
Abstract
The inorganic fraction of fine particles affects numerous physicochemical processes in the atmosphere. However, there is large uncertainty in its burden and composition due to limited global measurements. Here, we present observations from eleven different aircraft campaigns from around the globe and investigate how aerosol pH and ammonium balance change from polluted to remote regions, such as over the oceans. Both parameters show increasing acidity with remoteness, at all altitudes, with pH decreasing from about 3 to about −1 and ammonium balance decreasing from almost 1 to nearly 0. We compare these observations against nine widely used chemical transport models and find that the simulations show more scatter (generally R2 < 0.50) and typically predict less acidic aerosol in the most remote regions. These differences in observations and predictions are likely to result in underestimating the model-predicted direct radiative cooling effect for sulfate, nitrate, and ammonium aerosol by 15–39%.
Similar content being viewed by others

Introduction
Atmospheric aerosols affect human health, climate, cloud formation, nutrient availability for biota, and atmospheric chemistry1,2,3,4,5. Globally, submicron particulate matter (diameters <1 µm; PM1) accounts for an important fraction of aerosol mass concentration and radiative forcing5. Inorganic species are an important and highly variable fraction of the PM1 mass6,7. The inorganic PM1 is mostly secondary, formed from oxidation of precursors such as NOx (NO + NO2) and SO2 to form HNO3 and H2SO4, respectively, and partitioning of total ammonia (NHx = NH3,g + NH4,p+) between the gas- and aerosol-phases4. Sulfate is the dominant component of inorganic PM1, and, thus, has been most studied8,9. For polluted urban regions, there is still a debate about the chemistry that leads to the observed rapid sulfate formation and mass concentration10,11. Outside polluted urban regions, comparisons of chemical transport models (CTMs), with both each other and with observations, provide more insight into how well CTMs capture the processes that control sulfate. Generally, the processes controlling sulfate are better understood in these regions, and show better agreement with observations along with a reduced intermodel spread for sulfate7,10,11,12,13. Larger discrepancies are observed in both cases for the semi-volatile species nitrate and ammonium13,14.
These differences for PM1 nitrate and ammonium between observations and model predictions indicate a larger uncertainty in the emissions, chemistry, and lifetime that control their concentrations and that of their precursor gases, nitric acid (HNO3) and ammonia (NH3). However, the inorganic nitrate contribution to global PM1 aerosol is minor in most environments6,7. Exceptions to this include near combustion sources, such as biomass burning (BB) plumes15,16, urban areas17, as well as in deep convection over polluted regions18. This is due to the volatility of nitrate and the decrease of aerosol pH with distance from sources, which leads to partitioning of particle-phase nitrate into the gas-phase (Fig. 1c)17,19. Uncertainty in gas-phase ammonia and particle-phase ammonium impacts the models’ capability to predict important aerosol properties, such as aerosol pH10,14,20, and the concentration and composition of ammonium-sulfate salts (e.g., ammonium sulfate, letovicite, or ammonium bisulfate)21,22. Aerosol pH, one measure of aerosol acidity typically estimated with a thermodynamic model20, modulates numerous aerosol chemical and physical processes (Fig. 1). This includes key processes that control the oxidative capacity of the atmosphere (Fig. 1a), the formation of secondary organic aerosol (Fig. 1a), and the lifetime and fate of nitrogen oxides (Fig. 1c). The speciated composition of ammonium, sulfate, and nitrate ions in the aerosol controls the hygroscopic growth factor (HGF) (e.g., the increase in aerosol diameter due to water uptake)23, and, in turn, the radiative properties of the inorganic portion of the aerosol21,24.

See SI for references (Supplemental Table 1) and analytical equations. a Reaction probability of gas-phase epoxides (green circle and line) and HO2 (gray diamond and line) vs pH. b Fractional S(IV) species, SO2·H2O (dark red), HSO3 (red), and SO3 (light red) vs pH in equilibrium (left) and rates of oxidizing S(IV) to S(VI) through several mechanisms, O3 (solid black), H2O2 (dashed black), Fe (dashed-dot black), Mn (long-dashed black), and NO2 (long-short dashed black) vs pH (right). c Fraction of total nitrate (HNO3(g) + NO3‒(p)) (blue square), ammonia (NH3(g) + NH4+(p)) (orange diamond), chloride (HCl(g) + Cl−(p)) (pink triangle), and nitrite (HONO(g) + NO2−(p)) (dark yellow circle) in the particle phase vs pH. (d) Rate of dissolution of iron (Fe3+) to Fe2+ vs pH. e Measured phase separation relative humidity for different organic compounds (2MA = 2-methylglutaric acid (black), 3MA = 3-methylglutaric acid (gray), 2,2-BHBA = 2,2-bis(hydroxymethyl)butyric acid (light gray), 3,3-DMA = 3,3-dimethylglutaric acid (red), 1,2,6-HT = 1,2,6-hexanetriol (blue), and DEMA = diethylmalonic acid (dark yellow)) vs pH. f Molar abundance of imidazole-2-carboxaldehyde (“imidazole”) (brown), and its geminal diol form (“diol”) (orange) vs pH. Range of observation-based pH values for remote clean/continental boreal, remote polluted, SE US (southeastern United States), and CTMs’ (chemical transport models) (Supplementary Data 1) for the BL (boundary layer) is shown for reference.
There is large uncertainty in modeled ammonia and ammonium, also reflected in the disagreement of modeled concentrations with observations globally13. Outside the continental boundary layer (BL)13,20,25, observational constraints needed to improve CTMs are sparse. However, speciation of measured ammonium-salts is challenging with current analytical methods and requires a thermodynamic model, which can introduce additional uncertainties. Thus, a commonly used metric for comparing observations and CTMs is the fractional charge neutralization of nitrate, sulfate, and chloride by ammonium (herein referred to as “ammonium balance” or “NH4_Bal”), calculated as:
where nNH4+, nSO42−, nNO3−, and nCl− are the molar concentrations (moles per unit volume, or n) in the aerosol phase. Non-refractory chloride is typically a minor component of total PM16 and was found to be a minor component for the campaigns used here and is thus ignored. Reporting the charge balance in terms of ammonium is useful, as ammonium is the most abundant non-hydronium cation in PM126, and provides a qualitative, direct measure of acidity and whether the inorganic aerosol will behave more similarly to sulfuric acid, ammonium bisulfate, or ammonium sulfate21,24. The few studies that have investigated NH4_Bal in the remote atmosphere have reported inconsistent results, ranging from low (NH4_Bal < 0.4) to high values (NH4_Bal > 0.8)27,28,29. Remote polar regions have been shown to have low NH4_Bal30,31,32,33. Note, however, NH4_Bal is only predictive of pH under some conditions34, but generally not in other conditions (e.g., polluted BL), as NH4_Bal does not include the impacts of, e.g., aerosol liquid water and temperature20,35, and these may be conditions where aerosol pH is highly buffered36.
Here, observations of inorganic non-refractory PM1 from eleven different aircraft campaigns are used to investigate the evolution of inorganic aerosols, and associated intrinsic properties such as NH4_Bal and pH. The observations span data from very polluted to the most remote regions of the troposphere, both geographically (middle of the Pacific and Atlantic Oceans) and vertically (upper troposphere, defined here as between 400 and 250 hPa or ~7 to ~10 km). The observations from these campaigns are compared against nine widely used CTMs with different degrees of sophistication in their treatment of inorganic aerosols. The observations and simulations are evaluated along chemical coordinates, as this provides the most robust comparison by reducing the potentially confounding influence from transport and meteorology in different model runs and observations37,38. Finally, we performed several sensitivity simulations to explore ways to improve modeled pH and NH4_Bal relative to observations. Through these sensitivity simulations, we estimated the impact of acidity on the direct radiative effect estimated in a model, which was updated to best represent observations.
Results
Simulations show important differences in ammonium balance and pH versus observations
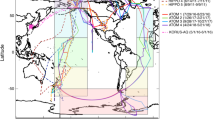
The wide spatial coverage of the eleven aircraft campaigns (Supplemental Fig. 1 and Supplemental Tables 2 and 3; using the Aerodyne Aerosol Mass Spectrometer (AMS)39) provides an opportunity to investigate the performance of nine CTMs for representing NH4_Bal and pH (“Methods” and Supplemental Table 4). The nine CTMs include four models that were part of a large collaborative model intercomparison study, used to investigate differences in model results with similar emissions, herein called the AeroCom-II models40, and five CTMs that were implemented and ran several years after the AeroCom-II study, herein called post-AeroCom-II models (see SI Supplemental Table 4 for more information). Regional CTMs have been used to investigate NH4_Bal and pH20,41,42 and have in general found a large spread in the predicted NH4_Bal and pH. As this study focuses on global observations and trends, only global models are used here. As shown in Fig. 2, the comparison between observations and post-AeroCom-II model simulations shows better agreement for sulfate (similar to prior studies7,12) than for nitrate and ammonium. The discrepancy for ammonium and nitrate increases over oceanic basins, as there are fewer observational constraints over the oceans versus over continental regions (especially in the northern hemisphere)7,13. This confirms on a global scale that there is more uncertainty in ammonium and nitrate, which will influence the comparisons between predicted and observed NH4_Bal and pH. There has been a long-standing over-prediction of nitrate in CTMs (e.g., Zakoura et al.43 and references therein); however, due to the negligible nitrate mass concentration observed during the ATom campaigns7, nitrate will have minimal influence on the calculated NH4_Bal and pH. Overall, the (older) AeroCom-II models13 show larger biases both in sulfate, ammonium, and nitrate (Supplemental Fig. 2). Since all concentrations tend to be underestimated in AeroCom-II models outside the BL, examining intensive properties, such as NH4_Bal and pH, should still be useful to at least assess source biases in these models.

Box plot for the ratios between post-AeroCom-II modeled and observed sulfate (SO4), nitrate (NO3), and ammonium (NH4) for BL (boundary layer, 800-surface hPa), FT (Free Troposphere, 400–800 hPa), and UT (Upper Troposphere, 250–400 hPa) for (a) non-urban focused continental campaigns (ARCTAS-A, ARCTAS-B, DC3, INTEX-B, and SEAC4RS) and (b) oceanic focused campaigns (ATom-1 and -2) evaluated here and post-AeroCom-II CTMs (chemical transport models; GEOS-Chem v10, GEOS-Chem v12, GEOS-Chem TOMAS, GEOS-5, and AM4.1). The blue horizontal line is the median ratio of the model-to-observations ensemble, and the boxes are the 25th and 75th percentiles. For AeroCom-II model comparisons, see Supplemental Fig. 2. For data used here, see Supplementary Data 1.
Curtain plots of models and measurements are shown in Fig. 3. The impact of year-to-year variability in emissions and meteorology on NH4_Bal and pH, as well as the impact of organics potentially being misattributed to total ammonium, nitrate, and sulfate (thus affecting the calculation of NH4_Bal and pH), are discussed in detail in “Methods” and SI. All these effects have a minimal impact on the results presented below.

Curtain plots of NH4_Bal (ammonium balance; a–f) and pH (g–l) for observations (a–c and f–h) and model (d–f and i–l) for Pacific Ocean (ATom-1 and -2), Atlantic Ocean (ATom-1 and -2), and above continents (other campaigns). Campaigns and their coordinates are listed in Supplemental Table 2. For ammonium balance, the model results are the averages of 9 CTMs (chemical transport models; Supplemental Table 4); whereas, for the pH, the model results are the averages of 3 CTMs that calculate pH on-line (Supplemental Table 4).
The observations show that the maxima in both NH4_Bal and pH occur at different locations in the two remote basins’ (Pacific and Atlantic) (Fig. 3). In the Pacific basin, the maximum is found north of 20°N, corresponding to Asian outflow44,45 and between 50°S and 30°S, likely corresponding to either the previously reported influence of pervasive BB46 (Supplemental Fig. 3) or oceanic NHx emissions29 (Supplemental Fig. 4). In the Atlantic basin, the maximum is found between 20°S and 30°N, corresponding to a mix of African BB47 and North American outflow48 (Supplemental Fig. 3). This particular region in the Atlantic basin is also associated with consistently high ammonia, as observed in some prior studies49,50. Thus, these features appear to be representative for the Atlantic basin. Outside of these regions of maximum NH4_Bal and pH, the typical tropospheric value over these two ocean basins is <0.3 (NH4_Bal) and 0 (pH).
Over the range of relative humidities (RH) typical for the troposphere (Supplemental Fig. 5), the observed NH4_Bal indicates that the aerosols in these regions generally have an HGF > 1.25, except in regions of BB and continental outflow (HGF 1.05–1.20) (Supplemental Figs. 6 and 7; see SI Sect. 2 for a description of the HGF calculation). This is due to more sulfuric-acid-like aerosol, which increases the water uptake. The higher HGF, and thus water content of the aerosol, along with lower pH, would indicate different chemical and physical processes (Fig. 1) than the lower HGF/higher pH in the regions that are influenced by continental-outflow, an important feature for CTMs to capture.
Finally, the low NH4_Bal and pH in the clean, remote marine boundary layer (MBL; here, defined from surface up to 800 hPa) suggest that both the Atlantic and Pacific Ocean basins, where sampled, have generally low local NHx emissions. Published NHx oceanic emission estimates range from 2 to 23 TgN yr−1, and a value of ~8 TgN yr−1 is typically used29. However, recent observationally constrained global study suggested that NHx oceanic emission estimates are closer to ~3 TgN yr−129,51, which is on the lower end of current emission inventories. On the other hand, ~20 Tg SO2 (as S) yr−1 is produced from dimethyl sulfide oxidation in oceanic environments8. Low marine NHx outgassing rates imply limited neutralization of nascent sulfate by ammonium (estimated mole ratio of 0.34N:S emitted from oceans for NHx and SO2). The observations in Fig. 3 support this imbalance of the oceanic emissions, with the emission and oxidation of sulfur being higher than the emissions of NHx, leading to fairly acidic (low NH4_Bal) conditions over the oceanic troposphere. Hence, oceanic emissions act to acidify marine submicron aerosols. CTMs often do not capture this effect. This is especially the case for those models that use too high oceanic NHx emission estimates.
The model averages generally show similar locations for the maxima in NH4_Bal as the observations for both oceanic basins. However, the spatial extent of the regions with higher NH4_Bal (e.g., >0.4) for the models is much larger than for the observations. Further, the model average does not indicate that the NH4_Bal gets much below 0.4 in either basin. In contrast, observations show large regions of the troposphere with NH4_Bal < 0.2. These contrasts are observed even at the coarse spatial resolution that both models and observations are averaged (100 hPa vertically and 5° latitude). As shown in Supplemental Fig. 7, NH4_Bal > 0.4 leads to a generally lower HGF (<1.3), which would bias the modeled chemical and physical aerosol processes.
Further, unlike the observational data, the model average maximum aerosol pH does not occur in the same regions as the model maximum NH4_Bal. In general, the model averages indicate the maximum aerosol pH occurs in the MBL, and it remains relatively uniform in the MBL in both oceanic basins. Also, the model average does not capture the maximum in aerosol pH in the outflow-influenced regions, especially the BB outflow-influenced regions, even though the models did capture the maximum in NH4_Bal for these regions. Another region where pH and NH4_Ba are at significant variance in the model average is the tropical Atlantic Ocean; NH4_Bal is relatively high, but there is large variability in aerosol pH (~−1.0 to 2.0). This can arise from the combination of RH and temperature, which impacts both the aerosol liquid water and the equilibrium distribution of semi-volatile compounds (nitrate and ammonium), impacting the aerosol pH19. Using a simple sensitivity study (see SI Sect. 3 for details), the predicted pH shows very high variability for the conditions when NH4_Bal starts decreasing from ~1.0 to ~0.5 (Supplemental Fig. 7) due to changes in RH. Thus, both aerosol composition and RH control the aerosol pH, making direct comparisons of NH4_Bal and pH when NH4_Bal is high in Fig. 3 complicated, as expected.
Unlike over remote oceanic basins, the observed NH4_Bal over the continents rarely drops below 0.3 (Fig. 3), in agreement with prior studies52,53,54. This is due to these regions having more ubiquitous and stronger sources of NHx, such as agriculture and BB55. Further, ammonia can be efficiently transported through convection56, which was observed during a few campaigns (e.g., DC3)57,58. The generally higher NH4_Bal observations result in HGF values that are lower than observed over most of the oceanic basins (Supplemental Fig. 7), indicating lower aerosol water content and hence smaller ambient aerosol diameters. Both will influence the physical and chemical properties of the aerosol compared to the more acidic aerosol observed over the oceanic basins.
Similar to the oceanic regions, the continental regions with higher NH4_Bal observations generally coincide with regions of higher aerosol pH.14 Overall, the variability of aerosol pH is not as extensive as for NH4_Bal, though, due to the modulation of pH by RH and temperature19. There are large spatial gradients observed in pH, for polluted versus cleaner/higher latitude regions (pH being generally >0.5 south of 50°N and less than 0 north of 50°N). Boreal forests are not a large source of ammonia except for BB events55, hence a low pH is observed, while air masses over the more polluted continental US, Mexico, and South Korea have an average pH ~2–3 units higher. This implies that aerosol processes between these regions would be very different (Fig. 1).
Unlike the oceanic basins, the averages of the CTMs over the continents predict a constant NH4_Bal regardless of location and altitude, and anions that are nearly always charge-balanced by ammonium (Fig. 3). The models miss the low NH4_Bal over boreal Canada and the upper troposphere over Canada and US, leading to a lower modeled HGF, and thus less modeled aerosol water. The consistently higher NH4_Bal for model output >60°N most likely arises from the models having too much ammonia throughout the troposphere, where it is 2–4 orders of magnitude higher than observationally constrained ammonia (Supplemental Fig. 9).
On the other hand, the model-averaged aerosol pH generally does better in capturing the observed aerosol pH maximum over the continental regions and the influence of convective transport56 on pH above the BL. As described above and shown in Supplemental Fig. 8, the variability in pH with nearly constant NH4_Bal is due to the non-linear response of pH in aerosol composition, RH, and temperature. Also, the models partially capture the differences in aerosol pH over polluted (< ~50°N) versus boreal (> ~50°N) continental regions. However, the models predict higher aerosol pH in the boreal forest BL, compared to observationally constrained pH. This could stem from overpredicted ammonia emissions from soils or BB, or from an underestimation of BB NOx emissions in the models. As this is a region of active biogenic organic photochemistry and secondary organic aerosol chemistry59 (e.g., organic epoxide uptake in Fig. 1a), differences in the aerosol pH of 0.5–1 pH unit can affect the uptake of organic gases to aerosol and the phase state of the aerosol, changing the predicted aerosol properties and chemistry. The difference in pH spans the sensitive region of potential organic phase separation (Fig. 1e), implying very different predicted versus observed physical properties for PM1 in this region. Note that not only pH, but also aerosol composition, including organic mixtures, can impact the phase separation60.
Ammonium balance and pH decrease with decreasing aerosol mass concentration
We use chemical coordinates, such as NH4_Bal or pH (y-axis) versus inorganic mass concentration (x-axis), to investigate potential reasons for the differences between the CTMs and the observations. Chemical coordinates provide a way to investigate chemical processes and emissions while minimizing the influence of the transport and other meteorological parameters (e.g., RH, T, and BL height)37,38.
For the observations, there is a robust correlation (R2 range for all fits is between 0.54–0.76) for NH4_Bal and inorganic dry PM1 mass concentration (with comparable results for pH). This result holds for all three tropospheric altitude regions (Fig. 4) and has not been previously reported, to our knowledge. This further supports that oceans promote acidifying submicron aerosols due to the imbalance between the emissions of NHx and those of sulfate precursors. The decrease in PM1 is a proxy for the gradual dilution and transformation of polluted air masses during global-scale transport and mixing. A recent study suggested that the two largest factors controlling aerosol pH were aerosol liquid water (potentially caused by different species concentrations) and temperature36. These factors create a buffer that maintains a relatively constant pH for a given region; however, this focused on areas near emission sources. Much of our study is for regions removed from emission sources. As shown in Supplemental Fig. 8, pH has a non-linear response to ammonium, RH, temperature, and aerosol liquid water. This indicates more factors control the pH away from emission sources. The simple parameterization suggested in Zheng et al.36 may not apply for the observations investigated here, in agreement with those authors’ conclusion that only ~40% of the continental surface was in the regime that buffered aerosol pH with aerosol liquid water. Finally, the slopes are statistically similar at all three levels for NH4_Bal and for aerosol pH at the 95% confidence level (Supplemental Tables 5 and 6).

Scatter plot of the average NH4_Bal (top row) and aerosol pH (calculated from E-AIM) (bottom row) versus inorganic PM1 (NH4 + SO4 + NO3) for boundary layer (a, d, defined as surface to 800 hPa), free troposphere (b, e, defined as 800–400 hPa), and upper troposphere (c, f, defined as 400–250 hPa). Each point represents the average observed value for each campaign at the specified pressure level and latitude zone (see Supplementary Data 1). These include ARCTAS-A (orange triangle), ARCATS-B impacted by BB (biomass burning; green open triangle), ARCTAS-B not impacted by BB (green solid triangle), CalNex (gray triangle), DC3 (light blue triangle), INTEX-B (light purple triangle), KORUS-AQ (dark green triangle), MILAGRO (black triangle), SEAC4RS impacted by BB (purple open triangle), SEAC4RS not impacted by BB (purple solid triangle), WINTER (pink triangle), and ATom-1 (blue) and -2 (red) >50°N (circle), 25°N to 50°N Pacific Ocean (square), 25°S to 25°N Pacific Ocean (upside-down triangle), 50°S to 25°S Pacific Ocean (left sideways triangle), >50°S (right sideways triangle), 50°S to 25°S Atlantic Ocean (sideways thin diamond), 25°S to 25°N Atlantic Ocean (thin diamond), and 25°N to 50°N Atlantic Ocean (normal diamond). a,bData filtered by BB markers (HCN < 350 pptv and CH3CN < 225 pptv = Not Biomass Burning and HCN > 350 pptv and CH3CN > 225 pptv = Biomass Burning)116.
Figure 4 shows important differences among campaigns that were not apparent in Fig. 3. First, there are clear differences in NH4_Bal and pH for aerosols influenced by BB (ARCTAS-B and SEAC4RS) versus not. NH4_Bal is a factor of 1.2–2.2 higher, and pH is 0.4–3 units higher for BB-dominated air masses versus non-BB-dominated air masses (see SI material). Note that BB is a source of organic acids (e.g., pyruvic acid)61, which can react with NHx to form salts62. Though the pH may be lower than the pKa values for various organic acids, the aerosol system is a non-ideal solution36,52,63. As there has been little research in regard to the partitioning and thermodynamics of these organic acids at low pH in non-ideal solutions, it is not certain whether these organics are present as ammonium salts or not. This may explain why the NH4_Bal exceeds 1.0 in some BB measurements (Fig. 4). Also, the possible presence of organic acids leads to some uncertainty in the estimated pH for the BB plumes, though, this effect is likely small64 due to offsetting effects (e.g., organic acids also increase aerosol liquid water and ion activity, leading to a negligible change in pH)65. The higher pH for aerosol influenced by BB emissions is similar to the results from Bougiatioti et al.64 and consistent with BB being a stronger source of NHx compared to other natural emissions (~2 TgN yr−1 for soils under natural vegetation versus ~5 TgN yr−1 for BB)55. Further, for regions influenced by BB, urban pollution, and deep convection, NH4_Bal and pH are higher in the free and upper troposphere than in BL regions without major sources, as convection can efficiently transport NHx to the free and upper troposphere56. In absence of deep convection near NHx emissions, ammonia quickly decreases with distance from sources (Supplemental Fig. 9), reducing the amount of ammonium in the aerosol phase relative to the amount of sulfuric acid produced from the oxidation of SO2 and dimethyl sulfide. The deep convection (continental observations between 15° and 50°N) observed during DC3, compared to SEAC4RS (similar location but less deep convection sampled), led to aerosol with large differences in NH4_Bal (0 versus 0.77 for SEAC4RS versus DC3, respectively) and pH (–0.93 versus 1.35 for SEAC4RS versus DC3, respectively) in the upper troposphere.
Unlike the observations, simulated NH4_Bal by the nine CTMs have a large spread in the correlation of NH4_Bal and inorganic PM1 (Fig. 5). Further, even for models that produce statistically similar slopes to observations for NH4_Bal (Supplemental Table 5), most of the trends show much lower correlation than observations (R2 < 0.5). This generally lower R2 suggests either uncertainty in NHx or nitrate for the post-AeroCom-II models or in NHx, nitrate, and sulfate for the AeroCom-II models. Though there is a large spread in model versus observed nitrate (Fig. 2 and Supplemental Fig. 2), the combination of generally low nitrate mass concentration due to low pH19 and NH4_Bal from models being higher than observations (Figs. 3 and 5d–f,) indicates that the spread and difference between models and observations are mostly due to uncertainty in NHx. This is further explored in the SI (SI Sect. S1 and Supplemental Fig. 9). In general, CTMs have higher ammonia mixing ratios than observationally constrained ammonia mixing ratios, further supporting models having too much ammonia.

Comparison of observations (black solid line) and averages of AEROCOM-II (red dashed-dot) and post-AEROCOM-II (blue dashed-dot) model results for (a–c) NH4_Bal versus log10(inorganic PM1) slopes, (d–f) normalized probability distribution function (PDF) of NH4_Bal, and (g–i) normalized PDF of estimated HGF for observations. For (g–i), the HGF values are from Supplementary Fig. 5, and for average values of RH from observations (~50%, ~35%, and ~35% for the boundary layer, free troposphere, and upper troposphere, respectively). For all data from models in comparison with observations, see Supplemental Figure 10. The model bands shown represent the range in the model results. The respective composite R2 are shown in (a–c).
Numerous factors could lead to these differences in NH4_Bal between observations and models. Observations of NH4_Bal above the BL previously used in the evaluation of CTMs have typically been based on aerosols collected onto Teflon filters and analyzed off-line21. However, as discussed in Nault et al. (and references therein)66, acidic aerosols collected onto filters will react with ammonia in the aircraft cabin, biasing the ammonium mass concentration and NH4_Bal. Another potential factor is overestimated oceanic29,51 and/or continental67 NHx emissions in models. Decreasing the oceanic emissions, from 8 Tg N yr−1 (GEIA68) to observationally constrained emissions of 2.4–3.2 Tg N yr−1 (Paulot et al.29,51), together with a reduction in the continental NHx emissions of 25%, better captures the observations in the BL and the acidification of submicron aerosol with remoteness (Supplemental Fig. 11). The improved BL probability distribution function (PDF) is due to the continued sulfuric acid production that occurs over remote oceans from the oxidation of SO28, with minimal NHx, leading to more acidic sulfate aerosol. An additional potential factor, as discussed in Bian et al.13, is that models may underestimate the pH-dependent wet deposition of NHx. As demonstrated in Supplemental Fig. 11, reducing the Henry’s constant of ammonia, which decreases the wet deposition of ammonia in GEOS-Chem (“increased NHx lifetime”), to make it more similar to other models13, shifts the BL NH4_Bal to higher values (more similar to CCSM4, GISS-MATRIX, and GISS-ModelE). Finally, there may be a temperature dependence on the strength of continental NHx emissions69 and there is a temperature dependence on the NHx partitioning to aerosol19; however, as most of the campaigns presented here focused on spring- and summer-time, exploration of this dependence was not possible.
The upper troposphere shows less sensitivity to NHx emissions and more sensitivity to increased NHx lifetime compared to the BL (Supplemental Fig. 11). This would imply that a shorter lifetime for NHx would be necessary to improve the agreement. However, the Henry’s law constant of ammonia, which strongly influences its wet deposition, already uses a high default value (3.3 × 106 M atm−1), limiting further removal of ammonia13. Thus, at this time, it is unclear what is needed to reconcile the differences in upper troposphere NH4_Bal between observations and CTMs, although errors in the spatiotemporal patterns of precipitation might play a role.
Post-AeroCom-II models show generally less deviation from observations for pH versus inorganic PM1 (Fig. 6), especially outside of the BL; whereas, the AeroCom-II models show large deviations throughout the troposphere. For the post-AeroCom-II models, the largest error occurs in the BL, specifically for GEOS-Chem v10 and v12 (Fig. 6d). Overall, the reduced error of models versus observations compared to those for NH4_Bal (Fig. 5 versus Fig. 6) (outside of the BL and not including AeroCom-II models) may partially stem from needing large changes in ammonia concentrations for effecting a unit change in pH14 and potentially from calculating aerosol pH similarly to observations for some models (Supplemental Table 4) (e.g., not including sea-salt).

Comparison of observations (black solid line) and averages of AEROCOM-II (red-dashed line) and post-AEROCOM-II (blue dashed-line) model results for (a–c) pH versus log10(inorganic PM1) slopes and (d–f) normalized probability distribution function (PDF) of pH. For all data from models in comparison with observations, see Supplemental Fig. 12. The model bands shown represent the range in the model results. The respective composite R2 are shown in (a–c).
It should be noted that the models that calculate the aerosol pH online use ISORROPIA (GEOS-Chem v10, GEOS-Chem v12, and AM4.1). ISORROPIA is not as explicit of a model as E-AIM20,70, but they generally produce similar results20.
The potential impact of non-volatile cations (NVC), specifically sodium from sea-salt and potassium from dust, on PM1 pH has been investigated and discussed in detail in the SI (Section S4 and Supplemental Figs. 13–15). To summarize, other aerosol measurements showed that there were two main aerosol populations: (1) fine aerosol, within the AMS size range, dominated by sulfate and organics, and with very little NVC, and (2) coarse aerosol, or larger particles, dominated by NVC (mostly sea-salt and dust), of which only a very small fraction is within the AMS size range (Supplemental Fig. 13). These two populations might have a different pH and thus different chemical and physical properties; however, the focus of this paper and comparisons with models are for population (1), the fine aerosol. The models generally calculate aerosol pH for fine aerosol internally mixed with submicron sea-salt as a single value, leading to higher pH. By not treating the particle populations separately, the models are missing different important chemical reactions due to missing these different populations. Further, as discussed in Hodzic et al.7 and Murphy et al.71, ATom-2 had significantly higher sea-salt than ATom-1 (~20% versus 2% of data in ATom-2 versus −1 had sea-salt comprising >20% of fine aerosol composition). Removing the ATom-2 observations from the results in Fig. 4 did not statistically change the slopes at a 95% confidence interval. Finally, as shown in Hodzic et al.7 and Murphy et al.71, sea-salt is negligible outside the MBL. Thus, NVCs are negligible for the sulfate-organic dominated fine mode (Supplemental Fig. 13) and hence do not impact fine mode aerosol pH.
A sensitivity run in GEOS-Chem, where sea-salt (accumulation mode, as coarse mode is already not included in the thermodynamic calculations72) was removed from the calculation of aerosol pH to be reflective of externally mixed sulfate-organic-dominated aerosol population described above. This leads to the model better representing the trend in pH versus inorganic PM1 (Supplemental Fig. 16a). Also, the exclusion of sea-salt from the GEOS-Chem aerosol pH calculation leads to a normalized distribution more similar to the observations (Supplemental Fig. 16b). Accumulation-mode sea-salt included in CTMs is mostly outside the AMS size-range (Supplemental Fig. 13), is closer to 1 µm in diameter, and is mostly externally mixed, similar to the conclusion of prior studies7. Thus, accumulation-mode sea-salt should be treated separately for pH calculations.
Impacts on modeled direct radiative effects due to uncertainty in ammonia emissions, aerosol composition, and inorganic phase
Prior studies have indicated that NH4_Bal is an important parameter in predicting the direct radiative effect (DRE) due to its impact on water uptake and HGFs21,22. However, this important parameter, NH4_Bal, may not be included in HGF estimations needed to determine the effective radius of the aerosol to in turn calculate the DRE in CTMs (e.g., see “Methods”). Further, as discussed above and shown in Supplemental Fig. 11, differences in lifetime and/or emissions of NHx impact NH4_Bal, which would impact the models’ predictions of the DRE. Thus, the impact of HGF related to RH and NH4_Bal and of NHx lifetime and emissions on predicted DRE is explored.
The impacts of acidity-dependent (e.g., NH4_Bal-dependent) HGF on DRE calculations compared to the base case (constant HGF per RH value and independent of acidity) are investigated with GEOS-Chem v12. The calculated annual average DRE becomes more negative (more cooling) for all updated cases explored here compared to the Base Case (Fig. 7, Supplemental Table 7). The main contribution to the decrease in DRE is due to the updated HGF table (Supplemental Fig. 5). As an example, the Base Case HGF at 50% RH is 1.17 (see “Methods”); however, as shown in Supplemental Fig. 5, the HGF ranges from 1.06 to 1.59, depending on NH4_Bal, at the same RH. Continental regions generally have higher ammonia emission rates than oceanic regions, leading to less acidic (higher NH4_Bal) aerosols over continents (Fig. 3). Hence, acidity-dependent HGFs lead to less cooling in polluted regions relative to the base-case (Supplemental Fig. 17). The higher HGF over oceanic regions due to lower ammonia emissions leads to more water uptake and thus a larger effective radius and more scattering. This leads to DRE becoming more negative for the updated cases.

Annual, global average direct radiative effect (DRE) for SO4 + NO3 + NH4 (and associated aerosol water) for all sky (a) and clear sky (b) for Base Case (blue) and the various updated cases (see Supplemental Table 7 for description of each updated case; gray). See Supplemental Fig. 17 for annually average DRE Base Case and absolute differences with each updated case. Here, increased/decreased NHx H refers to Henry’s law constant, which controls NHx wet deposition (and thus lifetime), and Cont. refers to continental.
Overall, the DRE estimates become 13–30% more negative for all sky and 25–39% more negative for clear sky compared to the base case. The strong cooling effect is related to the large areas of DRE becoming more negative over remote regions compared to the areas of DRE becoming more positive over polluted continental regions (Supplemental Fig. 17). The changes in DRE due to switching to acidity-dependent HGFs and NHx lifetime and emissions emphasizes the importance of properly predicting NH4_Bal and its properties in estimating and understanding DRE.
Summary
The inorganic fraction of PM1 affects many chemical and physical processes of ambient aerosol. However, there is large uncertainty in the chemical composition of inorganic PM1, due to uncertainty in emissions and lifetime of the precursor gases (specifically ammonia) and lack of measurements covering large swaths of the troposphere. Here, we use observations of the inorganic PM1 collected during eleven aircraft campaigns to investigate the trends of NH4_Bal and aerosol pH from polluted to the most pristine locations. We found a strong correlation of NH4_Bal and pH with inorganic PM1 mass concentration, indicating that as the air parcels are transported away from strong ammonia source regions (BB, agriculture, and anthropogenic activities), the continued production of sulfuric acid dominates the inorganic aerosol composition, leading to lower NH4_Bal and more acidic aerosol. However, the comparison of these observations with nine different CTMs indicates the models generally do not capture these trends due to numerous reasons, including (1) too high ammonia emissions in the CTMs, especially over oceanic environments, (2) inefficient removal of ammonia leading to modeled lifetimes that are too long, and/or (3) assumption of internal mixing state of inorganic aerosol with sea-salt. Note that another potential reason the CTMs may not capture these trends is due to model resolution20. However, (1) average values for a campaign are compared against the comparable averaged value from CTMs, (2) we do not expect many of the processes to be non-linear, and (3) none of the features we discuss here are small compared to the resolution of the CTMs. Thus, we do not believe any potential impacts from resolution affect the results here.
These uncertainties impact predicted aerosol properties (e.g., aerosol phase) and aerosol-related processes, including aerosol chemistry (e.g., epoxide uptake to aerosol) and the aerosol direct radiative effect. These uncertainties, along with assumptions and simplifications used by some models concerning HGF, can affect the predicted global annual average direct radiative impact of sulfate-nitrate-ammonium PM1, with 13 to 39% more cooling (more negative DRE) than in the base case. These uncertainties will be potentially more important in the future, where ocean acidification is predicted to further decrease oceanic NHx emissions51, leading to more acidic aerosol. We conclude that reducing the ammonia uncertainties will lead to better model predictions of inorganic aerosol composition and its chemical and physical properties.
Methods
Campaigns and Instrumentation
The campaigns used for this analysis are listed in Supplemental Table 2, along with the references that describe the campaigns, locations, and general goals. All the campaigns are airborne campaigns, either flown on the NASA DC-8 (ARCTAS-A and -B, DC3, SEAC4RS, KORUS-AQ, and ATom-1 and −2), NSF/NCAR C-130 (MILAGRO, INTEX-B, and WINTER), or NOAA P-3 (CalNex) research aircraft. In general, the MILAGRO, CalNex, WINTER, and KORUS-AQ campaigns sampled polluted, urban locations; the INTEX-B, ARCTAS-A and -B, DC3, and SEAC4RS campaigns sampled continental background locations (including some BB sampling for ARCTAS-B and SEAC4RS); and, ATom-1 and -2 and part of INTEX-B sampled remote oceanic background over the Pacific, Southern, Atlantic, and Arctic Oceans.
The instruments used for analysis are listed in Supplemental Table 3, along with references describing the instrument and its configuration and performance for each campaign. For the Aerodyne Aerosol Mass Spectrometers (AMS), the measurements were typically compared with other aerosol measurements to ensure confidence in the performance and mass concentrations for each campaign17,45,73,74,75,76,77,78. The effect of organic interference on total ammonium, nitrate, and sulfate is summarized below and described in detail in the SI. Nitric acid was measured with one of four methods: (a) CF3O− chemical ionization mass spectrometer (CIMS)79, (b) iodide CIMS80,81, (c) SiF5− CIMS82, or (d) mist chamber ion chromatography (MC/IC), which measures total nitrate (gas-phase HNO3 and particle-phase NO3−)83. The CF3O− CIMS and MC/IC flew on multiple campaigns together (ARCTAS-A and -B, DC3, SEAC4RS, KORUS-AQ, and ATom-1 and -2), as did the high-resolution time-of-flight AMS operated by the University of Colorado Boulder group. The agreement between the MC/IC and CF3O− CIMS varied for each campaign, due to differences in time response78 and potential instrument issues at high altitudes due to colder temperatures. Thus, as described below, both are used to calculate aerosol pH to investigate (and minimize) potential biases in the calculated aerosol pH.
Other measurements that were used in the analysis from the ATom campaigns include the NOAA Particle Analysis by Laser Mass Spectrometer84 for fraction of BB; the University of California, Irvine, Whole Air Sampler85 for methyl nitrate; the NOAA aerosol microphysical properties (AMP) suite of aerosol size spectrometers77,86 for particle number concentration; and, the NOAA single-particle soot photometer (SP2)87. The NASA Langley diode laser hygrometer (DLH)88 was used for water vapor to calculate RH and was used in all of the DC-8 campaigns listed.
Thermodynamic calculation of aerosol pH
In this work, we are studying the acidity of fine mode aerosol, in which sulfate and organics are typically internally mixed, versus the coarse mode, which includes sea-salt and dust and is typically externally mixed from the fine aerosol. The Extended Aerosol Inorganics Model (E-AIM) is the thermodynamic model89,90,91,92 used here to calculate gas-liquid equilibrium in the aqueous aerosol systems and pH for both observations and for CTMs that did not calculate aerosol pH on-line. Here, it is assumed that the aerosol remains in a metastable state below the deliquescence RH for typical tropospheric conditions14,19,20,93. E-AIM is considered one of the reference models for the thermodynamic predictions of aerosol pH20, as the model is based upon thermodynamic data for pure aqueous solutions and mixtures over a wide range of temperatures. Laboratory studies have shown that E-AIM pH predictions generally agree well with observed aerosol pH for synthetic aerosols94. In order to predict pH,, E-AIM calculates the ionic activities in terms of interactions between pairs and triplets of solute species94. E-AIM uses the Pitzer-Simonson-Clegg equations89,95,96 to calculate the solute activity coefficient, in single-ion values, and the solvents in the aqueous aerosol phase, on a mole fraction scale. Model IV was used in this work97 and included the following ions and gases in the calculation: H+, NH4+, SO42−, HSO4−, NO3−, HNO3, and NH3. Inputs into the model included SO42−, NH4+, total nitrate (HNO3 + NO3−), RH, temperature, and estimated H+ (from charge balance). Since gas-phase ammonia was not measured in most campaigns, similar to prior studies19,98, gas-phase ammonia was estimated by running the model iteratively until convergence (minimal changes in overall NHx) occurred. Depending on location and total aerosol mass concentration, about 20−50 iterations were needed. The model was run in the “forward” mode35. This has been shown to be the most stable mode and reduces the impact of measurement uncertainty in the calculation of pH35,99. Chloride (Cl−) was not included in the models, as (a) inclusion of Cl− limits the temperature range and the metastable assumption that can be used to calculate pH97 and (b) it composes a small fraction of the total inorganic PM1 mass concentration6 and is mostly associated with sea-salt100,101 (see Impacts of Nonvolatile Cations and Aerosol Mixing State for further discussion). Further descriptions about the chemical system and equilibria that are solved can be found in Pye et al.20 and references therein.
The H+ and inorganic aerosol liquid water calculated from E-AIM is used to calculate the aerosol pH for observations and models. To be consistent with the models that calculate aerosol pH on-line, and to be comparable with prior studies, the pHF definition is used, where pH is defined by the molality of H+, excluding activity \(({m}_{{\mathrm{H}}^{+}})\):
Here, \({\mathrm{H}}_{{\mathrm{air}}}^{+}\) (μg sm−3) is the hydronium ion mass concentration per volume air, Wi (μg sm−3) is the aerosol water concentration associated with the inorganic portion, and 1000 is a conversion factor. As shown in Pye et al.20, pHF may underestimate pH, depending on the atmospheric conditions (RH, temperature, and aerosol composition); however, this effect is generally smaller than 0.5 pH units. The ability to compare against prior studies and against CTMs, as both use the definition of pHF, is more important than a potential 0.5 pH unit difference to better evaluate the differences in observations versus models. Similar to prior studies19,52,93, organics were not included in the calculation of pH. Prior studies have shown the effect of organics on liquid water and hydronium molality is small102 and may prevent crystallization, ensuring aerosol remains metastable103,104. Further, the inclusion of organic aerosol has off-setting effects on pH and liquid water65. We expect the exclusion of organics will only lead to a small impact to the pH that is within the overall uncertainty of the calculated pH (±0.5 pH units)65,102.
The following limits are imposed for the calculation of pH to prevent reporting of values where E-AIM thermodynamic priors are outside the range that has been constrained and verified in laboratory studies. First, pH is not reported for ionic strengths >6–40 mol kg−1, depending on composition, as those are the highest ionic strengths for the laboratory solutions used to build E-AIM91,97. Second, the water supersaturation relative to ice is calculated, following Koop et al.105, and any data point where homogeneous ice nucleation is likely (defined as a(H2O)ice + 0.265, per Koop et al.) is ignored. Finally, only values calculated for pressure levels of 250 hPa or greater are reported here to further limit calculated pH to T and RH, where the E-AIM model has been verified in laboratory studies89,91,97. These acidic aerosols are expected to be aqueous (retain water) even at the lower temperatures in the UT106.
Finally, a detailed discussion pertaining to the evaluation of E-AIM results based on the partitioning of the semi-volatile species HNO3 + NO3− (Supplemental Figs. 18–20) and NH3 + NH4+ (Supplemental Figs. 21–24) can be found in SI Sect. 6. Briefly, E-AIM predicted the observed particle-phase nitrate and gas-phase nitrate for all but two campaigns (INTEX-B and ATom-2), and exclusion of those two campaigns did not change the slopes and R2 values reported in this study. Finally, in a sensitivity analysis for one campaign where gas-phase ammonia measurements were available, little variation in pH (~0.1 pH unit change, see SI) was found between the E-AIM model ran with total nitrate (gas- and particle-phase), total NHx (gas- and particle-phase), sulfate, RH, and temperature inputs versus the results from the full iterative model used for the other campaigns (note that the aerosol was on average fairly neutralized for that campaign, hence the sensitivity to NHx is highest). A similar difference (~0.2 pH units) between predicted pH for ISORROPIA ran either with gas-phase ammonia or with iterating the model for gas-phase ammonia convergence has been reported before,19 further supporting the robustness of running the E-AIM model in this configuration.
Investigation of and minimal impact due to changing emissions, changing meteorology, and organic fragmentation
The campaigns used in our study range over a period of 10 years; however, this generally does not impact the comparison of NH4_Bal and aerosol pH. A large change in the ammonia mixing ratio is necessary to change from ammonium sulfate-like aerosols into sulfuric acid-like aerosols and to change the aerosol pH10,14. Observations have shown small to minimal decreases in NH4_,Bal and aerosol pH per year during the past decade14,31,107 while ammonia has been constant or slightly increasing in the troposphere108,109. Although there is substantial uncertainty in the representation of this variable in CTMs13, several sensitivity runs for NH4_Bal and pH using GEOS-Chem showed no changes in the last decade, neither with changing (Supplemental Figs. 25 and 26) nor with constant (Supplemental Figs. 27 and 28) emissions, in agreement with observations.
An important aspect of the AMS measurements is that the functional group in organic nitrates, sulfates, and organic reduced nitrogen compounds (e.g., amines and pyridine) are by default assigned to inorganic nitrate, sulfate, and ammonium, although the extent of these organic interferences can be quantified or estimated110,111,112,113. A detailed analysis concerning each type of compound can be found in SI Sect. S5. Briefly, although the inclusion of these organics into the total nitrate, sulfate, and ammonium measurements can increase the scatter in aerosol pH (Supplemental Figs. 29 and 30) the effect is generally small and within the uncertainty of the predicted aerosol pH. Also, changes in NH4_Bal are minimal (typically <5%). These organics are a small amount of the total mass concentration, especially for ammonium (Supplemental Fig. 31 and Supplemental Table 8). Thus, for the observations used here, these small organic interferences do not change the trends, comparisons, and conclusions discussed.
Chemical transport models
The CTMs (atmospheric chemistry components of global climate models, such as AM4.1, which has been grouped with CTMs throughout the rest of paper for simplicity) used in this study are described in the SI (Supplemental Table 4). Here, the analysis approach for the models and the sensitivity experiments are discussed.
For the models, areas encompassing each campaign (Supplemental Table 2) were averaged for each tropospheric pressure zone (BL = surface to 800 hPa, FT = 800–400 hPa, and UT = 400–250 hPa). This was done instead of analyzing the models for the flight path of each campaign to minimize the influence of potential biases on the modeled transport of air masses versus the observations. Further, average monthly model results for the same months as the campaigns are compared. The average results were then used to compare the trends in the modeled NH4_Bal and aerosol pH versus inorganic mass concentration (see SI Material). This method of analysis further minimizes the impacts of transport and meteorology on the comparison of observations with modeled results37.
For models that did not calculate aerosol pH on-line (CCSM4, GISS-ModelE, GISS-MATRIX, GEOS-5, and GEOS-Chem-TOMAS), the outputs from the model were used to calculate the aerosol pH off-line with E-AIM, as described above. One model, TM4-ECPL-F, lacked the output necessary to calculate aerosol pH.
Direct radiative effect calculation
GEOS-Chem v12.1.1 was used to calculate the contribution of sulfate, nitrate, and ammonium to DRE. In the base case (the default in GEOS-Chem), GEOS-Chem calculates aerosol optical depth, single scattering albedo, and asymmetry parameter of each aerosol based on the pre-calculated Mie table with spherical shape assumption114. GEOS-Chem describes the hygroscopic growth of aerosols with 7 discrete RH bins, and prescribed HGF (wet/dry radius ratio) of sulfate-nitrate-ammonium are [1.0, 1.17, 1.34, 1.52, 1.86, 2.33, 3.95] at RH = [0%, 50%, 70%, 80%, 90%, 95%, 99%]. GEOS-Chem linearly interpolates the optical parameters when RH does not exactly match the look-up table RHs. The DRE of each aerosol is then calculated by GEOS-Chem using the rapid radiative transfer model for GCMs (RRTMG)114,115, for all-sky and clear-sky conditions. Calculations are based on the radiation difference between runs with and without aerosol species included of interest while other conditions (e.g., meteorological conditions, gases, and aerosols) are the same.
For the updated cases (Supplemental Table 7), the basic calculation remains the same, except new HGFs are used based on Supplemental Fig. 5, with a new pre-calculated Mie parameter table for each corresponding HGF in Supplemental Fig. 5 grid spaces, not accounting for differences in ammonium sulfate versus ammonium nitrate. Since the HGF sensitivity to temperature is smaller than those for RH and NH4_Bal, the parametrization for the GEOS-Chem HGF calculation only includes the latter (Supplemental Fig. 5).
Code availability
Aircraft observations and chemical transport model output can be found at https://doi.org/10.3334/ORNLDAAC/1857. The data used to estimate condensational sink and volume contribution between non-volatile and all aerosol can be found at https://doi.org/10.3334/ORNLDAAC/1671.
References
Abbatt, J. P. D. et al. Solid ammonium sulfate aerosols as ice nuclei: a pathway for cirrus cloud formation. Science 313, 1770–1773 (2006).
Meskhidze, N., Chameides, W. L., Nenes, A. & Chen, G. Iron mobilization in mineral dust: Can anthropogenic SO2 emissions affect ocean productivity? Geophys. Res. Lett. 30, 2085 (2003).
Cohen, A. J. et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet 389, 1907–1918 (2017).
Seinfeld, J. H. & Pandis, S. N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. 1232 (John Wiley & Sons, Inc., 2006).
Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. 1535 (Cambridge University Press, 2013).
Jimenez, J. L. et al. Evolution of organic aerosols in the atmosphere. Science 326, 1525–1529 (2009).
Hodzic, A. et al. Characterization of organic aerosol across the global remote troposphere: a comparison of ATom measurements and global chemistry models. Atmos. Chem. Phys. 20, 4607–4635 (2020).
Faloona, I. Sulfur processing in the marine atmospheric boundary layer: a review and critical assessment of modeling uncertainties. Atmos. Environ. 43, 2841–2854 (2009).
Chin, M. et al. A global three-dimensional model of tropospheric sulfate. J. Geophys. Res. 101, 18667–18690 (1996).
Guo, H., Weber, R. J. & Nenes, A. High levels of ammonia do not raise fine particle pH sufficiently to yield nitrogen oxide-dominated sulfate production. Sci. Rep. 7, 12109 (2017).
Song, S. et al. Possible heterogeneous chemistry of hydroxymethanesulfonate (HMS) in northern China winter haze. Atmos. Chem. Phys. 19, 1357–1371 (2019).
Spracklen, D. V. et al. Aerosol mass spectrometer constraint on the global secondary organic aerosol budget. Atmos. Chem. Phys. 11, 12109–12136 (2011).
Bian, H. et al. Investigation of global particulate nitrate from the AeroCom phase III experiment. Atmos. Chem. Phys. 17, 12911–12940 (2017).
Weber, R. J., Guo, H., Russell, A. G. & Nenes, A. High aerosol acidity despite declining atmospheric sulfate concentrations over the past 15 years. Nat. Geosci. 9, 282–285 (2016).
Akagi, S. K. et al. Evolution of trace gases and particles emitted by a chaparral fire in California. Atmos. Chem. Phys. 12, 1397–1421 (2012).
Paulot, F. et al. Gas-aerosol partitioning of ammonia in biomass burning plumes: Implications for the interpretation of spaceborne observations of ammonia and the radiative forcing of ammonium nitrate. Geophys. Res. Lett. 44, 8084–8093 (2017).
DeCarlo, P. F. et al. Fast airborne aerosol size and chemistry measurements above Mexico City and Central Mexico during the MILAGRO campaign. Atmos. Chem. Phys. 8, 4027–4048 (2008).
Höpfner, M. et al. Ammonium nitrate particles formed in upper troposphere from ground ammonia sources during Asian monsoons. Nat. Geosci. 12, 608–612 (2019).
Guo, H. et al. Fine particle pH and the partitioning of nitric acid during winter in the northeastern United States. J. Geophys. Res. D: Atmos. 121, 355–10,376 (2016).
Pye, H. O. T. et al. The acidity of atmospheric particles and clouds. Atmos. Chem. Phys. 20, 4809–4888 (2020).
Wang, J., Hoffmann, A. A., Park, R. J., Jacob, D. J. & Martin, S. T. Global distribution of solid and aqueous sulfate aerosols: effect of the hysteresis of particle phase transitions. J. Geophys. Res. 113, 1770 (2008).
Wang, J., Jacob, D. J. & Martin, S. T. Sensitivity of sulfate direct climate forcing to the hysteresis of particle phase transitions. J. Geophys. Res. 113, 13791 (2008).
Brock, C. A. et al. Aerosol optical properties in the southeastern United States in summer—Part 1: Hygroscopic growth. Atmos. Chem. Phys. 16, 4987–5007 (2016).
Colberg, C. A., Luo, B. P., Wernli, H., Koop, T. & Peter, T. A novel model to predict the physical state of atmospheric H2SO4/NH3/H2O aerosol particles. Atmos. Chem. Phys. 3, 909–924 (2003).
Van Damme, M. et al. Atmospheric chemistry and physics global distributions, time series and error characterization of atmospheric ammonia (NH3) from IASI satellite observations. Atmos. Chem. Phys. 14, 2905–2922 (2014).
Dentener, F. J. & Crutzen, P. J. A three-dimensional model of the global ammonia cycle. J. Atmos. Chem. 19, 331–369 (1994).
Dibb, J. E. et al. Airborne sampling of aerosol particles: comparison between surface sampling at Christmas Island and P-3 sampling during PEM-Tropics B. J. Geophys. Res. 108, 8230 (2002).
Dibb, J. E. et al. Aerosol chemical composition in Asian continental outflow during the TRACE-P campaign: comparison with PEM-West B. J. Geophys. Res.: Atmos. 108, 8815 (2003).
Paulot, F. et al. Global oceanic emission of ammonia: constraints from seawater and atmospheric observations. Global Biogeochem. Cycles 29, 1165–1178 (2015).
Weller, R., Legrand, M. & Preunkert, S. Size distribution and ionic composition of marine summer aerosol at the continental Antarctic site Kohnen. Atmos. Chem. Phys. 18, 2413–2430 (2018).
Quinn, P. K., Bates, T. S., Schulz, K. & Shaw, G. E. Decadal trends in aerosol chemical composition at Barrow, Alaska: 1976–2008. Atmos. Chem. Phys. 9, 8883–8888 (2009).
Willis, M. D. et al. Aircraft-based measurements of High Arctic springtime aerosol show evidence for vertically varying sources, transport and composition. Atmos. Chem. Phys. 19, 57–76 (2019).
Ricard, V. et al. Two years of continuous aerosol measurements in northern Finland. J. Geophys. Res. D: Atmos. 107, 4129 (2002).
Schueneman, M. K. et al. Aerosol pH indicator and organosulfate detectability from Aerosol Mass Spectrometry measurements. Atmos. Meas. Tech. Discuss. 14, 2237–2260 (2021).
Hennigan, C. J., Izumi, J., Sullivan, A. P., Weber, R. J. & Nenes, A. A critical evaluation of proxy methods used to estimate the acidity of atmospheric particles. Atmos. Chem. Phys. 15, 2775–2790 (2015).
Zheng, G. et al. Multiphase buffer theory explains contrasts in atmospheric aerosol acidity. Science 369, 1374–1377 (2020).
Woody, M. C. et al. Understanding sources of organic aerosol during CalNex-2010 using the CMAQ-VBS. Atmos. Chem. Phys. 16, 4081–4100 (2016).
Cohen, R. C. et al. Quantitative constraints on the atmospheric chemistry of nitrogen oxides: an analysis along chemical coordinates. J. Geophys. Res. 105, 24283–24304 (2000).
DeCarlo, P. F. et al. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 78, 8281–8289 (2006).
Tsigaridis, K. et al. The AeroCom evaluation and intercomparison of organic aerosol in global models. Atmos. Chem. Phys. 14, 10845–10895 (2014).
Tao, W. et al. Aerosol pH and chemical regimes of sulfate formation in aerosol water during winter haze in the North China Plain. Atmos. Chem. Phys. 20, 11729–11746 (2020).
Zakoura, M., Kakavas, S., Nenes, A. & Pandis, S. N. Size-resolved aerosol pH over Europe during summer. Atmos. Chem. Phys. Discuss. https://doi.org/10.5194/acp-2019-1146 (2020).
Zakoura, M. & Pandis, S. N. Overprediction of aerosol nitrate by chemical transport models: the role of grid resolution. Atmos. Environ. 187, 390–400 (2018).
Hudman, R. C. et al. Ozone production in transpacific Asian pollution plumes and implications for ozone air quality in California. J. Geophys. Res. D: Atmos. 109, (2004).
Dunlea, E. J. et al. Evolution of Asian aerosols during transpacific transport in INTEX-B. Atmos. Chem. Phys. 9, 7257–7287 (2009).
Schill, G. P. et al. Widespread biomass burning smoke throughout the remote troposphere. Nat. Geosci. 13, 422–427 (2020).
Talbot, R. W. et al. Chemical characteristics of continental outflow over the tropical South Atlantic Ocean from Brazil and Africa. J. Geophys. Res. D: Atmos. 101, 24187–24202 (1996).
Li, Q. et al. North American pollution outflow and the trapping of convectively lifted pollution by upper-level anticyclone. J. Geophys. Res. 110, D10301 (2005).
Someya, Y., Imasu, R., Shiomi, K. & Saitoh, N. Atmospheric ammonia retrieval from the TANSO-FTS/GOSAT thermal infrared sounder. Atmos. Meas. Tech. 13, 309–321 (2020).
Khan, M. A. H. et al. Global and regional model simulations of atmospheric ammonia. Atmos. Res. 234, 104702 (2020).
Paulot, F., Stock, C., John, J. G., Zadeh, N. & Horowitz, L. W. Ocean ammonia outgassing: modulation by CO2 and anthropogenic nitrogen deposition. J. Adv. Model. Earth Syst. 12, e2019MS002026 (2020).
Guo, H., Nenes, A. & Weber, R. J. The underappreciated role of nonvolatile cations in aerosol ammonium-sulfate molar ratios. Atmos. Chem. Phys. 18, 17307–17323 (2018).
de Sá, S. S. et al. Urban influence on the concentration and composition of submicron particulate matter in central Amazonia. Atmos. Chem. Phys. 18, 12185–12206 (2018).
Xu, L., Suresh, S., Guo, H., Weber, R. J. & Ng, N. L. Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates. Atmos. Chem. Phys. 15, 7307–7336 (2015).
Sutton, M. A. et al. Towards a climate-dependent paradigm of ammonia emission and deposition. Philos. Trans. R. Soc. Lond. B Biol. Sci 368, 20130166 (2013).
Ge, C., Zhu, C., Francisco, J. S., Zeng, X. C. & Wang, J. A molecular perspective for global modeling of upper atmospheric NH3 from freezing clouds. Proc. Natl Acad. Sci. USA 115, 6147–6152 (2018).
Yang, Q. et al. Aerosol transport and wet scavenging in deep convective clouds: a case study and model evaluation using a multiple passive tracer analysis approach. J. Geophys. Res. D: Atmos. 120, 8448–8468 (2015).
Fairlie, T. D. et al. Estimates of Regional Source Contributions to the Asian Tropopause Aerosol Layer Using a Chemical Transport Model. J. Geophys. Res. D: Atmos. 125, 18,607 (2020).
Slowik, J. G. et al. Characterization of a large biogenic secondary organic aerosol event from eastern Canadian forests. Atmos. Chem. Phys. 10, 2825–2845 (2010).
You, Y., Renbaum-Wolff, L. & Bertram, A. K. Liquid–liquid phase separation in particles containing organics mixed with ammonium sulfate, ammonium bisulfate, ammonium nitrate or sodium chloride. Atmos. Chem. Phys. 13, 11723–11734 (2013).
Veres, P. et al. Measurements of gas-phase inorganic and organic acids from biomass fires by negative-ion proton-transfer chemical-ionization mass spectrometry. J. Geophys. Res. 115, D23302 (2010).
Updyke, K. M., Nguyen, T. B. & Nizkorodov, S. A. Formation of brown carbon via reactions of ammonia with secondary organic aerosols from biogenic and anthropogenic precursors. Atmos. Environ. 63, 22–31 (2012).
Liu, T., Clegg, S. L. & Abbatt, J. P. D. Fast oxidation of sulfur dioxide by hydrogen peroxide in deliquesced aerosol particles. Proc. Natl Acad. Sci. USA 117, 1354–1359 (2020).
Bougiatioti, A. et al. Particle water and pH in the eastern Mediterranean: source variability and implications for nutrient availability. Atmos. Chem. Phys. 16, 4579–4591 (2016).
Battaglia, M. A., Jr., Weber, R. J., Nenes, A. & Hennigan, C. J. Effects of water-soluble organic carbon on aerosol pH. Atmos. Chem. Phys. 19, 14607–14620 (2019).
Nault, B. A. et al. Interferences on aerosol acidity quantification due to gas-phase ammonia uptake onto acidic sulfate filter samples. Atmos. Meas. Tech. 13, 6193–6213 (2020).
Dammers, E. et al. NH3 emissions from large point sources derived from CrIS and IASI satellite observations. Atmos. Chem. Phys. 19, 12261–12293 (2019).
Bouwman, A. F. et al. A global high-resolution emission inventory for ammonia. Global Biogeochem. Cycles 11, 561–587 (1997).
Shah, V. et al. Chemical feedbacks weaken the wintertime response of particulate sulfate and nitrate to emissions reductions over the eastern United States. Proc. Natl Acad. Sci. USA 115, 8110–8115 (2018).
Fountoukis, C. & Nenes, A. ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+–Ca2+–Mg2+–NH4+–Na+–SO42−–NO3−–Cl−–H2O aerosols. Atmos. Chem. Phys. 7, 4639–4659 (2007).
Murphy, D. M. et al. The distribution of sea-salt aerosol in the global troposphere. Atmos. Chem. Phys. 19, 4903–4104 (2019).
Pye, H. O. T. et al. Effect of changes in climate and emissions on future sulfate-nitrate-ammonium aerosol levels in the United States. J. Geophys. Res. 114, 1097 (2009).
Schroder, J. C. et al. Sources and Secondary Production of Organic Aerosols in the Northeastern US during WINTER. J. Geophys. Res. D: Atmos. https://doi.org/10.1029/2018JD028475 (2018).
Nault, B. A. et al. Secondary organic aerosol production from local emissions dominates the organic aerosol budget over Seoul, South Korea, during KORUS-AQ. Atmos. Chem. Phys. 18, 17769–17800 (2018).
Liu, X. et al. Airborne measurements of western U.S. wildfire emissions: comparison with prescribed burning and air quality implications. J. Geophys. Res. D: Atmos. 122, 6108–6129 (2017).
Bahreini, R. et al. Gasoline emissions dominate over diesel in formation of secondary organic aerosol mass. Geophys. Res. Lett. 39, L06805 (2012).
Brock, C. A. et al. Aerosol size distributions during the Atmospheric Tomography Mission (ATom): methods, uncertainties, and data products. Atmos. Meas. Tech. 12, 3081–3099 (2019).
Aknan, A. NASA Airborne Science Data for Atmospheric Composition. TAbMEP2 POLARCAT Preliminary Assessment Reports http://www-air.larc.nasa.gov// (2015).
Crounse, J. D., McKinney, K. A., Kwan, A. J. & Wennberg, P. O. Measurement of gas-phase hydroperoxides by chemical ionization mass spectrometry. Anal. Chem. 78, 6726–6732 (2006).
Slusher, D. L., Huey, L. G., Tanner, D. J., Flocke, F. M. & Roberts, J. M. A thermal dissociation-chemical ionization mass spectrometry (TD-CIMS) technique for the simultaneous measurement of peroxyacyl nitrates and dinitrogen pentoxide. J. Geophys. Res.: Atmos. 109, D19315–D19315 (2004).
Lee, B. H. et al. An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: application to atmospheric inorganic and organic compounds. Environ. Sci. Technol. 48, 6309–6317 (2014).
Neuman, J. A. et al. Fast-response airborne in situ measurements of HNO3 during the Texas 2000 Air Quality Study. J. Geophys. Res. 107, 4436 (2002).
Talbot, R. W. et al. Large-scale distributions of tropospheric nitric, formic, and acetic acids over the western Pacific basin during wintertime. J. Geophys. Res.: Atmos. 102, 28303–28313 (1997).
Froyd, K. D. et al. A new method to quantify mineral dust and other aerosol species from aircraft platforms using single-particle mass spectrometry. Atmos. Meas. Tech. 12, 6209–6239 (2019).
Blake, N. J. et al. The seasonal evolution of NMHC’s and light alkyl nitrates at middle to northern latitudes during TOPSE. J. Geophys. Res. D: Atmos. 108, 8359 (2003).
Kupc, A., Williamson, C., Wagner, N. L., Richardson, M. & Brock, C. A. Modification, calibration, and performance of the Ultra-High Sensitivity Aerosol Spectrometer for particle size distribution and volatility measurements during the Atmospheric Tomography Mission (ATom) airborne campaign. Atmos. Meas. Tech. 11, 369–383 (2018).
Katich, J. M. et al. Strong contrast in remote black carbon aerosol loadings between the Atlantic and Pacific basins. J. Geophys. Res. D: Atmos. 123, 151 (2018).
Diskin, G. S., Podolske, J. R., Sachse, G. W. & Slate, T. A. In Open-path Airborne Tunable Diode Laser Hygrometer (ed. Fried, A.) vol. 4817 196 (International Society for Optics and Photonics, 2002).
Clegg, S. L., Brimblecombe, P. & Wexler, A. S. Thermodynamic model of the system H+−NH4+−SO42−−NO3−−H2O at tropospheric temperatures. J. Phys. Chem. A 102, 2137–2154 (1998).
Massucci, M., Clegg, S. L. & Brimblecombe, P. Equilibrium partial pressures, thermodynamic properties of aqueous and solid phases, and Cl2 production from aqueous HCl and HNO3 and their mixtures. J. Phys. Chem. A 103, 4209–4226 (1999).
Clegg, S. L. & Brimblecombe, P. Equilibrium partial pressures and mean activity and osmotic coefficients of 0–100% nitric acid as a function of temperature. J. Phys. Chem. 94, 5369–5380 (1990).
Wexler, A. S. & Clegg, S. L. Atmospheric aerosol models for systems including the ions H+, NH4+, Na+, SO42−, NO3−, Cl−, Br−, and H2O. J. Geophys. Res. 107, 4207 (2002).
Guo, H. et al. Fine particle pH and gas–particle phase partitioning of inorganic species in Pasadena, California, during the 2010 CalNex campaign. Atmos. Chem. Phys. 17, 5703–5719 (2017).
Jang, M., Sun, S., Winslow, R., Han, S. & Yu, Z. In situ aerosol acidity measurements using a UV–Visible micro-spectrometer and its application to the ambient air. Aerosol Sci. Technol. 54, 446–461 (2020).
Pitzer, K. S. & Simonson, J. M. Thermodynamics of multicomponent, miscible, ionic systems: theory and equations. J. Phys. Chem. 90, 3005–3009 (1986).
Clegg, S. L., Pitzer, K. S. & Brimblecombe, P. Thermodynamics of multicomponent, miscible, ionic solutions. Mixtures including unsymmetrical electrolytes. J. Phys. Chem. 96, 9470–9479 (1992).
Friese, E. & Ebel, A. Temperature dependent thermodynamic model of the system H+−NH4+−Na+−SO42−−NO3−−Cl−−H2O. J. Phys. Chem. A 114, 11595–11631 (2010).
Ibikunle, I. et al. Fine particle pH and sensitivity to NH3 and HNO3 over summertime South Korea during KORUS-AQ. Atmos. Chem. Phis. Discuss. In Review, (2020).
Song, S. et al. Fine-particle pH for Beijing winter haze as inferred from different thermodynamic equilibrium models. Atmos. Chem. Phys. 18, 7423–7438 (2018).
Gaston, C. J. et al. The cloud nucleating properties and mixing state of marine aerosols sampled along the southern California coast. Atmosphere 9, 52 (2018).
Gaston, C. J., Pratt, K. A., Qin, X. & Prather, K. A. Real-Time detection and mixing state of methanesulfonate in single particles at an inland urban location during a phytoplankton bloom. Environ. Sci. Technol. 44, 1566–1572 (2010).
Guo, H. et al. Fine-particle water and pH in the southeastern United States. Atmos. Chem. Phys. 15, 5211–5228 (2015).
Murray, B. J. Inhibition of ice crystallisation in highly viscous aqueous organic acid droplets. Atmos. Chem. Phys. 8, 5423–5433 (2008).
Bodsworth, A., Zobrist, B. & Bertram, A. K. Inhibition of efflorescence in mixed organic-inorganic particles at temperatures less than 250 K. Phys. Chem. Chem. Phys. 12, 12259–12266 (2010).
Koop, T., Luo, B., Tsias, A. & Peter, T. Water activity as the determinant for homogeneous ice nucleation in aqueous solutions. Nature 406, 611–614 (2000).
Murray, B. J. & Bertram, A. K. Inhibition of solute crystallisation in aqueous H+–NH4+–SO42−–H2O droplets. Phys. Chem. Chem. Phys. 10, 3287 (2008).
Tao, Y. & Murphy, J. G. The sensitivity of PM2.5 acidity to meteorological parameters and chemical composition changes: 10-year records from six Canadian monitoring sites. Atmos. Chem. Phys. 19, 9309–9320 (2019).
Warner, J. X. et al. Increased atmospheric ammonia over the world’s major agricultural areas detected from space. Geophys. Res. Lett. 44, 2875–2884 (2017).
Warner, J. X., Wei, Z., Larrabee Strow, L., Dickerson, R. R. & Nowak, J. B. The global tropospheric ammonia distribution as seen in the 13-year AIRS measurement record. Atmos. Chem. Phys. 16, 5467–5479 (2016).
Huffman, J. A. et al. Chemically-resolved aerosol volatility measurements from two megacity field studies. Atmos. Chem. Phys. 9, 7161–7182 (2009).
Fry, J. L. et al. Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011. Atmos. Chem. Phys. 13, 8585–8605 (2013).
Farmer, D. K. et al. Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry. Proc. Natl Acad. Sci. USA 107, 6670–6675 (2010).
Chen, Y. et al. Response of the aerodyne aerosol mass spectrometer to inorganic sulfates and organosulfur compounds: applications in field and laboratory measurements. Environ. Sci. Technol. 53, 5176–5186 (2019).
Heald, C. L. et al. Contrasting the direct radiative effect and direct radiative forcing of aerosols. Atmos. Chem. Phys. 14, 5513–5527 (2014).
Iacono, M. J. et al. Radiative forcing by long-lived greenhouse gases: calculations with the AER radiative transfer models. J. Geophys. Res. 113, 233 (2008).
Hudman, R. C. et al. Surface and lightning sources of nitrogen oxides over the United States: magnitudes, chemical evolution, and outflow. J. Geophys. Res. 112, D12S05 (2007).
Acknowledgements
This work was supported by NASA grants NNX15AH33A, NNX15AJ23G, 80NSSC19K0124, 80NSSC18K0630, NNX15AG61A, NSF grants 1360745, 1652688, and DOE (BER/ASRprogram) DE-SC0016559. We would like to acknowledge high-performance computing support from Cheyenne (doi:10.5065/D6RX99HX) provided by NCAR’s Computational and Information Systems Laboratory, sponsored by the National Sciences Foundation. We thank Charles Brock, Edward Dunlea, Karl Froyd, Daniel Murphy, and Joshua Schwarz for the use of their measurements.
Author information
Authors and Affiliations
Contributions
B.A.N., P.C.-J. and J.L.J. designed the experiment and wrote the paper. B.A.N., P.C.-J., D.A.D., J.C.S., H.M.A., R.B., D.R.B., J.D.C., M.J.C., P.F.D., J.E.D., G.S.D., W.H., J.M.K., M.J.K., A.K., F.D.L.-H., A.M.M., J.A.N., J.B.N., B.B.P., G.P.S., E.S., J.A.T., P.O.W., C.J.W. and J.L.J. collected and analyzed the campaign data. D.S.J., H.B., M.C., P.R.C., A.H., J.K.K., E.A.M., F.P., J.R.P. and K.T. ran the CTMs and provided the CTM output. B.A.N., P.C.-J., S.L.C. and J.L.J. ran and analyzed the E-AIM model and results for both campaign and CTM data. All authors reviewed the paper.
Corresponding authors
Ethics declarations
Competing interests
All authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nault, B.A., Campuzano-Jost, P., Day, D.A. et al. Chemical transport models often underestimate inorganic aerosol acidity in remote regions of the atmosphere. Commun Earth Environ 2, 93 (2021). https://doi.org/10.1038/s43247-021-00164-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-021-00164-0
This article is cited by
-
Thermodynamical framework for effective mitigation of high aerosol loading in the Indo-Gangetic Plain during winter
Scientific Reports (2023)
-
A central arctic extreme aerosol event triggered by a warm air-mass intrusion
Nature Communications (2022)
-
Synergistic HNO3–H2SO4–NH3 upper tropospheric particle formation
Nature (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
