
Sensing performance
The cellular AgNW network assembled by directional freezing ensured an efficient electrical conductive pathway in the CCAP hydrogel. Together with excellent compressibility, the CCAP hydrogel exhibited a remarkable variation in compression-dependent resistance. The pressure-sensitivity (S) of the CCAP hydrogel was defined as S = (ΔR/R0)/ΔP, where ΔR is the resistance change (R0 − R), R0 and R are the initial resistance and resistance at a certain pressure, respectively, and ΔP is the change in applied pressure (P). As plotted in Fig. 4a, the CCAP hydrogel showed a sharply increased resistance variation ratio in response to the loaded pressure with a sensitivity of 0.22 kPa−1 for P < 4 kPa, which was more sensitive than graphene-based47, polyelectrolyte48 and ionic-conductive35 hydrogels (0.02–0.1 kPa−1) and more sensitve than the 0.00345 kPa−1 reported for a carbon nanofibre aerogel sensor19 in a low-pressure regime. The subsequent densification of the hydrogel network contributed to a nonlinear regime with S = 0.016 kPa−1. In contrast, no rapid increase in pressure sensitivity was detected for the comparative Ag/PAM hydrogel (Supplementary Fig. 20a). At a maximum strain of 50%, the loading curve of the CCAP showed a high response region with the resistance decreasing exponentially at ε ≤ 20%, and a stable response region with gradually varying resistance at ε > 20% (Supplementary Fig. 20b). When the loading was removed, the ΔR/R0 returned to the initial zero, and a fine closed loop was formed by the loading and unloading profiles.

a, Normalized resistance change (ΔR/R0) of the CCAP hydrogel versus pressure in air and water. b, ΔR/R0 during five compression cycles at different strains in air. c, ΔR/R0 at 50% strain during 1,000 compression cycles in air. d, Schematic illustrations of the cumulative deformation and structural recovery under sliding motion in the x direction. Δl and Δh are the instantaneous tensile deformation and compressive deformation, respectively, during the sliding. e, Schematic illustrations showing resistance change dependent on the cumulative tensile strain in the x direction and compressive deformation when pressing the hydrogel. Positions 1 and 2 are selected to show structural evolution before and after the pressing. l and h are the transverse and vertical dimensions at position 1. f,g, ΔR/R0 in response to sliding on the CCAP surface at different velocities in the x (f) and y (g) directions. The green and blue dotted lines show the beginning and the end of the movements on the hydrogel surface. h, ΔR/R0 when pressing the CCAP surface to different depths with a glass rod. i, ΔR/R0 versus strain during cyclic compression in water. j, ΔR/R0 in response to bending and relaxing cycles of a finger in water at various bending angles. The inset images show the monitoring of the finger motion in water. k, ΔR/R0 in response to gradual finger bending in water from 0° to 90° and relaxing to 0°.
This tendency to change resistance was attributed to contact spot variation during the process of structural deformation under compression. During the initial compression stage, electrical resistance reduced dramatically due to the rapid increase in the number of temporary contacts caused by bending and buckling of the cell walls and struts. Upon further compression, the interface electrical resistance arising from the increased contact area partly offset the reduced resistance from the newly formed contact spots and the overall variation in resistance was small. It was noted that free rotation and twining of AgNWs under compression, which recovered only very slightly as the force was released, were greatly limited by the PAM coating29. Therefore, the PAM-coated AgNW nodes could return to their predeformed configurations, thereby removing the extra contact spots, allowing the hydrogel to recover its original structure when the load was removed. Additionally, the CCAP hydrogel demonstrated a stable and consistent response to broad compressive strains from a tiny 0.3% to a large 50% as ΔR/R0 increased from 7% to 82%, respectively, during five loading–unloading cycles (Fig. 4b and Supplementary Fig. 21a). Long-term fatigue tests were further performed to evaluate the stability of the CCAP hydrogel as a pressure sensor (Fig. 4c and Supplementary Fig. 21b). Negligible degradation and identical amplitude of electrical signals were delivered for 2,000 compressive cycles at an initial 20% strain and a subsequent 50% strain, highlighting the hydrogel’s robust structure and reliable stability in practical application as a sensor.
Furthermore, the change in resistance of the CCAP hydrogel in reponse to tensile strain (ΔRT) was investigated, where the stretching direction parallel to the current was defined as x and the orthogonal direction was defined as y (Supplementary Fig. 22a). In the x direction, resistance was increased and the corresponding resistance change was decreased (ΔRTx < 0) (Supplementary Fig. 22b) due to the increased electron transport and reduced cross-sectional area caused by the geometric effect. However, the resistance change in the y direction was negligible (ΔRTy ≈ 0). We attributed this anisotropic response to tensile deformations in directions orthogonal to the stable honeycomb network of AgNWs combined with polymer chains (Supplementary Fig. 23). When stretched, this honeycomb network can be elongated to relax the tension effectively. In addition, the share of external force in the integrated PAM-coated AgNW architecture resulted in much smaller tensile strain (λ′) on the AgNW unit than the applied λ, which further dissipated the crack energy and protected the conductive paths. Therefore, the conductivity from the joined AgNWs remained constant for both stretching directions and the variation in resistance during the stretch was the result of the geometric effect.
The above distinct responses to tensile strains combined with robust compressibility give the CCAP hydrogel a remarkable capability to sense movement direction. A glass rod sliding on the hydrogel surface simultaneously produced compressive deformation and cumulative tensile deformation vertical to the sliding direction (Fig. 4d and Supplementary Fig. 24). However, omnidirectional tensile deformations were produced, in addition to compressive deformation, when pressing the hydrogel surface (Fig. 4e). When the glass rod moved along the x direction, ΔRx of the hydrogel was the combination of the compressive strains (ΔRC) and cumulative tensile strains in the y direction (ΔRTy), that is, ΔRx = ΔRC + ΔRTy, where ΔRC > 0 and ΔRTy ≈ 0. Therefore, the final resistance change was increased (ΔRx > 0) (Fig. 4f). In contrast, moving the glass rod along the y direction decreased the resistance change (ΔRy < 0) (Fig. 4g). This can be represented by the equation ΔRy = ΔRC + ΔRTx, where ΔRC > 0 and ΔRTx < 0, which was confirmed by the pressing experiment. The resistance change was decreased upon pressing the glass rod (ΔRp < 0) (Fig. 4h). Moreover, the deeper the rod was pressed and the thicker the rod employed (Supplementary Fig. 25), the greater the change in resistance. Considering that tensile strains non-parallel to the current direction had tiny effects on the final resistance and the compressive strain led to the increased resistance change (ΔRC > 0), the decreased ΔRp was attributed to the cumulative tensile strain along the current (ΔRTx < 0). Therefore, the resistance changes of the hydrogel with both pressing (ΔRp < 0) and sliding vertical to the current (ΔRy < 0) were the predominant responses to the cumulative tensile deformation along the current (ΔRTx < 0).
In addition to the motion direction, the movement speeds produced characteristic signals as also revealed by the sliding experiments. Increasing the velocity in both the x and y directions caused the peak of the cumulative deformation-dependent resistance change to start earlier with a steeper slope and a bigger height (Fig. 4f,g), which was caused by the opposing effects of deformation generation and relaxation. At an intrinsically fixed relaxation velocity, a faster motion rapidly yielded a sharper peak. Furthermore, at the end of sliding on the hydrogel surface in the x direction, the resistance nearly recovered its initial value at a low velocity of 2 mm s−1 and the unrecovered resistance arose from the residual deformation under high-speed movement. Notably, the recovered resistances were investigated when the sliding finished in the y direction at velocities from 2 to 8 mm s−1, which might be the net result of the compressive and tensile deformations in the simultaneous generation and relaxation processes.
To explore the potential of the CCAP hydrogel as a flexible and wearable sensor, its electromechanical behaviour under water was investigated. The pressure-dependent ΔR/R0 curve (Fig. 4a) showed two distinctive regimes with the similar sensitivities in air, indicating the hydrogel’s structural stability in water. When compressed to 50% strain, ΔR/R0 was increased, following a steep slope at ε ≤ 20% and a subsequent smooth slope at ε > 20% (Fig. 4i). When the compression was released, the hydrogel delivered a good recovery path for ΔR/R0. In addition, the hydrogel’s outstanding fatigue resistance in water was indicated by the absence of decay in ΔR/R0 for 1,000 cycles at 20% strain and a subsequent 1,000 cycles at 50% strain (Supplementary Fig. 26). We further mounted the hydrogel on a finger joint to monitor actual human motions in water. It was found that the signal of the resistance change ratio fluctuated continuously and repeatedly with an identical amplitude over bending cycles at a fixed bending angle, and a higher bending angle resulted in a larger resistance change (Fig. 4j). When gradually relaxing the bending angle from 90° to 0°, the ΔR/R0 values were decreased stepwise and returned to zero (Fig. 4k). These results revealed the great potential of the CCAP hydrogel for use as an underwater sensor with high sensitivity, good reproducibility and environmental adaptability.
Serviceability in harsh environments
To further extend the adaptability of the hydrogel in harsh environments, including organic solvents and extreme temperatures (Fig. 5), an organohydrogel strategy was developed by in situ polymerization of an oleophilic polymer network on the hydrophilic CCAP network (Fig. 5a). Due to its robust hierarchical framework with reinforced-concrete-type constituents, the CCAP hydrogel maintained a good appearance with a normalized weight decrease of 28.6% and a volume decrease of 27.6% after soaking in ethanol, in sharp contrast to the results with pure PAM hydrogel (Supplementary Fig. 27), indicating that the hydrogel network maintained an extended conformation in a water–ethanol co-solvent environment with a moderate ethanol content49. In this favourable co-solvent environment the soaked CCAP hydrogel has good affinity with both hydrophilic chains and oleophilic monomers. Ultraviolet irradiation of the CCAP hydrogel infused with the polymerizable organogel precursors showed that the CCAP-based organohydrogel exhibited an endoplasmic-reticulum-like structure with high porosity and well-maintained cell walls (Supplementary Fig. 28), demonstrating the formation of an interpenetrating oleophilic–hydrophilic network. This interpenetrating network enabled the switching of hydrophilic and oleophilic chains to be exposed on the gel surface in water and organic solvents, rendering the organohydrogel highly compressible in both water and hydrophobic organic solvents (Fig. 5b)50. To evaluate the mechanical properties of the gel, long-term compression cycles were conducted in water and different kinds of hydrophobic organic liquids. The nearly overlapping compressive profiles during 1,000 cycles at a high strain of 50% indicated the remarkable structural elasticity and fatigue resistance of the gel in these liquids (Fig. 5c,d and Supplementary Fig. 29). Figure 5e shows a minor decay in the mechanical property in liquids, with a high stress retention of 85–96% after 1,000 compression cycles. Furthermore, the organohydrogel exhibited high sensitivity to external pressure in organic liquids. For example, applying compression to the organohydrogel in n-heptane resulted in its ΔR/R0 increasing sharply at low pressures (≤2 kPa) with a high sensitivity of 0.23 kPa−1 (Fig. 5f), comparable to that of the CCAP hydrogel in air (Fig. 4c). In addition, the sensing stability of the organohydrogel was further estimated by measuring the ΔR/R0 values for loading–unloading cycles at strains from 0.2% to 50% (Fig. 5g). ΔR/R0 was increased when compressing the organohydrogel to maximum strain, which was vividly reflected by the brighter bulb during the compression (Fig. 5k). Once the compression had been removed, the resistance returned to its initial value. During 100 cycles to 50% compressive strain, there was negligible change in both profile shape and ΔR/R0 value, demonstrating outstanding sensing reliability in hydrophobic organic liquids (Fig. 5h and Supplementary Fig. 30). Hence, this CCAP-based organohydrogel provides excellent electromechanical performance in harsh organic solvents.

a, Schematic illustrations for the synthesis of organohydrogel with an interpenetrating oleophilic–hydrophilic network by soaking the CCAP hydrogel in ethanol solution containing BMA, LMA, EGDMA and DEAP and the subsequent ultraviolet-irradiation-induced polymerization. b, Schematic illustrations of the adaptive mechanism of the organohydrogel in water and hydrophobic organic solvents. In water, the swollen hydrophilic polymer chains are exposed on the gel surface. In organic solvents, the oleophilic chains are switched on the surface. c,d, Compressive stress–strain curves in water (c) and in n-heptane (d) under cyclic compression. The photographs show the recovered organohydrogels after releasing compression. e, Maximum stress retention after 1,000 compression cycles in water and different hydrophobic organic solvents. f, Normalized resistance change (ΔR/R0) of the organohydrogel versus pressure in n-heptane. g, ΔR/R0 in n-heptane during nine compression cycles at different strains. h, ΔR/R0 during 100 compression cycles at 50% strain in n-heptane. i, Stress–strain curves for 100 compression cycles at −50 °C. The inserted photographs show good recovery of the organohydrogel after the compression. j, ΔR/R0 during 100 compression cycles at 50% strain at −50 °C. k,l, Photographs showing the bulb getting brighter when compressing the organohydrogel in n-heptane (k) and liquid nitrogen (l).
In addition, the interpenetrating oleophilic–hydrophilic network enabled the CCAP-based organohydrogel to endure a high compression strain at −50 °C (Fig. 5i). Typical loading–unloading profiles were recorded and 80% of maximal stress was retained after 100 cycles at 50% strain. This antifreezing behaviour originated from the reduction in continuous ice crystallizing in the polymer network due to the heterogeneous hydrophobic/hydrophilic composites therein50—the interpenetrated oleophilic network resulted in discontinuous crystal domains in the hydrophilic CCAP network, and therefore provided the organohydrogel with softness and elasticity at temperatures below the freezing point. Benefiting from its strong tolerance to low temperatures, the organohydrogel continued to be conductive in liquid nitrogen and the connected bulb became even brighter upon pressing the gel (Fig. 5l). A further electromechanical test demonstrated a fast and sensitive response in the ΔR/R0 value when compressing the gel to 50% strain at −50 °C (Fig. 5j). Upon cancelling the pressure, its resistance returned to its original value, indicating a good recovery in structure. Additionally, the ΔR/R0 profile in the 100th compression cycle was consistent with that in the first cycle, confirming the gel’s structural resilience at subzero temperature and its great potential as a pressure sensor at extreme temperatures.
Conclusions
We have used the shaping–distortion–stabilization effect to fabricate a type of highly compressible and fatigue-resistant CCAP hydrogel with a high-tortuosity interconnected cellular network by a combined self-assembly and two-stage in situ cryopolymerization and r.t. polymerization process. Due to their high hierarchically structured deformation, reinforced-concrete-type components, and rapid and anisotropic swelling behaviour, the CCAP hydrogels exhibit strong resistance to high compression upon heavy cyclic loadings by delivering negligible residual strains at a large compressive strain of 50% for 3 × 104 cycles in air and 5 × 104 cycles in water. These CCAP hydrogels give a sensitive and reliable response to human motions in water, suggesting their possible use as an underwater sensor. Impressively, the movement direction and speed on the hydrogel surface can be distinguished without a complex circuit due to an endoplasmic-reticulum-structure-enabled distinct response to the tensile strains in varied directions. Furthermore, the CCAP-engineered organohydrogels with interpenetrating oleophilic–hydrophilic networks show great serviceability in harsh environments, including a wide variety of organic solvents and cryogenic temperatures. The design concept and fabrication method in this work open avenues to access highly elastic and conductive materials with useful functionality by controlling the constituents and hierarchical structures, offering opportunities for wearable electronics in extreme conditions.
Methods
Synthesis of the AgNWs
The AgNWs were synthesized by the NaCl-assisted polyol method51. Typically, 5.86 g of polyvinylpyrrolidine was dissolved into 190 ml of glycerol with heating at 110 °C. After cooling to room temperature, 1.58 g of AgNO3 and NaCl solution containing 59 mg of NaCl, 10 ml of glycerol and 0.5 ml of deionized water were added into the above solution with gentle stirring and heating to 210 °C. After 30 min, the solution turned greyish-green and the reaction was complete. After washing three times by centrifugation to remove residual polyvinylpyrrolidine, the AgNWs were obtained for further use.
Preparation of CCAP hydrogel
In a typical procedure, 120 mg ml−1 of AM as monomer, 0.4 mg ml−1 of N,N′-methylenebisacrylamide (MBAA) as crosslinker and 4 mg ml−1 of potassium persulfate as initiator were dissolved into 30 mg ml−1 AgNW dispersion in sequence under magnetic stirring in an ice-water bath. Note that once the catalyst of N,N,N′,N′-tetramethylethylenediamine (TEMED) was added, the mixed solution was rapidly transferred to a metal plate at −120 °C with a liquid nitrogen bath for 5 min. The CCAP hydrogel was fabricated by two-stage in situ polymerization by placing the above directionally freezing monolith in a fridge at −18 °C for 12 h followed by room temperature for 2 h. For comparison, an Ag/PAM hydrogel was prepared by two-stage polymerization of the same constituents as the CCAP hydrogel without the directional freezing. The other comparative DF-PAM hydrogel was prepared without AgNWs by a similar directional freezing and two-stage polymerization method.
Preparation of CuNW/PAM, CNT/PAM, SiO2/PAM, GO/PAM and PEDOT:PSS/PAM hydrogels
The synthesis procedures were similar to those used for the CCAP hydrogel by a directional freezing assembly at −120 °C and two-stage in situ polymerization at −18 °C for 12 h followed by room temperature for 2 h. Specifically, the contents of CuNWs, CNTs, SiO2NWs, GONSs and PEDOT:PSS were 12 mg ml−1, 4.8 mg ml−1, 20 mg ml−1, 2.2 mg ml−1 and 0.9 wt%, respectively.
Preparation of CCAP-based organohydrogel
After immersing in ethanol for solvent change (three times), the CCAP hydrogel was soaked in a polymerizable solution containing 10 ml of n-butyl methacrylate (BMA), 10 ml of lauryl methacrylate (LMA), 190 μl of ethylene glycol dimethacrylate (EGDMA), 95 mg of 2,2-diethoxyacetophenone (DEAP) and 10 ml of ethanol for 10 h. Irradiating the CCAP hydrogel infused with the polymerizable organogel precursors under ultraviolet light for 30 min resulted in synthesis of the organohydrogel with an interpenetrating network, which was washed with ethanol to remove the impurities.
Pressure sensor fabrication
To sense motion direction and velocity, a glass rod with 6 mm in diameter was used to slide on the surface of a CCAP hydrogel with dimensions of 42 mm × 25 mm × 2 mm; copper tape was attached to both ends of the gel to serve as the electrodes. Movement speed control was achieved by fixing the glass rod on an Instron 5965 mechanical testing system. For underwater body motion sensing, the pressure sensor was fabricated by sandwiching a CCAP hydrogel piece of 10 mm × 10 mm × 2 mm between two polydimethylsiloxane films connected with two copper tapes on each side.
Materials characterization
Structural and compositional analyses of the samples were performed by scanning electron microcopy images and elemental mappings on a Zeiss Merlin Compact field emission scanning electron microscope equipped with an Oxford Inca energy instrument at an acceleration voltage of 5 kV. The volume of water displaced by wholly submerging bare AgNW aerogel prepared by the directional freezing assembly method in water was equal to the aerogel volume, which was regarded as the volume of the AgNWs in the CCAP hydrogel. Cryo-Raman spectra were recorded on a LabRAM-HR (Horiba JY) confocal laser microRaman spectrometer by using a Linkam heating and cooling stage to control the temperature with a cooling rate of 10 K min−1. DSC measurements were performed on a Netzsch DSC214 differential scanning calorimeter in temperature-modulated mode with a modulation period of 60 s and a heating rate of 5 K min−1. X-ray photoelectron spectroscopy measurements were conducted on an ESCALAB Mk II X-ray photoelectron spectrometer with an excitation source of Mg Kα radiation. The mechanical tests were carried out on an Instron 5965 mechanical testing system with a 100 N load cell. The hysteresis curves were recorded at the strain ramp rate of 0.5 mm s−1. In situ structural evolution of the hydrogel upon compression was investigated by a Keyence VK-X250 three-dimensional laser scanning microscope. For the electromechanical tests, the resistance variations of the gel and sensor were measured on a Keithley 4200 SCS and the compression and stretching movements were controlled by an Instron 5965 mechanical testing system. Note that two pieces of copper sheets connected with copper wires were attached on the top and bottom surfaces of the gel column during the compression process and two ends of the gel piece during the stretching process, respectively.
Data availability
The data supporting the findings of this study are available within the paper and its Supplementary Information. Source data are provided with this paper.
References
Seliktar, D. Designing cell-compatible hydrogels for biomedical applications. Science 336, 1124–1128 (2012).
Chortos, A., Liu, J. & Bao, Z. Pursuing prosthetic electronic skin. Nat. Mater. 15, 937–950 (2016).
Zhang, Y.-Z. et al. Mxenes stretch hydrogel sensor performance to new limits. Sci. Adv. 4, eaat0098 (2018).
Huxley, A. F. Reflections on Muscle (Princeton Univ. Press, 1980).
Kamata, H., Akagi, Y., Kayasuga-Kariya, Y., Chung, U.-I. & Sakai, T. ‘Nonswellable’ hydrogel without mechanical hysteresis. Science 343, 873–875 (2014).
Meyers, M. A., Chen, P.-Y., Lin, A. Y.-M. & Seki, Y. Biological materials: structure and mechanical properties. Prog. Mater Sci. 53, 1–206 (2008).
Schaedler, T. A. et al. Ultralight metallic microlattices. Science 334, 962–965 (2011).
Qiu, L., Liu, J. Z., Chang, S. L. Y., Wu, Y. Z. & Li, D. Biomimetic superelastic graphene-based cellular monoliths. Nat. Commun. 3, 1241 (2012).
Si, Y., Yu, J., Tang, X., Ge, J. & Ding, B. Ultralight nanofibre-assembled cellular aerogels with superelasticity and multifunctionality. Nat. Commun. 5, 5802 (2014).
Wu, C., Huang, X., Wu, X., Qian, R. & Jiang, P. Mechanically flexible and multifunctional polymer-based graphene foams for elastic conductors and oil-water separators. Adv. Mater. 25, 5658–5662 (2013).
Sheng, L. et al. Bubble-decorated honeycomb-like graphene film as ultrahigh sensitivity pressure sensors. Adv. Funct. Mater. 25, 6545–6551 (2015).
Meza, L. R., Das, S. & Greer, J. R. Strong, lightweight, and recoverable three-dimensional ceramic nanolattices. Science 345, 1322–1326 (2014).
Yeo, S. J., Oh, M. J. & Yoo, P. J. Structurally controlled cellular architectures for high-performance ultra-lightweight materials. Adv. Mater. 31, 1803670 (2019).
Cao, A., Dickrell, P. L., Sawyer, W. G., Ghasemi-Nejhad, M. N. & Ajayan, P. M. Super-compressible foamlike carbon nanotube films. Science 310, 1307–1310 (2005).
Suhr, J. et al. Fatigue resistance of aligned carbon nanotube arrays under cyclic compression. Nat. Nanotechnol. 2, 417 (2007).
Xu, M., Futaba, D. N., Yamada, T., Yumura, M. & Hata, K. Carbon nanotubes with temperature-invariant viscoelasticity from −196° to 1000°C. Science 330, 1364–1368 (2010).
Gui, X. et al. Carbon nanotube sponges. Adv. Mater. 22, 617–621 (2010).
Wang, H. et al. Ultra-lightweight and highly adaptive all-carbon elastic conductors with stable electrical resistance. Adv. Funct. Mater. 27, 1606220 (2017).
Yu, Z.-L. et al. Superelastic hard carbon nanofiber aerogels. Adv. Mater. 31, 1900651 (2019).
Li, C. et al. Temperature-invariant superelastic and fatigue resistant carbon nanofiber aerogels. Adv. Mater. 32, 1904331 (2020).
Gao, H.-L. et al. Super-elastic and fatigue resistant carbon material with lamellar multi-arch microstructure. Nat. Commun. 7, 12920 (2016).
Zhu, C. et al. Highly compressible 3D periodic graphene aerogel microlattices. Nat. Commun. 6, 6962 (2015).
Hu, H., Zhao, Z. B., Wan, W. B., Gogotsi, Y. & Qiu, J. S. Ultralight and highly compressible graphene aerogels. Adv. Mater. 25, 2219–2223 (2013).
Wu, Y. et al. Three-dimensionally bonded spongy graphene material with super compressive elasticity and near-zero poisson’s ratio. Nat. Commun. 6, 6141 (2015).
Qiu, L. et al. Extremely low density and super-compressible graphene cellular materials. Adv. Mater. 29, 1701553 (2017).
Si, Y. et al. Ultralight biomass-derived carbonaceous nanofibrous aerogels with superelasticity and high pressure-sensitivity. Adv. Mater. 28, 9512–9518 (2016).
Yao, H.-B., Huang, G., Cui, C.-H., Wang, X.-H. & Yu, S.-H. Macroscale elastomeric conductors generated from hydrothermally synthesized metal–polymer hybrid nanocable sponges. Adv. Mater. 23, 3643–3647 (2011).
Liang, H. W. et al. Macroscopic-scale template synthesis of robust carbonaceous nanofiber hydrogels and aerogels and their applications. Angew. Chem. Int. Ed. 51, 5101–5105 (2012).
Kim, K. H., Oh, Y. & Islam, M. F. Graphene coating makes carbon nanotube aerogels superelastic and resistant to fatigue. Nat. Nanotechnol. 7, 562 (2012).
Sun, H. Y., Xu, Z. & Gao, C. Multifunctional, ultra-flyweight, synergistically assembled carbon aerogels. Adv. Mater. 25, 2554–2560 (2013).
Zhuo, H. et al. A supercompressible, elastic, and bendable carbon aerogel with ultrasensitive detection limits for compression strain, pressure, and bending angle. Adv. Mater. 30, 1706705 (2018).
Hua, M. et al. Strong tough hydrogels via the synergy of freeze-casting and salting out. Nature 590, 594–599 (2021).
Gibson, L. J. & Ashby, M. F. Cellular Solids: Structure and Properties (Cambridge Univ. Press, 1997).
Gibson, L. J., Ashby, M. F. & Harley, B. A. Cellular Materials in Nature and Medicine (Cambridge Univ. Press, 2010).
Si, Y. et al. Ultrahigh-water-content, superelastic, and shape-memory nanofiber-assembled hydrogels exhibiting pressure-responsive conductivity. Adv. Mater. 29, 1700339 (2017).
Song, P., Qin, H., Gao, H.-L., Cong, H.-P. & Yu, S.-H. Self-healing and superstretchable conductors from hierarchical nanowire assemblies. Nat. Commun. 9, 2786 (2018).
Yao, S. & Zhu, Y. Nanomaterial-enabled stretchable conductors: strategies, materials and devices. Adv. Mater. 27, 1480–1511 (2015).
Matsuhisa, N., Chen, X., Bao, Z. & Someya, T. Materials and structural designs of stretchable conductors. Chem. Soc. Rev. 48, 2946–2966 (2019).
Li, W. L., Lu, K. & Walz, J. Y. Freeze casting of porous materials: review of critical factors in microstructure evolution. Inter. Mater. Rev. 57, 37–60 (2012).
Quinn, F. X., Kampff, E., Smyth, G. & McBrierty, V. J. Water in hydrogels. 1. A study of water in poly(n-vinyl-2-pyrrolidone/methyl methacrylate) copolymer. Macromolecules 21, 3191–3198 (1988).
Men, Z. et al. Raman spectra of proton order of thin ice Ih film. J. Raman Spectrosc. 46, 388–391 (2015).
Cerveny, S., Colmenero, J. & Alegría, A. Dielectric investigation of the low-temperature water dynamics in the poly(vinyl methyl ether)/H2O system. Macromolecules 38, 7056–7063 (2005).
Giguère, P. A. Bifurcated hydrogen bonds in water. J. Raman Spectrosc. 15, 354–359 (1984).
Katayama, S. & Fujiwara, S. NMR study of the freezing/thawing mechanism of water in polyacrylamide gel. J. Phys. Chem. 84, 2320–2325 (1980).
Ye, G. et al. Dynamic Ag–N bond enhanced stretchable conductor for transparent and self-healing electronic skin. ACS Appl. Mater. Interfaces 12, 1486–1494 (2020).
Du, G. et al. Tough and fatigue resistant biomimetic hydrogels of interlaced self-assembled conjugated polymer belts with a polyelectrolyte network. Chem. Mater. 26, 3522–3529 (2014).
Zhou, H., Yao, W., Li, G., Wang, J. & Lu, Y. Graphene/poly(3,4-ethylenedioxythiophene) hydrogel with excellent mechanical performance and high conductivity. Carbon 59, 495–502 (2013).
Ding, B., Wang, M., Wang, X., Yu, J. & Sun, G. Electrospun nanomaterials for ultrasensitive sensors. Mater. Today 13, 16–27 (2010).
Wu, S. & Shanks, R. A. Solubility study of polyacrylamide in polar solvents. J. Appl. Polym. Sci. 93, 1493–1499 (2004).
Gao, H. et al. Adaptive and freeze-tolerant heteronetwork organohydrogels with enhanced mechanical stability over a wide temperature range. Nat. Commun. 8, 15911 (2017).
Yang, C. et al. Silver nanowires: from scalable synthesis to recyclable foldable electronics. Adv. Mater. 23, 3052–3056 (2011).
Acknowledgements
This work is supported by the National Natural Science Foundation of China (grant numbers 21922104 (H.P.C.), 51732011 (S.-H.Y. and H.-P.C.), 22171066 (H.-P.C.) and U1932213 (S.-H.Y.)), the National Key Research and Development Program of China (grant numbers 2021YFA0715700 (S.-H.Y.) and 2018YFE0202201 (S.-H.Y.)), the Fundamental Research Funds for the Central Universities (grant numbers JZ2019HGPB0104 (H.-P.C.), and JZ2021HGPA0064 (H.Q.)), the University Synergy Innovation Program of Anhui Province (grant number GXXT-2019-028 (S.-H.Y. and H.-P.C.)) and the Anhui Provincial Natural Science Foundation (grant number 1908085J05 (H.-P.C.)).
Author information
Authors and Affiliations
Contributions
H.-P.C. and S.-H.Y. supervised the project, conceived the idea and designed the experiments. Y.W. and H.Q. planned and performed the experiments, and collected and analysed the data. Z.L. helped with the synthesis and characterization of the organohydrogel. J.D. contributed to the structural characterization. Y.W., H.Q., H.-P.C. and S.-H.Y. wrote the paper, and all authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks Mingjie Liu Yong Zhu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary handling editor Peter Seavill, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary methods, Figs. 1–30 and Table 1.
Source data
Source Data Fig. 3
Source data for the mechanical performance presented in Fig. 3.
Source Data Fig. 4
Source data for the sensing performance presented in Fig. 4.
Source Data Fig. 5
Source data for the environmentally adaptive performance presented in Fig. 5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Qin, H., Li, Z. et al. Highly compressible and environmentally adaptive conductors with high-tortuosity interconnected cellular architecture. Nat. Synth 1, 975–986 (2022). https://doi.org/10.1038/s44160-022-00167-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44160-022-00167-5
This article is cited by
-
Rapid and inexpensive synthesis of liter-scale SiC aerogels
Nature Communications (2024)
-
Highly cross-linked carbon tube aerogels with enhanced elasticity and fatigue resistance
Nature Communications (2023)
-
Supercompressible conductive hydrogels in harsh environments
Science China Materials (2023)
