
Abstract
Quantum chemical calculations are mainly regarded as a method for mechanistic studies in organic chemistry, whereas their use for the simulation of unknown reactions could greatly assist in reaction development. Here we report a strategy for developing multicomponent reactions on the basis of the results of computational reaction simulations. In silico screening of multicomponent reactions with difluorocarbene using the artificial force induced reaction method suggested that cycloadditions between an azomethine ylide and a variety of coupling partners would proceed to generate the corresponding α,α-difluorinated N-heterocyclic compounds. The predicted reaction was successfully realized experimentally, leading to a multicomponent N-difluoroalkylative dearomatization of pyridines involving a pyridinium ylide-mediated 1,3-dipolar cycloaddition with a diverse range of electrophiles such as aldehydes, ketones, imines, alkenes and alkynes. Moreover, the performance of the cycloaddition could be explained by comparing the energy barrier of the desired pathway with that of the competitive undesired pathway, which was also identified by the artificial force induced reaction search.

Similar content being viewed by others
Main
The discovery and development of chemical reactions have been primarily led by empirical and intuitive investigations, in which huge numbers of experiments are often required to obtain prospective results, including serendipitous findings1,2. To avoid such a high volume of experiments, a technology for predicting the results of chemical reactions would be particularly desirable in the realm of methodology development. In this context, a variety of strategies have been reported using experimental or chemoinformatic techniques such as high-throughput experimental methods or machine learning, enabling the discovery of new reactions or catalysts3,4. However, these methods still require large numbers of experiments or the collection of data.
Over the years, computational chemistry has become essential for analysing the structure and properties of molecules5. In organic chemistry, quantum chemical calculations play an important role in obtaining mechanistic insight into chemical reactions. Substantial progress has been made in computational methods, as well as in computing power, allowing for deep understanding of chemical reactivities and selectivities, but less progress has been made in reaction design or prediction, in which all conceivable reaction pathways starting from the input molecules must be considered6,7,8,9,10. In this context, we have developed the artificial force induced reaction (AFIR) method, which leverages an artificial force on the input molecules to induce molecular transformations during the calculations to explore all of the theoretically possible reaction pathways11,12. This method is potentially applicable not only for mechanistic studies of known chemical reactions, but also for predictions of reactions in which the products or pathways are uncertain. As an example of the latter, we recently reported a synthetic method for α,α-difluoroglycine derivatives that was discovered using computational retrosynthetic disconnection and a subsequent search for the reaction pathways to the possible products (which included not only the target molecule but also the other possible side products) using the AFIR method13,14. The computationally proposed multicomponent reaction of amines, difluorocarbene and CO2 towards the target compound was then successfully demonstrated experimentally.
Difluorocarbene—which contains only three atoms, but is potentially reactive towards both nucleophiles and electrophiles due to its singlet carbene character—is valuable in synthetic and medicinal chemistry for the incorporation of the difluoromethylene group into organic compounds to alter their biological properties15. Its small size makes it particularly suitable for AFIR calculations, enabling a smooth computational process, which inspired us to envision that the AFIR method could be leveraged for computational design of unexplored reactions with difluorocarbene. Here we demonstrate in silico combinatorial screening using the AFIR method, which predicts multicomponent cycloadditions with difluorocarbene, leading to the discovery of diverse N-difluoroalkylative dearomatization reactions of pyridines to afford α,α-difluorinated N-heterocyclic compounds that have previously been difficult to access.
Results and discussion
In silico reaction screening
We used the AFIR method to explore and identify multicomponent reactions with difluorocarbene and pairs of fragments that bear unsaturated bonds. The strategy is outlined in Fig. 1, and involved four processes that were performed sequentially: (1) setting the substrate combinations and calculation conditions for the combinatorial searches; (2) a computational reaction simulation of each multicomponent reaction using the single component-artificial force induced reaction (SC-AFIR) method, which generates the reaction path network—including equilibrium structures and transition states for conformational changes, as well as bond-forming or -cleaving reactions—by iterative exploration of reaction pathways from input molecules and generated equilibrium structures. The yield of each product is estimated using the rate constant matrix contraction (RCMC) method, which is a kinetics-based simulation in the generated reaction path network under the designated reaction time and temperature16; (3) evaluation of the calculated results obtained as a reaction path network that includes reaction pathways to all possible products with the associated computational yields; and (4) implementation of experiments with available substrates having comparable reactivity with those used in the combinational search to realize the predicted reaction mode. To achieve the combinatorial screening in a practical calculation time, we used formaldehyde, methanimine, ethylene and acetylene as representative components bearing C=O, C=N, C=C or C≡C bonds, as they are the simplest compounds of these types that exhibit the inherent nucleo- or electrophilicity of these functionalities, thus enabling more efficient calculations compared with using larger compounds alongside these unsaturated bonds.

The strategy for computational screening to identify unexplored multicomponent reactions of a pair of unsaturated organic compounds with difluorocarbene is shown. Each reaction is simulated computationally by quantum chemical calculations using the AFIR method. After all of the reactions are calculated, the results are obtained as a reaction path network, which includes the estimated products, each reaction mechanism with the energy barrier (ΔG‡), and the computational yields. The valuable multicomponent reactions among these networks are investigated experimentally with available organic substrates to realize the computationally predicted reaction modes.
The calculations to search for the reaction pathways from each input component were performed in a pairwise manner with difluorocarbene using the SC-AFIR method at the ωB97X-D/LanL2DZ level of theory with the CPCM(THF) solvation model17,18,19,20. In each calculation, 200 random Cartesian coordinates of the initial structures were generated automatically to avoid dependence of the calculation results on the initial structure. The theoretically possible reaction pathways that could proceed within 1 s were explored under kinetics-based navigation by the RCMC method.
After the automatic calculations for all pairs of unsaturated bonds with difluorocarbene were performed in the GRRM program, the comprehensive reaction path networks—which include all possible reaction intermediates, products and reaction pathways, as well as each energy diagram—were obtained (Figs. 2 and 3). At all of the discrete path points along all obtained pathways, single-point energy calculations were performed at the ωB97X-D/Def2-SVP/CPCM(THF) level of theory21. The computational yields of all of the predicted products in the reaction path network were obtained by simulation of the product propagation from the initial components using the RCMC method at reaction temperatures of 200 K, 300 K and 400 K, and a reaction time of 1 s16. The results indicated that seven reactions can be expected to preferentially form the five-membered difluorinated structures A1–7; α,α-difluorinated N-heterocycles A1–4 were probably obtained from the formation of azomethine ylide by methanimine and difluorocarbene, followed by a cycloaddition with the remaining component, whereas α,α-difluorinated O-heterocycles A5–7 potentially formed via a concerted three-component cycloaddition. The formation of the analogous cyclic products B8, E9 and C10 derived from ethylene and/or acetylene was also suggested, but their computational yields are expected to be low and the pathways to three-membered motifs A8–10 were assumed to be the major products in each reaction.

All calculations were performed using the developer version of the GRRM program combined with the Gaussian 16 program11,22. Automatic searches for the pathways were performed using the SC-AFIR method at the ωB97X-D/LanL2DZ/CPCM(THF) level of theory, and the Grid=FineGrid option was adopted. The reaction path networks were obtained, with all possible products shown as blue circles, and the top five products A–E in terms of the computational yields are presented for each reaction. For products with computational yields of less than 0.01, chemically reasonable products were chosen from the ones in the network. EQs, equilibrium structures.

As with Fig. 2, but with O=CH2/H2C=CH2, O=CH2/HC≡CH, 2 × H2C=CH2, H2C=CH2/HC≡CH and 2 × HC≡CH as the unsaturated compound pairs.
Reaction design and realization
Encouraged by the computational results that predicted the multicomponent cycloadditions with difluorocarbene shown in Figs. 2 and 3, we considered the reaction conditions to realize them experimentally. In particular, among the ten simulated reactions, four out of the seven reactions expected to furnish major products involve imines leading to α,α-difluorinated N-heterocyclic compounds, which would be valuable as drug candidates as fluorinated structures often have an influence on the pharmacological activity by improving metabolic stability, protein-binding ability and permeability23. Although N-heterocycles that bear two fluorine atoms at the β- and γ-positions are prevalent in pharmaceuticals such as the DPP-4 (dipeptidyl peptidase) inhibitors gosogliptin or gemigliptin (Fig. 4a), the analogous α,α-difluorinated N-heterocyclic motif has not been used for these applications, presumably due to the lack of synthetic methods24,25,26,27,28,29,30,31,32,33,34,35. In this context, we sought to realize the multicomponent reaction of a C=N bond, a C=O bond and a difluorocarbene to afford N-α,α-difluorooxazolidines A1 (Fig. 4b). The classical approach to prepare the structures is the electrochemical oxidative fluorination of organic compounds such as oxazolidines, where unselective fluorination proceeds, leading to perfluorinated products24,25. However, despite the substantial progress in methods using difluorocarbene, there have been only two reports on the preparation of isolable α,α-difluorinated N-heterocycles via multicomponent cycloadditions with difluorocarbene; these reactions require a stoichiometric amount of toxic lead for the reduction of dibromodifluoromethane to generate difluorocarbene, and the substrate scope is quite narrow, with only five examples using specific ketimines, ketones and fumaronitrile under ultrasonic irradiation36,37. To develop a practical multicomponent reaction with a broad substrate scope, we chose reaction conditions using less toxic reagents for the generation of difluorocarbene and explored two substrates that were suitable for the predicted reaction mode shown in Fig. 4b. To this end, we used 1a and 1b as imine sources (which have been widely employed as typical substrates in organic synthesis), and benzaldehyde 3a or acetophenone 3b as carbonyl electrophiles (Fig. 4c). Bromo(trimethylsilyl)difluoromethane (Me3SiCF2Br) and tetrabutylammonium difluorotriphenylsilicate (Ph3SiF2·NBu4)—which have been proven to be effective for the multicomponent reaction of tert-amines, difluorocarbene and CO2—were used to generate difluorocarbene in situ13,14. However, the reactions using 1a and 1b did not provide the desired cyclic products when using benzaldehyde 3a. To clarify the reason for this failure, we investigated the barriers of the desired cycloaddition pathways by optimizing the transition-state geometries using the ωB97X-D functional and the Def2-SVP basis set with the CPCM(THF) solvation model, revealing values of 6.5 kcal mol−1 (1a) and 4.2 kcal mol−1 (1b) for the formation of the iminium ylide. Although these values are relatively small, the barrier for the undesired dimerization of two molecules of difluorocarbene to generate tetrafluoroethylene is 1 kcal mol–1, and this reaction would therefore preferentially take place in the presence of these imines38,39. We therefore investigated the barrier of ylide formation with other available nitrogen nucleophiles to find a suitable substrate. Pyridines 2a and 2b, which bear a more nucleophilic nitrogen atom on their structures than those of the imines, exhibited low barriers (1.0 and 1.3 kcal mol−1, respectively) that are comparable to that of difluorocarbene dimerization. Encouraged by these calculations and by previous reports using pyridines and difluorocarbene40,41, we conducted the reaction with 2a, which resulted in an N-difluoroalkylative cycloaddition that involves the dearomatization of the pyridine skeleton to provide the desired fluorinated heterocyclic compound in 70% yield in the crude mixture42. However, despite intensive attempts, the product could not be isolated, presumably due to the elimination of the fluoride assisted by the nitrogen atom, which would trigger its further decomposition. On the other hand, the reaction with 2b, which bears two electron-withdrawing ester groups, successfully afforded the isolable product 4 in 75% isolated yield. Moreover, we could also demonstrate the reaction with acetophenone 3b to give the desired product 5, which bears a quaternary carbon centre, in 51% yield. These reactions provided the products in both endo- and exo forms, with nuclear Overhauser effect (NOE) analyses showing that the major components in both cases were the endo-products.

a, Fluorinated N-heterocycles and associated drugs. b, Experimental conditions for realizing the predicted reaction mode. c, Experimental and computational investigation that identified the N-difluoroalkylative dearomatization. The reactions were performed with 0.30 or 0.50 mmol of 1 or 2, 3 (2 equiv.), Me3SiCF2Br (1.1 equiv.), Ph3SiF2∙NBu4 (1.1 equiv.) and THF at 0 °C for 2 h. The energy barriers for the ylide formation and dimerization of two molecules of difluorocarbene were investigated by optimizing the transition-state geometries at the ωB97X-D/Def2-SVP/CPCM(THF) level of theory. aThe yield of the product from pyridine 2a was measured using 19F NMR spectroscopic analysis with benzotrifluoride as the internal standard. bIsolated yields are shown. The ratios of endo- to exo-isomers were measured using 1H NMR spectroscopic analysis; Boc, tert-butoxycarbonyl; THF, tetrahydrofuran; NOE, nuclear Overhauser effect.
Scope of the N-difluoroalkylative dearomative cycloaddition
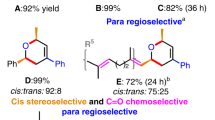
With these promising results in hand, we explored the scope of the N-difluoroalkylative dearomative cycloadditions. As demonstrated in Table 1, a broad range of aldehydes, ketones and pyridines underwent the three-component cycloaddition reaction. The reaction of aromatic aldehydes including heterocycles proceeded smoothly to give the dearomative products in synthetically useful yields (6–11). Alkenyl and enolizable aliphatic aldehydes were also employed to afford the desired cyclic products (12–16). Even sterically hindered pivalaldehyde could react to provide 17. Moreover, a variety of ketones that are less electrophilic than aldehydes can also serve as the coupling partner for the cycloaddition to deliver the corresponding products bearing quaternary carbon stereocentres (18–21). The pyridine scope was also investigated with i-butyraldehyde as the coupling partner. A series of bioactive nicotine derivatives underwent the cycloaddition, and substrates with a variety of functionalities such as ester, nitro, nitrile, ketone, amide, bromide and boronic ester groups are well tolerated (22–35). The cycloadditions proceeded preferentially at the less-hindered carbon atom adjacent to the nitrogen atom on the pyridine substrates. Notably, the reaction could also be applied on the gram scale to a bioactive compound with a complex scaffold (namely, tocopherol nicotinate) to afford dearomatized product 35 in 89% yield.
We further explored the pyridinium ylide-mediated cycloadditions with other coupling partners (Fig. 5) suggested by the AFIR search (Figs. 2 and 3). The reaction of two methanimines and difluorocarbene gave fluorinated cyclic product A2 in the simulations. After an investigation to determine suitable reactants and their protecting groups, we found that N-Boc-protected imines are suitable for the proposed cycloaddition to provide the difluorinated products (36–39); the major components were confirmed to be endo-isomers as well as the products from aldehydes or ketones via NOE analysis of 39. The multicomponent cycloaddition of methanimine, difluorocarbene and ethylene as an alkene source (which was predicted to generate cyclic product A3 in the AFIR search) was realized using alkenes such as acrylonitrile or methyl methacrylate derivatives, affording dihydroindolizine products (40–48) bearing quaternary carbon stereocentres. On the other hand, although the kinetics simulation based on the reaction path network proposed that similar cycloadditions with acetylene would proceed to generate the corresponding cyclic product A4, the competitive alkylative difluoromethylation of the C=N bond to give the corresponding linear product B4 was also predicted. The reaction with methyl propionate provided both cyclic product 49 and difunctionalized product 50; 49 would be formed through the predicted cycloaddition and subsequent elimination of hydrogen fluoride. Based on the fact that the formation of 50 was proposed to occur through the deprotonation of the terminal alkyne, we attempted the reactions using a dimethyl or diethyl acetylenedicarboxylate, which bear two substituents on the alkyne moiety. Gratifyingly, the reactions proceeded smoothly to provide products 51 and 52 in 51% and 68% yield, respectively. The solid-state structures of products 40 and 51, which are derived from the alkene and alkyne, were determined via single-crystal X-ray diffraction analysis. As described above, computational predictions based on an AFIR search are able to provide potential reaction modes that can guide the development of the diverse N-difluoroalkylative dearomatization.

a, Reaction realization and scope of imines based on the AFIR prediction for the reaction of two molecules of methanimine and difluorocarbene. b, Reaction realization and scope of alkenes based on the AFIR prediction for the reaction of methanimine, ethylene and difluorocarbene. c, Reaction realization and scope of alkynes based on the AFIR prediction for the reaction of methanimine, acetylene, and difluorocarbene. Reactions were performed under the conditions shown in each section. Isolated yields are given in all cases. The ratios of endo- to exo-isomers were measured using 1H NMR spectroscopic analysis, and the endo-form of 41 was confirmed using NOE analysis. n.d., not detected.
Cycloadditions toward α,α-difluorinated O-heterocycles
Although the computationally predicted cycloaddition via the azomethine ylide intermediates resulted in diverse dearomative cycloadditions with pyridines, we also considered the application of the cycloaddition to give α,α-difluorinated O-heterocyclic compounds A5–7 (Figs. 2 and 3). The computational results suggested that the multicomponent reactions of two formaldehydes and difluorocarbene would proceed via concerted multicomponent cycloadditions to form A5 (ΔG‡ = 2.2 kcal mol−1), whereas the energy barriers for the similar cycloadditions to generate A6 and A7 were 6.7 kcal mol−1 and 8.3 kcal mol−1, respectively (Fig. 6). As the energy barrier of the dimerization of difluorocarbenes (1 kcal mol−1; Fig. 4c) was lower than that of the latter two reactions, we attempted to perform the reaction to experimentally provide the structural motif of A5 using available carbonyl compounds and difluorocarbene. After intensive attempts, however, we unable to obtain the desired difluorinated O-heterocycles using representative carbonyl compounds such as benzaldehyde or acetone. To further investigate these results, we conducted additional calculations to evaluate these barriers by optimizing the transition-state geometries using the ωB97X-D functional and the Def2-SVP basis set with the CPCM(THF) solvation model. The calculated ΔG‡ values (3a, 19.1 kcal mol−1; 3c, 13.5 kcal mol−1) were much higher than that for the dimerization of difluorocarbene to generate tetrafluoroethylene, which was clearly observed in the experiment (see Supplementary Fig. 3 for details). This side reaction would thus be problematic in the reaction to form the α,α-difluorinated O-heterocyclic motif using the multicomponent cycloadditions at this stage.

a, The energy barriers for the formation of A5–A7 obtained in the calculations shown in Figs. 2 and 3. b, Experimental investigations with 3a and 3c, and their energy barriers for the cycloaddition obtained by optimizing the transition-state geometries at the ωB97X-D/Def2-SVP/CPCM(THF) level of theory.
Understanding the competitive undesired pathways
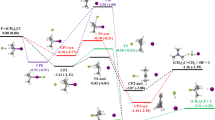
With the N-difluoroalkylative dearomatization of pyridines using a broad scope of coupling partners such as aldehydes, ketones, imines, alkenes and alkynes in hand, we further envisioned that the reaction performance could be generally explained by the barriers of the desired cycloaddition pathways via the transition states TSII and those of competitive side reactions. When the reaction provided the desired product in low yield, the pyridine substrates were almost completely consumed, and several unidentifiable minor side products were observed. Based on these experimental results and the computational finding that the reaction of pyridine and difluorocarbene to form the pyridinium ylide I was facile (TSI, 1 kcal mol−1; Fig. 4c), the undesired competitive pathways seemed to be derived from the unstable intermediate I. To identify possible competitive pathways, we systematically explored the reaction pathways using the SC-AFIR method with two molecules of I as initial structures (Fig. 7a). The exploration was conducted at the same level of theory as the combinatorial screening with the kinetics-based navigation shown in Figs. 2 and 3, but a shorter reaction time (10–6 s) was chosen instead of 1 s as the decomposition pathways would probably proceed within this time scale. The computation identified 8,464 pathways with 909 equilibrium structures (see Supplementary Figs. 1 and 2 for details). The transition-state geometries corresponding to the first steps starting from the initial structures among the reaction path network were optimized at the ωB97X-D/Def2-SVP/CPCM(THF) level of theory. The calculations suggested that the pathway for the dimerization to form IIIa via bond formation between one of the difluoromethyl anions and the C2 position on the other I was the most facile undesired reaction (TSIIIa, ΔG‡ = 3.1 kcal mol−1). Once unstable IIIa forms, further decomposition would provide the other possible undesired side products, which is consistent with the experimental result that the reaction of pyridine 2a without the coupling partner under the standard conditions shown in Fig. 4b furnished a complex mixture of fluorinated side products.

a, The desired and undesired pathways of the pyridinium ylide-mediated cycloaddition. The possible undesired pathways were suggested by a systematic search using the AFIR method at the ωB97X-D/LanL2DZ/CPCM(THF) level of theory (see the Supplementary Information for details). The pathway with the lowest energy barrier among the pathways for the first step from two molecules of I, which was obtained by optimizing the transition-state geometries at the ωB97X-D/Def2-SVP/CPCM(THF) level of theory, is shown. b, Comparison of the energy barriers for each coupling partner with the experimental results. The energy barriers for the cycloadditions were evaluated at the same level of theory. Reaction conditions: 2a (0.30 mmol, 1 equiv.), coupling partner (0.60 mmol, 2 equiv., or under a 1 atm ethylene atmosphere with a balloon), Me3SiCF2Br (0.33 mmol, 1.1 equiv.), Ph3SiF2·NBu4 (0.33 mmol, 1.1 equiv.) and THF (3 ml) at 0 °C for 2 h. Yields were determined using 19F NMR spectroscopic analysis with benzotrifluoride as the internal standard.
With the possible competitive pathways to the desired cycloadditions in hand, we wondered whether the reaction performance could be predicted by comparing the energy barriers of the desired cycloaddition to form the desired product II with those of the undesired pathways. To this end, we evaluated the energy barriers by optimizing the transition-state geometries TSIIa–g with the representative coupling partners (Fig. 7b). Although the electrophilic partners such as benzaldehyde, tert-butyl benzylidenecarbamate, acrylonitrile and dimethyl acetylenedicarboxylate exhibited lower ΔG‡ values for their transition states (TSIIa–d, 0.7–2.2 kcal mol−1) than the barrier of the undesired pathway (ΔG‡ = 3.1 kcal mol−1), those of the partners ethylene, styrene and diphenylacetylene were much higher (TSIIe, 9.1 kcal mol−1; TSIIf, 6.8 kcal mol−1; TSIIg, 11.8 kcal mol−1). The combinations of the first four reactions successfully provided the corresponding products (IIa–d) in experiments, whereas the other four did not, and the coupling partners remained after the reaction, except for ethylene due to its gaseous character. These results showcase the fact that the performance of the cycloaddition can be predicted based on the energy barriers of the desired cycloaddition pathway and the competitive pathway that was obtained using the AFIR search.
Conclusion
Recent progress in computational methods has enabled us not only to gain deep mechanistic insights into chemical reactions, but also to predict the reaction pathways that can theoretically occur from the input molecules. In this study we showed how to use the AFIR method for in silico reaction simulations to develop multicomponent reactions with difluorocarbene, as described below:
-
1.
Multicomponent reactions of difluorocarbene and pairs of components with unsaturated bonds were simulated combinatorially using quantum chemical calculations. The systematic exploration of all possible reaction pathways in each reaction was performed using the AFIR method.
-
2.
After obtaining the comprehensive reaction path networks, the target reactions were selected from among the numerous reaction pathways based on the reaction mechanism and the computational yield. In this study, the three-component cycloadditions to provide the medicinally important α,α-difluorinated N- or O-heterocyclic motifs were the target reaction to be developed.
-
3.
To realize the target reaction mode, experimental investigations to identify suitable substrates were conducted with the aid of calculations, which suggested that pyridine was the key substrate for the formation of the corresponding ylide intermediate.
-
4.
By understanding the competitive unproductive pathways systematically explored using the AFIR method and comparing their energy barriers to those of the desired reaction pathways, the reaction performance could generally be predicted, whether it proceeded or not.
Using this workflow, we developed a diverse N-difluoroalkylative dearomatization of pyridines with a broad range of electrophiles, including aldehydes, ketones, imines, alkenes and alkynes to provide α,α-difluorinated N-heterocycles, which are valuable as drug candidates but have hitherto been inaccessible via previously reported synthetic routes. The combination of reaction simulation using the AFIR method with experimental investigation thus represents a powerful strategy for methodological development.
Methods
Computational methods
All of the calculations were performed with the developer version of the GRRM program combined with the Gaussian 16 program11,22. The reaction pathways were explored using the SC-AFIR method with the ωB97X-D functional and LanL2DZ basis set using the Grid=FineGrid option17,18. In the SC-AFIR search, the force-induced reaction pathways were relaxed using the locally updated planes (LUP) method43,44 (denoted by LUP paths). Single-point energy calculations were performed using the ωB97X-D functional and Def2-SVP basis set at all discrete path points along all of the LUP paths. All of the transition-state optimizations were performed using the ωB97X-D functional and the Def2-SVP basis set21. To obtain transition-state geometries, the paths were relaxed using the LUP method43,44 until the highest energy point converged to the first-order saddle point. The transition states were confirmed to connect the reactants and products using intrinsic reaction coordinate calculations. In all of the calculations, the solvation of THF was taken into account using the CPCM model19,20. The Gibbs free energy values at 300 K and 1 atm were estimated by assuming ideal-gas, rigid-rotor and harmonic-vibrational models, in which all of the harmonic frequencies smaller than 50 cm−1 were set as 50 cm−1, as recommended in the literature45. All of the reaction barriers shown in this paper were obtained by optimizing transition-state geometries. The transition states were confirmed to connect the reactants and products using intrinsic reaction coordinate calculations. See the Supplementary Information for details.
Experimental methods
An oven-dried round-bottom flask was charged with Ph3SiF2∙NBu4 (297 mg, 0.55 mmol, 1.1 equiv.) and a pyridine derivative (0.50 mmol, 1 equiv.). After the addition of THF (5 ml), the reaction mixture was cooled to 0 °C (in the reactions with aldehydes, ketones, or imines) or −40 °C (in the reactions with alkynes or alkynes), and the coupling partner (aldehydes, ketones or imines (1 mmol, 2 equiv.); alkenes or alkynes (2.50 mmol, 5 equiv.)) and Me3SiCF2Br (86 μl, 0.55 mmol, 1.1 equiv.) were added. After the resulting mixture was stirred at the same temperature for the indicated time, the solvent was evaporated to give the crude mixture. The approximate yield of the product was determined using 19F NMR spectroscopy with benzotrifluoride as an internal standard. The crude mixture was purified by column chromatography on silica gel to afford the products. If necessary, GPC purification was performed to provide a pure sample.
Data availability
Crystallographic data are deposited at the Cambridge Crystallographic Data Centre under deposition nos. CCDC 2101564 (11), 2101565 (33), 2101566 (40) and 2101567 (51). All of the other data supporting the findings of this study are available in the main text or Supplementary Information.
References
Eleuterio, H. S. Olefin metathesis: chance favors those minds that are best prepared. J. Mol. Catal. 65, 55–61 (1991).
Fischer, K., Jonas, K., Misbach, P., Stabba, R. & Wilke, G. The “nickel effect”. Angew. Chem. Int. Ed. 12, 943–953 (1973).
Collins, K. D., Gensch, T. & Glorius, F. Contemporary screening approaches to reaction discovery and development. Nat. Chem. 6, 859–871 (2014).
Nandy, A. et al. Computational discovery of transition-metal complexes: from high-throughput screening to machine learning. Chem. Rev. 121, 9927–10000 (2021).
Houk, K. N. & Liu, F. Holy Grails for computational organic chemistry and biochemistry. Acc. Chem. Res. 50, 539–543 (2017).
Maeda, S., Ohno, K. & Morokuma, K. Systematic exploration of the mechanism of chemical reactions: the global reaction route mapping (GRRM) strategy using the ADDF and AFIR methods. Phys. Chem. Chem. Phys. 15, 3683–3701 (2013).
Wang, L.-P. et al. Discovering chemistry with an ab initio nanoreactor. Nat. Chem. 6, 1044–1048 (2014).
Grambow, C. A. et al. Unimolecular reaction pathways of a γ‑ketohydroperoxide from combined application of automated reaction discovery methods. J. Am. Chem. Soc. 140, 1035–1048 (2018).
Dewyer, A. L., Argüelles, A. J. & Zimmerman, P. M. Methods for exploring reaction space in molecular systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 8, e1354 (2018).
Simm, G. N., Vaucher, A. C. & Reiher, M. Exploration of reaction pathways and chemical transformation networks. J. Phys. Chem. A 123, 385–399 (2019).
Maeda, S. et al. Implementation and performance of the artificial force induced reaction method in the GRRM17 program. J. Comput. Chem. 39, 233–251 (2018).
Maeda, S. & Harabuchi, Y. Exploring paths of chemical transformations in molecular and periodic systems: an approach utilizing force. Wiley Interdiscip. Rev. Comput. Mol. Sci. 11, e1538 (2021).
Mita, T., Harabuchi, Y. & Maeda, S. Discovery of a synthesis method for a difluoroglycine derivative based on a path generated by quantum chemical calculations. Chem. Sci. 11, 7569–7577 (2020).
Hayashi, H. et al. Synthesis of difluoroglycine derivatives from amines, difluorocarbene, and CO2: computational design, scope, and applications. Chem. Eur. J. 27, 10040–10047 (2021).
Dilman, A. D. & Levin, V. V. Difluorocarbene as a building block for consecutive bond-forming reactions. Acc. Chem. Res. 51, 1272–1280 (2018).
Sumiya, Y. & Maeda, S. Rate constant matrix contraction method for systematic analysis of reaction path networks. Chem. Lett. 49, 553–564 (2020).
Chai, J.-D. & Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008).
Hay, P. J. & Wadt, W. R. Ab initio effective core potentials for molecular calculations. potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 82, 299–310 (1985).
Barone, V. & Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 102, 1995–2001 (1998).
Cossi, M., Rega, N., Scalmani, G. & Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 24, 669–681 (2003).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Frisch, M. J. et al. Gaussian 16 Revision B.01 (Gaussian, 2016).
Gillis, E. P., Eastman, K. J., Hill, M. D., Donnelly, D. J. & Meanwell, N. A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 58, 8315–8359 (2015).
Young, J. A., Simmons, T. C. & Hoffmann, F. W. Fluorocarbon nitrogen compounds. I. Perfluorocarbamic acid derivatives, amides and oxazolidines. J. Am. Chem. Soc. 78, 5637–5639 (1956).
Abe, T., Hayashi, E., Baba, H. & Fukaya, H. The electrochemical fluorination of nitrogen-containing carboxylic acids. fluorination of dimethylamino- or diethylamino-substituted carboxylic acid derivatives. J. Fluorine Chem. 48, 257–279 (1990).
Hayashi, H., Sonoda, H., Fuchigami, K. & Nagata, T. 2,2-Difluoro-1,3-dimethylimidazolidine (DFI). A new fluorinating agent. Chem. Commun. 7, 1618–1619 (2002).
Novikov, M. S., Voznyi, I. V., Khlebnikov, A. F., Kopf, J. & Kostikov, R. R. Unprecedented 1,3-dipolar cycloaddition of azomethine ylides to ester carbonyl. J. Chem. Soc., Perkin Trans. 1, 1628–1630 (2002).
Voznyi, I. V., Novikov, M. S., Khlebnikov, A. F. & Kostikov, R. R. Reactions of 1,5-π-cyclization of gem-difluoro-substituted azomethine ylides involving an aromatic ring. Russ. J. Org. Chem. 42, 689–695 (2006).
Tajima, T., Nakajima, A. & Fuchigami, T. Electrolytic partial fluorination of organic compounds. 83. Anodic fluorination of N-Substituted pyrroles and its synthetic applications to gem-difluorinated heterocyclic compounds. J. Org. Chem. 71, 1436–1441 (2006).
Hugenberg, V., Fröhlich, R. & Haufe, G. Oxidative desulfurization–fluorination of thioethers. Application for the synthesis of fluorinated nitrogen containing building blocks. Org. Biomol. Chem. 8, 5682–5691 (2010).
Prakash, G. K. S. et al. N‑Difluoromethylation of imidazoles and benzimidazoles using the Ruppert–Prakash reagent under neutral conditions. Org. Lett. 16, 54–57 (2014).
Liu, P., Gao, Y., Gu, W., Shen, Z. & Sun, P. Regioselective fluorination of imidazo[1,2‑a]pyridines with selectfluor in aqueous condition. J. Org. Chem. 80, 11559–11565 (2015).
Petko, K. I., Vlasenko, Y. G. & Kachkovskii, O. D. Unexpected “reverse” cyclization of 2-mercaptobenzimidazole with chlorotrifluoroethylene and hexafluoropropene. J. Fluor. Chem. 185, 168–172 (2016).
Ilin, E. A. et al. ortho-Dialkylamino arylboranes as efficient reagents for difluorocarbene trapping. Chem. Commun. 56, 7140–7142 (2020).
Ye, F., Ge, Y., Spannenberg, A., Neumann, H. & Beller, M. The role of allyl ammonium salts in palladium-catalyzed cascade reactions towards the synthesis of spiro-fused heterocycles. Nat. Commun. 11, 5383 (2020).
Novikov, M. S., Khlebnikov, A. F., Shevchenko, M. V., Kostikov, R. R. & Vidovic, D. 1,3-Dipolar cycloaddition of difluoro-substituted azomethine ylides. Synthesis and transformations of 2-fluoro-4,5-dihydropyrroles. Russ. J. Org. Chem. 41, 1496–1506 (2005).
Gaisina, K. R., Khlebnikov, A. F. & Novikov, M. S. Non-pericyclic cycloaddition of gem-difluorosubstituted azomethine ylides to the C=O bond: computational study and synthesis of fluorinated oxazole derivatives. Org. Biomol. Chem. 15, 4579–4586 (2017).
García-Domínguez, A. et al. Difluorocarbene generation from TMSCF3: kinetics and mechanism of NaI-mediated and Si-induced anionic chain reactions. J. Am. Chem. Soc. 142, 14649–14663 (2020).
Li, L. et al. TMSCF3 as a convenient source of CF2=CF2 for pentafluoroethylation, (aryloxy)tetrafluoroethylation, and tetrafluoroethylation. Angew. Chem. Int. Ed. 56, 9971–9975 (2017).
Kobylianskii, I. J., Novikov, M. S. & Khlebnikov, A. F. Formation and reactivity of gem-difluoro-substituted pyridinium ylides: experimental and DFT investigation. J. Fluor. Chem. 132, 175–180 (2011).
Zhou, S. et al. Metal-free difunctionalization of pyridines: selective construction of N‑CF2H and N‑CHO dihydropyridines. Org. Lett. 23, 2205–2211 (2021).
Dong, S., Fu, X. & Xu, X. [3+2]-Cycloaddition of catalytically generated pyridinium ylide: a general access to indolizine derivatives. Asian J. Org. Chem. 9, 1133–1143 (2020).
Choi, C. & Elber, R. Reaction path study of helix formation in tetrapeptides: effect of side chains. J. Chem. Phys. 94, 751–760 (1991).
Ayala, P. Y. & Schlegel, H. B. A combined method for determining reaction paths, minima, and transition state geometries. J. Chem. Phys. 107, 375–384 (1997).
Ribeiro, R. F., Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Use of solution-phase vibrational frequencies in continuum models for the free energy of solvation. J. Phys. Chem. B 115, 14556–14562 (2011).
Acknowledgements
We thank B. List for carefully reading and giving fruitful comments on this manuscript. We gratefully acknowledge K. Higashida for performing X-ray crystallographic analyses. This work was financially supported by JST-ERATO (grant no. JPMJER1903), as well as by the JSPS-WPI and JSPS KAKENHI Grants-in-Aid for Challenging Research (Exploratory) (grant no. 21K18945), Scientific Research (B) (grant no. 22H02069) and Young Scientists (grant no. 20K15284). H.H. thanks the Central Glass Award in Synthetic Organic Chemistry, Japan, and the NOASTEC foundation for financial support. T.M. thanks the Akiyama Life Science Foundation, the Ube Industries Foundation, the Fugaku Trust for Medical Research, the Uehara Memorial Foundation and the Naito Foundation for financial support.
Author information
Authors and Affiliations
Contributions
H.H., T.M. and S.M. conceived and designed the calculations and experiments. H.H., Y.H. and S.M. performed the calculations and analysed the data. H.H., H.K., H.T. and T.M. performed experiments and analysed the data. H.H. and T.M. co-wrote the manuscript with the support of H.K., H.T., Y.H. and S.M. All authors contributed to discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks Clémence Corminboeuf, Yu-hong Lam and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling editor: Thomas West, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–3. computational methods, experimental methods, data for characterization and references.
Supplementary Data 1
Crystallographic data for compound 11; CCDC no. 2101564.
Supplementary Data 2
Crystallographic data for compound 33; CCDC no. 2101565.
Supplementary Data 3
Crystallographic data for compound 40; CCDC no. 2101566.
Supplementary Data 4
Crystallographic data for compound 51; CCDC no. 2101567.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hayashi, H., Katsuyama, H., Takano, H. et al. In silico reaction screening with difluorocarbene for N-difluoroalkylative dearomatization of pyridines. Nat. Synth 1, 804–814 (2022). https://doi.org/10.1038/s44160-022-00128-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44160-022-00128-y
