
Abstract
Long-lived basaltic volcanic eruptions are a globally important source of environmentally reactive, volatile metal pollutant elements such as selenium, cadmium and lead. The 2018 eruption of Kīlauea, Hawai’i produced exceptionally high discharge of metal pollutants, and was an unprecedented opportunity to track them from vent to deposition. Here we show, through geochemical sampling of the plume that volatile metal pollutants were depleted in the plume up to 100 times faster than refractory species, such as magnesium and iron. We propose that this rapid wet deposition of complexes containing reactive and potentially toxic volatile metal pollutants may disproportionately impact localised areas close to the vent. We infer that the relationship between volatility and solubility is an important control on the atmospheric behaviour of elements. We suggest that assessment of hazards from volcanic emissions should account for heterogeneous plume depletion of metal pollutants.
Similar content being viewed by others

Introduction
More than 29 million people live within 10 km of active volcanoes, and around 800 million live within 100 km1 (2011 population data), where they may be exposed to environmental and air pollution hazards from gas- and particulate-rich emissions. Throughout this work we will refer to volcanic ‘emissions’, and unless otherwise stated our intended meaning is gas and particulate (including ash) emissions. Effusive basaltic eruptions can emit gas and particulate matter (PM) into the lower troposphere, elevating pollutant concentrations at ground level. Sustained emissions from such eruptions can last months (e.g. Holuhraun 2014–20152,3, Kīlauea 20184,5), to decades (e.g. Masaya 1993–present6, Kīlauea 1983–20187); to potentially hundreds or thousands of years during the eruption of flood basalts in Earth’s geological past8. Alongside major gas species (e.g. H2O, SO2, CO2 etc.) and ash9, basaltic volcanoes emit volatile trace metals and metalloids, many of which are collectively classified as ‘metal pollutants’ by environmental and health protection agencies (e.g. Cu, Zn, As, Pb, Se)10,11,12. Emission rates of metal pollutants during periods of intense degassing can be comparable to total anthropogenic fluxes from populous industrialised countries (Fig. 1). Metal pollutants can cause harm, in particular through chronic or frequent exposure to contaminated water and food, and/or by inhalation13,14,15.

Volcanic sources shown are basaltic volcanoes with ash-poor emissions. Note that metals are typically measured during a single period of (often elevated) activity and so represent a snapshot of activity rather than long-term averages. The legend should be read down each column and then across. Data sources: volcanoes2,6,22,23,24,26 (described in full in Supplementary Methods 1 and Supplementary Table 1); Europe84; China85; USA12. Metal pollutant emission rates from Kīlauea in 2018 are shown as two data bars: the darker red bar is calculated using an SO2 emission rate of 39 ± 11 kt day−1 26, as measured during our campaign, and the pale pink bar is calculated using the maximum SO2 emission rate during the eruption (~200 kt day−1 5) to demonstrate the potential maximum trace element emission rates during the 2018 eruption, assuming that X/SO2 ratios remained approximately constant. Error bars represent the full range of emission rates, where the upper and lower limits are calculated using maximum and minimum values (of X/SO2 ratios and SO2 emission rate), respectively. No error/uncertainty estimates are available for the anthropogenic emission data. Where bars are not visible, a measurement for this element is not available.
In addition to factors such as the tectonic setting, magma composition and the bulk gas emission rate of an eruption16, emission rates of volcanogenic metal pollutants depend strongly on element-specific volatility16,17,18 which is a measure of the extent to which an element partitions from the melt into the gas phase. The volatility of metals and metalloids during magmatic degassing has been studied at several volcanoes worldwide (e.g. 2,6,18,19,20,21,22,23,24) leading to wide recognition that volatile elements such as Se, As, Te and Re are emitted as gases from high-temperature volcanic vents. Volatility depends on a range of pre-eruptive parameters such as magma temperature, pressure, oxidation state, and the concentration of other volatiles16. It can be described using emanation coefficients: ε \(=\left({[X]}_{i}-{[X]}_{f}\right)/{[X]}_{i}.\) In this study, \({[X]}_{i}\) and \({[X]}_{f}\) are defined as the concentration of element \(X\) in the magma just prior to syn-eruptive degassing, and the final degassed concentration of element \(X\) in the magma, respectively (originally defined by Lambert et al.25). Emanation coefficients can be expressed as either a fraction or a percentage. Here, we define ‘volatile’ elements as those with ε ≥ 10−3 %; and ‘refractory’ as those with ε < 10−3 %, using data presented in Mason et al.26. As magmatic gases cool and oxidise after emission, these trace gases condense rapidly into solid or aqueous PM27 and may also adsorb to the surface of ash (e.g. 28).
Previous studies of the dispersal of volcanogenic metals have shown accumulation of metal pollutants in soils, rain, snow, and plants in the immediate vicinity (within ~10 km) of active volcanic vents19,29,30,31,32, and have detected their presence in airborne PM3,33 or adsorbed to ash34 10–1000 s of km downwind. However, detailed studies of downwind changes in concentrations of toxic and/or environmentally reactive metal pollutants (e.g. Se, As, Cd, and Pb) are rare. Compared to more abundant volcanogenic elements such as sulphur, much remains unknown regarding the details of the atmospheric dispersion, lifetimes, and deposition rates of metal pollutants, and also their impacts on air quality, the environment, and health35,36,37. This study seeks to examine the progressive changes in metal pollutant load during a large basaltic fissure eruption, the 2018 Lower East Rift Zone eruption of Kīlauea volcano, following the plume in the lower troposphere from the active vent to more than 200 km distance.
Kīlauea volcano on the Island of Hawai’i erupted near-continuously between 1983 and 20187, degrading air quality (operationally monitored pollutants: SO2 gas and PM2.5—PM ≤2.5 µm diameter) across much of the island38 (Fig. 2), and resulting in damage to agriculture and infrastructure by acid rain39. Negative health impacts have also been reported in the exposed communities40,41. The most significant and recent escalation of Kīlauea’s activity took place between May and August 2018 when the locus of lava effusion shifted to Leilani Estates, a populated area in the south-east of the island4 (Fig. 3). The shift in eruption location was accompanied by an increase in both eruptive rate4 and SO2 emission rate, which sometimes exceeded 200 kt day−1 5. Over the course of the 2018 eruptive episode an estimated 7.1–13.6 Mt SO2 was released5. The plume was transported in the lower troposphere due to the low altitude of the emission source (<200 m above sea level) and the predominantly effusive nature of the eruption. During the summer months, in which all of our atmospheric sampling was carried out, east-northeast trade winds dominate the wind field over the Island of Hawai’i. Orographic lifting of persistent east-northeast winds gives rise to high mean rainfall on Hawai’i’s windward mountain slopes (i.e. the ‘Hilo side’, Fig. 3), and low rainfall prevails in leeward lowlands (i.e. the ‘Kona side’, Fig. 3) and on the upper slopes of the highest mountains (Supplementary Fig. 1 from the Rainfall Atlas of Hawai’i)42.

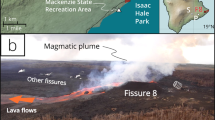
a Kīlauea 2018 eruption plume near-source (residential buildings for scale). The plume condenses rapidly after emission and the fallout of plume components begins close to the active vent, with a typical appearance of a fog-like cloud. Photo: Harry Durgin. b Volcanic plume of Kīlauea 2018 in the far-field (looking ~N from Mauna Loa towards Mauna Kea), manifesting as a ground-hugging haze locally known as ‘vog’. Photo: Evgenia Ilyinskaya/USGS. c The same view as in (b) showing a vog-free atmosphere in 2019, a year after Kīlauea stopped erupting. Photo: Evgenia Ilyinskaya/USGS.

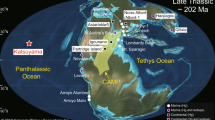
a Island of Hawai’i with the typical volcanic plume dispersion pattern and average plume age (hours). Fraction of sulphur (XS) in the gas phase is shown here as a proxy for the plume’s chemical maturity at variable distances from source. For Pāhala, the XS value should be viewed as an approximation due to saturation of the gas samples (Methods M2). The plume dispersion pattern and the map’s spatial domain are based on the operational volcanic air pollution forecast in Hawaii (VMAP) for 23 July 2018 and is representative of typical trade wind conditions which dominated during the eruption. The inset map in the top right shows the location of the Leilani sampling station close to Fissure 8. The red outline shows the final extent of lava flows at the end of the eruption in early August 2018 (adapted from Neal et al.4). The yellow area shows the typical plume transport direction from Fissure 8 during our campaign (indicative only). Sampling sites: F8 – Fissure 8 (lava flows shown in red); V – Volcano village, PH – Pāhala, OV – Ocean View, K – Kailua-Kona, ML – Mauna Loa Observatory, L – Leilani. Elevation of each sampling site is given in metres above sea level. Plume age calculations in Methods M6. b Comparison of PM concentrations (µg m−3) of sulphate, chloride, and metal pollutants during the eruption (2018) and during background conditions (2019). Error bars represent the standard deviation on n samples for each element (n given for each sampling location in grey bar at the bottom of the figure). The average sampling time (in hours) at each location is also given here (t(h)). At-source F8 concentrations of volatile elements (Se, Zn, Cu, As Cd, Pb) were 1–3 orders of magnitude higher than in the far-field (up to ten times higher for refractory Al and Fe) and are not shown on this figure but can be found in Mason et al.26,43. All data are from filter packs; sulphate and chloride presented here is that measured by ion chromatography (Methods M4).
We collected direct samples of Kīlauea’s gas and PM in July 2018 (Table 1, Methods M1–M5) to assess the dispersion of trace elements and quantify their depletion rates from the volcanic source into the far-field (up to ~240 km downwind). Near-source samples were collected at the main eruptive vent (‘Fissure 8’) at ground level, and just above it (<300 m above ground level using an Unoccupied Aircraft System (UAS)) and are presented in Mason et al.26,43. A time-series of far-field samples (typically 48–72 h time resolution) was collected in six locations on Hawai’i Island (Fig. 3). The eruptive activity ended in early August 2018 and the emissions dropped to some of the lowest levels since 19834. In June–July 2019, we repeated the field campaign to sample the local atmosphere unperturbed by the volcanic activity (Table 1). The field campaigns were designed to capture the composition of the volcanic plume and the background atmosphere at, or near ground level, where populations are exposed. The characteristics of the plume may have been different at higher altitudes in the atmosphere, due to vertical stratification of the plume in the troposphere; in particular, SO2 and other gas species have been observed to separate from PM44. Therefore, the plume sampled downwind is unlikely to represent the bulk plume composition integrated through the troposphere. Due to the high condensation temperatures of metal-pollutant bearing complexes, it is reasonable to assume that most metal pollutants will be transported as PM when detected at downwind stations45. Thus, except where indicated otherwise, all elements are discussed based on their measurements in PM (as opposed to in the gas phase), and where relevant in our discussion, we note the impact any potential plume stratification may have on our results.
We show the influence of volcanic emissions on the local composition of the atmosphere and identify significant variability in the atmospheric lifetimes of different volcanogenic species, including metal pollutants. We demonstrate that the dispersion patterns and environmental impacts of volcanogenic metal emissions do not necessarily match those of more abundant elements such as sulphur. We provide an improved understanding of the atmospheric lifetime of environmentally reactive trace elements in volcanic emissions from Kīlauea, and we believe our results can assist the generation of first-order dispersion maps and population exposure assessments for volcanogenic metal pollutants. These results could also be used to inform our understanding of tropospheric basaltic eruptions, and associated hazards, elsewhere.
Results and discussion
Local atmospheric change during the eruption
Under trade wind conditions, which were dominant during the 2018 eruption of Kīlauea (and the background sampling campaign in 2019), the plume was youngest and least chemically mature at the eruption site (‘Fissure 8’); and was oldest on the island’s west side (‘Kona side’), where local wind circulation returned the plume onshore (Fig. 3, Methods M6). The time-dependent chemical maturity of the plume is represented in Fig. 3 as mass fraction of sulphur (XS) in the gas phase (as measured in filter packs, see Methods M2). More than 90% of sulphur degassed is typically emitted as sulphur dioxide gas (SO2)46,47 and converts to sulphate PM (SO42−, typically speciated as sulphuric acid and/or a sulphate salt) at a rate that depends on multiple parameters including relative humidity, ambient temperature, solar flux, and interactions with other particulates48. The S conversion rate (also sometimes called an SO2 loss rate) is important for estimating downwind abundances of SO2 gas and SO42− PM for air quality forecasts, hazard assessments, and the effects of volcanic eruptions on the atmosphere’s radiative balance. This conversion rate in volcanic plumes has been found to be highly variable49 and spans at least three orders of magnitude for Kīlauea50. The calculation and comparison of S conversion rates in volcanic plumes is subject to significant uncertainties and caveats, and a full discussion of these is beyond the scope of this work. Therefore, here we provide only a qualitative description of the changes in S speciation we observed along the plume path (we include a quantification of the S conversion rate, with full uncertainties and caveats explained, in Supplementary Methods 2). In the ~3 h of plume transport between Fissure 8 and Volcano village we would expect any SO2 loss to be minimal51. However, at Volcano village, while concentrations of SO2 gas and sulphate are both much lower than at-source (assumed to be caused by dilution of the plume by background atmosphere), SO2 gas concentrations have decreased much more than sulphate (80% versus 10% of source concentration, respectively). This may be due to vertical stratification of S species in the plume44—if SO2 is transported at higher altitudes than sulphate, we might expect SO2 to be less concentrated at ground-level sampling stations. Beyond Volcano village SO2 concentrations continue to decrease consistently, but from Pāhala onwards, sulphate concentrations begin to increase as processes of sulphur conversion increase the proportion of S present as sulphate in the plume (Fig. 3).
The importance of volcanic emissions as a source of both sulphur and metal pollutants in the Hawaiian atmosphere (in gas and PM) was recognised during the 1983–2018 activity of Kīlauea19,38,40,47. We found that metal pollutant concentrations (Supplementary Data 1) were typically 1–3 orders of magnitude higher in plume-impacted areas in 2018 compared to the volcanically-unperturbed background atmosphere in 2019 (Supplementary Data 2; Fig. 3). An important exception is particulate chlorine (measured as Cl-), which did not show a significant difference between 2018 and 2019 in the far-field areas. This is attributed to its high concentrations (as marine aerosol) in the background Hawaiian atmosphere (sea-spray sample reported in Supplementary Data 2), which may overwhelm the relatively small amounts of volcanic Cl degassing at Kīlauea52.
The concentration of metal pollutants decreased in the plume-impacted areas with distance from source (Fig. 3), from Volcano village (~40 km downwind) through to Kailua-Kona (~240 km downwind). However, while Pāhala, Ocean View and Kailua-Kona were consistently impacted by the grounding volcanic plume under trade wind conditions, Volcano village received only occasional exposure to the plume (Supplementary Fig. 2 shows a time-series of SO2 concentrations measured at the Volcano village sampling site, supported by back-trajectory calculations Methods M6). These plume advection events at Volcano village typically lasted for a few hours at a time. Due to this, in only one sample was the PM composition significantly above the 2019 background, and therefore we consider only this sample in our analysis (Methods M7, Supplementary Data 1, Supplementary Data 2). Over the course of the 2018 eruption, it is possible that the communities in Pāhala, Ocean View and Kailua-Kona were exposed to higher cumulative concentrations of metal pollutants than Volcano village, due to more sustained exposure.
Volcanic emissions increased the concentrations of certain metal pollutants (e.g. Se, As, Fe) in the far-field populated areas on Hawai’i (~1000–12,000 residents per community)38 to levels comparable to populous cities (~2–8 million per city53) on the mainland USA (reported in PM2.5 size fraction54; Fig. 4). There are some important differences in the relative concentrations of pollutants in the volcanogenic and urban PM, which likely reflect the significant differences in their typical formation mechanisms, i.e. high-temperature magmatic gas condensation (volcanic)55, versus urban anthropogenic activities such as fuel combustion56. For example, volcanogenic PM is particularly enriched in Se compared to urban PM, in agreement with the high volatility and volcanic emission rate of this pollutant (Fig. 1). Concentrations of As, Fe, Al, Mn, and Cu were at comparable levels between Hawai’i and some US cities. Zn and Pb are lower in Hawai’i, as these elements are typically dominated by vehicle fuel emissions in urban areas56,57.

Average mass concentrations (µg m−3) of selected metal pollutants in PM2.5 (particulate matter with a diameter <2.5 µm) in populated areas on the Island of Hawai’i during the 2018 eruption, during a period of lower volcanic emissions 2002–2005 (Zn, Cu, Pb, Fe only)40, and in populous cities on mainland USA 2004–200554. Elements shown here are those reported from the cities54, and those above detection limits on Hawai’i in 2018. Cadmium is not reported in the cities by the same network54, but it is likely that PM dominated by a volcanic source will be relatively high in this pollutant compared to anthropogenic pollution (Fig. 1). Our 2018 Pāhala, Ocean View and Kailua-Kona data shown here are all cascade impactor totals to allow closer comparison with the other data sets, which also use impactors.
A subset of airborne metal pollutants was measured on Hawai’i in 2002–200540 in similar locations to our study (sampled in 2-week time averages)—Ocean View and Pāhala areas (reported together in the original study) and Kailua-Kona (Fig. 4). During that period, Kīlauea’s SO2 emission rate was 1–2 orders of magnitude lower than in 2018 (average 2 kt day−1 40 versus 50–200 kt day−1 4,5). As shown by Mason et al.26, the metal pollutant/SO2 ratio in Kīlauea’s emissions is relatively consistent between eruption periods, thus we infer that the metal emission rate would have been 1–2 orders of magnitude lower in 2002–2005 than in 2018. While the limited number of reported elements from 2002 to 2005 precludes a detailed analysis, we note that the airborne pollutant concentrations are perhaps higher than expected compared to 2018 based on the difference in metal emission rates between the two time periods. Several factors could contribute to this. Firstly, the non-volcanic atmospheric pollution levels may have changed. This is particularly plausible for Pb, which decreased in atmospheric concentrations in the USA by 98% between 1980 and 2014, primarily due to a reduction in the use of leaded gasoline12,57. Secondly, the location of the active volcanic vent has changed between the two time periods, with the main emission source moving ~15 km eastwards in 2018, i.e. further away from these sampling sites, as well as to lower elevation (by ~500 m). However, this increase is relatively small compared to the average plume path distance to the three sites (90, 160 and 240 km, respectively, in 2018). Thus, it is unlikely to fully account for the relatively small difference in the observed pollutant concentrations between 2002–2005 and 2018. Thirdly, it is possible that the atmospheric lifetime of volcanogenic pollutants does not scale linearly with their emission rate and distance from source, i.e. there is a mechanism that depletes some elements from volcanic plumes faster than others—we explore the potential mechanisms for this in the following sections.
We also present the size-distributions of volcanogenic PM, which are strongly related to element-specific volatility and mechanisms of particle formation (Fig. 5)58. Size-distributions are important when considering atmospheric lifetimes, as particle diameter has a strong effect on dry sedimentation/deposition rates. However, as well as sedimentation in the absence of precipitation (dry deposition), PM can be removed from volcanic plumes through scavenging by hydrometeors, e.g. cloud droplets and rain (wet deposition), where PM composition and solubility plays an important role55. The relative importance of these two modes of deposition depends on a combination of in-plume and ambient factors, especially atmospheric water availability59,60.

(No size-resolved data from Volcano village available during plume advection events). Elements shown on the figure are those above detection limits in size-resolved samples at Fissure 826. The elements’ mass concentration in each size fraction (with particle diameter denoted as D) is shown as % of total mass concentration measured in the bulk particulate matter (PM). Water-solubility (blue circles) is shown as % fraction of the bulk PM, and is calculated as outlined in Methods M4. Weighted ash fractions (WAFs; red squares) are shown as percentages and calculated as outlined in Mason et al.26. Error bars for WAF and water solubility are standard deviations of multiple samples at each location. Sulphur and chlorine were measured only in water-soluble sample extractions so no water-solubility data is available for them. Sulphur (PM) presented here is that measured by ion chromatography (Methods M4). The size-resolved data is not corrected for 2019 background concentrations.
At-source (Fissure 8), volatile species including sulphur and chlorine (measured as SO42- and Cl- respectively in PM) are found almost exclusively in the finest resolved PM size fraction (diameter, D < 0.25 µm), while refractory elements are found in coarser size fractions (D > 2.5 µm)26. The contrasting size distributions of volatile versus refractory elements is generally preserved as the plume is advected away from the source, although both refractory and volatile elements become progressively more prevalent in the coarsest size fractions. Tb and Ho were found in the finest size fraction at Kailua-Kona but were above detection limits in only one sample, so we refrain from making conclusions about this apparent change. Chlorine PM concentrations and dominant size bin increase rapidly with distance from the active vent (Fig. 5, Fig. 6). This is likely caused by increasing downwind influence of Cl-bearing aerosol of marine origin, abundant in the background Hawaiian lower troposphere as evidenced by samples collected in 2019 (Fig. 3 and Supplementary Data 2).

The average concentration at each sampling site (six samples each for Pāhala, Ocean View and Kailua-Kona; one sample only for Volcano village, FP_07_01, during advection event) is normalised to the at-source F8 concentration (UAS sample26) for ease of visual comparison. Elements are coloured by their emanation coefficient, as calculated in Mason et al.26, and are joined between different sampling stations by lines of the same colour. (x) and (x’) are descriptive (qualitative) lines, to highlight the dramatically different depletion rates between Fissure 8 and Volcano village, versus Volcano village and the other further downwind stations. (x) denotes a rapid decrease and large fractionation in element concentration between F8 and Volcano. (x’) denotes a slower decrease in element concentration and negligible fractionation from Volcano to Kailua-Kona (this is described in more detail in the main text and explored further in Fig. 7). The elements shown are those with concentrations above detection limits and above atmospheric background levels at F8 and at least 3 far-field sampling sites. Sulphur and chlorine are the only elements to increase in PM concentration with distance between Volcano village and Kailua-Kona through sulphur gas-to-PM conversion, and likely addition from downwind Cl-bearing sources (e.g. sea-spray), respectively. Only rare earth elements (REEs) La and Ce are shown here for clarity, however the other REEs plot in the same area as other refractory (purple) elements. Sulphate and chloride (PM) presented here is that measured by ion chromatography (Methods M4).
We use the measured concentrations of water- versus acid-based filter extractions (Methods M5) to determine the at-source and far-field water-solubility of refractory and volatile elements. At-source, the volatile elements are more water-soluble than refractory elements (Fig. 5), with the exception of Bi. As shown by a previous study on Kīlauea’s emissions in 200819, Bi becomes water-soluble when extracted using weak environmental acids. Therefore, it is likely that Bi is more soluble under natural conditions in the volcanic plume than is captured by our extraction method. In the far-field, the general difference between volatile (higher water solubility) and refractory elements (lower water solubility) is preserved. However, the water-solubility of refractory elements increases in the aged plume as seen at the Ocean View and Kailua-Kona sites. We attribute this to an increase in sulphuric acid concentration through sulphur gas-to-particle conversion as the plume matures chemically (Methods M5 and Fig. 6). Particles are likely to be progressively leached by the acid, resulting in a water-soluble surface layer of elements61. We note that this increase in refractory element solubility does not appear in data at Pāhala—we suggest that this may be due to the relatively dry conditions encountered between between Volcano village and Pāhala (e.g. the Ka‘ū Desert) which may limit production of leaching acids. However, further work would be required to assess this hypothesis comprehensively.
The contrasting size distribution and water-solubility patterns of refractory versus volatile elements may be explained by the differences in their PM formation mechanisms. In low-explosivity volcanic emissions, refractory elements predominantly derive from small silicate tephra particles (~1–100 µm), which become airborne through mechanical fragmentation of magma, and subsequent quenching, near the magma/atmosphere interface62,63. Volatile elements associated with volcanic gases form PM by gas-to-particle conversion, resulting in fine particle sizes62,63. This is supported by an independent observation reported in Mason et al.26 at Kīlauea in 2018: nearly 100% of the mass of refractory elements was found to be associated with silicate ash (‘weighted ash fraction’), compared with 0.001–5% for volatile elements. The importance of PM formation mechanisms and water solubility for the in-plume lifetimes of metal pollutants is explored further in the following sections.
Potential sources of variability in local atmospheric compositions
The broad spatial coverage of our sampling locations revealed markedly different rates of compositional change along different sections of the plume path:
-
Between the emission source (Fissure 8) and the first downwind sampling site (Volcano village, average plume path distance and age of 40 km and 3 h, respectively), the concentration of all elements decreased by around one order of magnitude or more. We denote this rapid initial change as x in Fig. 6 and Fig. 7. We note than during stage x, while measured refractory element concentrations in the local atmosphere decreased to 6–18% of their concentration at-source, volatile element concentrations decreased to an even greater extent – to 0.1–1.6% of their concentration at-source.
Fig. 7: Mixing models of volcanic and background PM concentrations of elements to reproduce concentrations measured during the 2018 eruption. 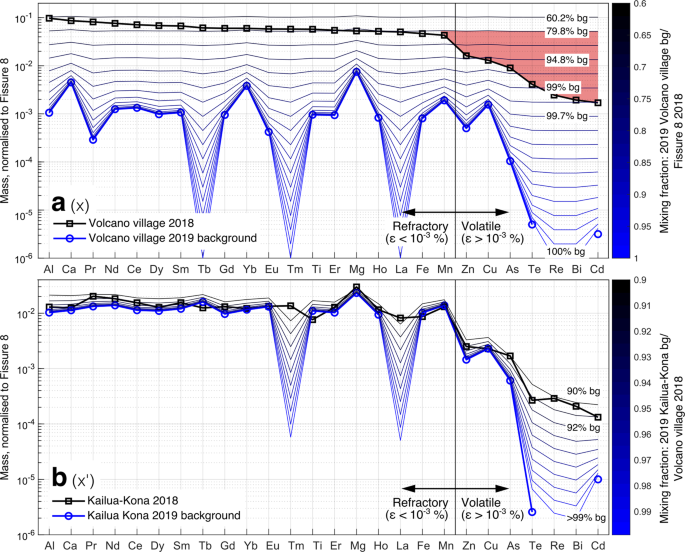
a To reproduce the plume composition measured in Volcano village in 2018 (solid black line with open black squares), we mixed the plume composition at-source (Fissure 8) with Volcano village 2019 background atmosphere (‘bg’) to create the blue to black mixing lines. The mismatch between the model and the observed concentrations is highlighted (pale red). Concentrations of Tb, Tm, La were below detection limits in 2019 at Volcano village and assumed to be 0 in the mixing model. b To reproduce the plume composition measured in Kailua-Kona in 2018 (solid black line with open black squares), we mixed the plume composition in Volcano village in 2018 with the Kailua-Kona 2019 background atmosphere to create the blue to black mixing lines. Concentrations of Tm and La were below detection limits in 2019 at Kailua-Kona and assumed to be 0 in the mixing model. Elements in both figures are arranged in order of increased depletion rate at Volcano. Pb, Se and In were below detection limits in 2018 at Volcano village and are excluded from the mixing model. Re and Bi were not measured in 2019. Full compositional data for 2018 and 2019 in43 and Supplementary Data 1 and Supplementary Data 2, respectively.
-
Between Volcano village and the three subsequent downwind sites (plume path distance: 90, 160 and 240 km, and plume age: 6, 11 and 19 h, respectively), the concentrations of all elements decreased more slowly (~50% decrease over an equivalent distance to x). We denote this slower change as x’ in Fig. 6 and Fig. 7. Over this distance, refractory and volatile element concentrations in the atmosphere decrease at comparable rates (as denoted by rate constant k in Fig. 8).
Fig. 8: Depletion rate of element PM concentrations in Kīlauea’s plume, emanation coefficients (ε). 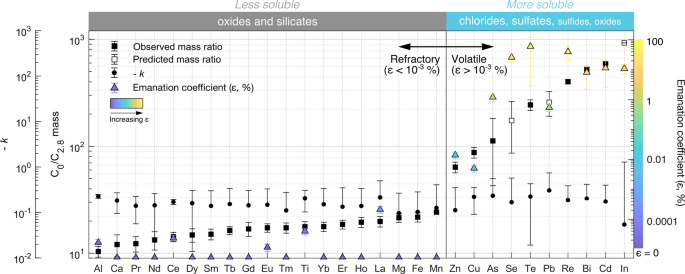
The elements shown are those with concentrations above detection limits and above atmospheric background levels at-source and at least 3 far-field sampling sites, and elements are coloured by emanation coefficient. The rapid initial depletion rate (x on Figs. 6 and 7) is quantified using the ratio of the mass concentration at-source (plume age 0 h, C0) and the first downwind sampling site (C2.8, plume age 2.8 h). Error bars for C0/C2.8 are propagated errors (Supplementary Data 3).The initial depletion rate of volatile elements (ε ≥ 10−3 %) is 3–100 times faster than for refractory elements (ε < 10−3). The slower depletion rate (x’ on Figs. 6 and 7) between the subsequent downwind sampling sites (Volcano village to Kailua-Kona, plume age 2.8–19 h) is expressed as the exponential depletion rate constant k and is similar for all elements. Error bars for k are the 95% confidence bounds of the exponential fit function. C2.8 was below detection limits for Pb, Se and In due to a relatively low sampling volume and is predicted by extrapolating the exponential curve fitted through Pāhala, Ocean View and Kailua-Kona. Depletion rate calculations are described in Methods M7; and ε values, errors and calculation methods can be found in Mason et al.26. The broad typical speciation of refractory versus volatile elements is labelled at the top of the figure18,20,21,67,68,69.


We next consider possible atmospheric processes both at-source and downwind that could provide mechanisms for these observed differences along the plume path.
Firstly, any variability of the composition of gas and particulate emissions at-source could affect the compositions we measure at downwind stations. However, the consistent SO2 emission rates during July–August 20185 suggest that trace metal emission rates would also have remained relatively stable during this time period, and the long sampling periods we used at downwind stations (~48–72 h) are likely to have averaged-out any short-term source variability.
A major process occurring from the point of emission onwards is the dilution of volcanic emissions by background air, which begins near-instantaneously in all types of eruptions64,65. The background atmosphere contains particulates and gases of non-volcanic sources, at much lower concentrations and with different relative abundances of refractory and volatile elements to that found in Kīlauea’s volcanic plume (Supplementary Data 2). A high level of dilution by the background atmosphere may therefore fractionate element abundances downwind relative to the volcanic source. Acting in isolation, this mechanism would yield a consistent background dilution factor for all elements. Using a mixing model of the at-source volcanic emission (2018 data26) and the local volcanic-free background (2019 data, this study) we investigated the predicted decrease in element concentrations over two different plume path distances (x and x’ on Fig. 6; mixing model in Fig. 7). The model considers elements found exclusively in the PM phase at ambient atmospheric conditions to exclude the effects of gas-to-particle conversion processes (i.e. S and Cl were not included in the model, Fig. 7).
-
We first consider the initial, rapid decrease (x in Fig. 6) observed between the emission source and the first far-field sampling site, Volcano village (plume path distance 0–40 km from source, ~3 h of atmospheric transport). For this we model a mixture of the at-source emission composition at Fissure 8 in 2018, and the 2019 Volcano village background atmospheric composition. The composition of the plume measured at Volcano village could not be reproduced by a consistent dilution factor for all elements (Fig. 7; mismatch between model and observed concentrations highlighted in red). The refractory elements required dilution by 60% background, while the volatile elements required dilution from 95 to >99% background.
-
Second, we consider the observed slower decrease between Volcano village and Kailua-Kona (x’ on Fig. 6), which is the furthest far-field site (plume path distance 40–240 km from source, ~19 h of atmospheric transport, Fig. 6). For this, we model a mixture of the atmospheric composition measured at Volcano village in 2018 with the 2019 background atmospheric composition at Kailua-Kona. The complete composition of the plume in Kailua-Kona could be reproduced by a comparable background dilution factor for all elements (90–92%; Fig. 7). This suggests that mixing with background atmosphere is likely the dominant mechanism for the decrease in element concentrations from Volcano village onwards.
The less substantial depletion of refractory elements compared to the volatile elements between Fissure 8 and Volcano village could also have been caused by the plume mixing with other source(s) of these elements in the initial 3 h of plume transport. For all the data presented here, unless otherwise indicated, the contribution from sources not related to the 2018 eruptive activity was quantified and subtracted from the 2018 samples using samples collected in the same locations during a non-eruptive period in 2019 (Supplementary Data 1, Supplementary Data 2). Therefore, background sources are unlikely to be able to explain the trend of the downwind fractionation. An additional atmospheric source in 2018, rich in refractory elements, could have been ash26 from Kīlauea’s summit crater, which produced periodic small explosions concurrent with the Fissure 8 eruption66. However, based on the following lines of evidence we concluded that addition of ash was unlikely to be contributing to concentrations measured downwind. Back-trajectory plume dispersion simulations showed a low likelihood of ash contribution from Kīlauea’s summit mixing into the sample collected in Volcano village as the summit was upwind of Fissure 8 and Volcano village during this period, which was also typical for most of the 2018 eruption (Methods M6, Supplementary Data 4). The simulations also showed that samples collected at subsequent sampling sites, Pāhala, Ocean View and Kailua-Kona may have received ash from Kīlauea’s summit. However, there is no significant enrichment in refractory and volatile elements at these stations compared to Volcano village, suggesting that any contribution of summit ash was also relatively small. Proximal to the source and further downwind, ash-volatile interactions could affect the volcanic contribution to local atmospheric compositions that we measured. Adsorption of volatile-bearing particles or gases onto the surfaces of ash particles has been reported in plumes from explosive eruptions (e.g. 28). However, we expect these processes to be limited in the Fissure 8 plume given the low abundance of ash produced. This was evidenced by visual observations of the plume during our campaign, as well as optical observations and modelling of the 2018 plume, which suggest a low to moderate contribution from volcanic ash5. Further, this mechanism, in which refractory and volatile element-bearing particles/gases are coupled, cannot not provide an explanation for the 1–3 orders of magnitude faster depletion rate of volatile elements versus refractory elements between Fissure 8 and Volcano village (Fig. 6).
We therefore conclude that there is another, previously unrecognised mechanism operating in the plume that preferentially depletes volatile elements within the first 3 h of plume transport after emission (Fig. 6, Fig. 8). We propose that element-specific volatility, and the speciation and therefore solubility of volcanic particulates plays an important role in determining depletion rates of refractory versus volatile elements in Kīlauea’s volcanic plume, and we discuss this mechanism in the following sections.
Rapid early deposition of volatile metal pollutants
Element-specific volatility is known to be an important control on particle-phase speciation at-source. This has been shown for a range of volcanic plumes18,19,20,21,67,68, including Kīlauea during an earlier eruption period in 200819. When volcanic emissions are at magmatic, or near-magmatic temperatures (~1000 °C), thermodynamic data shows that volatile elements are largely present in the gas phase (e.g. 6). However, during cooling, oxidation and quenching of the magmatic gases as they mix with the local atmosphere, trace gases condense rapidly into non-silicate particulates in the plume45. Particle-phase speciation is controlled by the composition, temperature and oxygen fugacity conditions of the magmatic gas-air mixture18,20,21,67,68,69, and speciation determines the water-solubility of particulates19. Refractory elements are found predominantly in small silicate ash particles62,63; or oxide compounds formed by gas-to-particle conversion at high temperatures18,20,21,67,68,69. During the initial cooling of magmatic gases as they mix with background air, but before the oxygen fugacity (fO2) increases to the extent that reduced phases are not able to form69, gas-to-particle conversion of chalcophile (i.e. ‘sulphur-loving’) volatile element-bearing gases leads to the condensation of sulphide phases18,20,21,67,68,69. Progressive mixing with background air causes a further drop in temperature and a rise in fO2, forming higher proportions of volatile element-bearing oxide and halide salt phases (Fig. 8)18,20,21,67,68,69. Particles can undergo subsequent speciation change at ambient or near-ambient temperatures, e.g. with sulphuric acid displacing halides to form a higher proportion of volatile element-bearing sulphate salts70. The difference in particle-phase speciation of refractory versus volatile elements is supported by the contrasting size distributions of the two groups (Fig. 5).
Elements complexing with chloride, sulphate and sulfide are more readily soluble in water than oxides, and silicates19,61,71 (Fig. 5), which we propose explains the observed fractionation in the plume lifetimes of volatile and refractory elements, respectively. The water-solubility of particles is an important factor for controlling in-cloud wet scavenging rates60,72 and wet deposition rates73,74 in and from volcanic plumes, respectively. Soluble particles in accumulation (0.1–1.0 µm diameter) and coarse modes (>1.0 µm diameter) are scavenged more efficiently by liquid cloud droplets than insoluble particles in the same size fractions60,72. We suggest that the observed lack of preferential depletion of volatile over refractory elements between Ocean View and Kailua-Kona (Fig. 8) may be explained in part by the progressively smaller differences in water-solubility between these groups of elements (Fig. 5), caused by changes in speciation, and in turn leading to more comparable rates of in-cloud scavenging and wet deposition in the plume further downwind. We note that this change in water solubility is not evident until after Pāhala and suggest that this may be due to dryer atmospheric conditions between Volcano village and Pāhala, as discussed above. We also note that within the volatile elements, there is at least an order of magnitude variability in their depletion rate between Fissure 8 and Volcano village. We propose that this could also relate to differences in the speciation, and therefore solubility of complexes bearing these elements in Kīlauea’s volcanic plume downwind from the source. Understanding these mechanisms would require modelling of the complex low temperature chemistry within the plume during downwind transport, which is beyond the scope of the current study.
Tropospheric volcanic plumes can become an environment with efficient in-cloud scavenging and wet deposition due to the high abundance of magmatic water vapour (~80% by gas volume in Kīlauea’s rift zone eruptions75) and ~100% relative humidity, observed as a rapid condensation of droplets after emission (Fig. 2). In 2018, there was anecdotal evidence from communities in the Leilani Estates and surrounding areas, that rainfall was higher than in previous years, as shown in community rainfall data in Supplementary Fig. 3 (CoCoRaHS data). In addition, the high relative humidity and high dew point, the atmospheric temperature below which water condenses, of the background atmosphere in Hawai’i (average ~73%, depending on elevation and time of day, and ~17 °C, respectively; from NCEI-ASOS data) will enhance these processes for Kīlauea’s plumes. Further, due to orographic lifting of the persistent east-northeast trade winds, some of the highest mean rainfall typically occurs over the ‘Hilo side’ of the Island of Hawai’i, including the region between and including Leilani Estates and Volcano village (up to around 6000 mm per year; Supplementary Fig. 1 from the Rainfall Atlas of Hawai’i)42. In contrast, substantially less rain falls over the plume path between Volcano village and the stations further downwind (up to around 3000 mm per year over Pāhala, Ocean View and Kailua-Kona, Supplementary Fig. 1).
The combination of the high-water content of the volcanic gas emissions, the high relative humidity of the background atmosphere in Hawai’i, and these orographic rain effects, creates an ideal environment for high degrees of wet deposition along the plume path between Fissure 8 and Volcano village. Differences in local tropospheric temperature, humidity and precipitation may lead to substantial changes in the rate at which metal pollutants are deposited from volcanic plumes. This should be further considered when simulating the dispersion of volcanogenic pollutants at different volcanoes, especially in low- versus high-latitude volcanic environments (e.g. Hawai’i versus Iceland). It should also be considered when assessing hazards from eruptions at different locations on the Island of Hawai’i itself—plumes from Kīlauea summit eruptions may not be subject to the same extent of wet deposition due to the natural pattern of rainfall over the island (Supplementary Fig. 1).
Implications: exposure to metal pollutants in populations and the environment
Our results have important implications for environmental and health impacts of volcanic emissions. Eight of the 12 volatile, rapidly-deposited elements (Zn, Cu, As, Pb, Se, Cl, Cd, S) are classified by environmental agencies as metal pollutants, in contrast to only 2–3 out of 20 detected refractory elements (Fe, Mn and by some agencies, Al)11,12. Therefore, the preferential rapid deposition of volatile elements places disproportionate environmental pressures on the populated areas in the vicinity of the active vent and, in turn, reduces the impacts on far-field communities. The preferential deposition mechanism may explain why the metal pollutant concentrations in the far-field communities of Pāhala, Ocean View and Kailua-Kona were not orders of magnitude higher in 2018 compared to 2002–2005 (Fig. 4) despite the relative increase in their emission rate. The high water-solubility of volatile metal pollutant-bearing particles, such as those from the 2018 eruption of Kīlauea, (Fig. 9) makes them labile in the environment, increasing their potential impacts and toxicity. Volatile metals and metalloids in natural volcanic emissions display comparable water-solubility to anthropogenic emissions, such as those associated with urban environments and fuel combustion (Fig. 9). In contrast, more refractory elements in volcanic emissions, such as Mg and Fe, display water-solubility intermediate between anthropogenic sources and desert dust. Volcanic emissions may be an underestimated source of volatile metal pollutants in environmental and population exposure assessments, not only due to the high emission rates over long periods of time (Fig. 1), but also due to the high environmental availability of important pollutants (Fig. 5) and the effects of local atmospheric conditions and rainfall on their deposition.

Water-solubility (fraction of total measured mass) of selected refractory and volatile volcanogenic elements compared to their water-solubility in particulates from other natural and anthropogenic sources. The elements shown here were selected based on data availability for other PM sources56.
Further, metal and metalloid pollutants (e.g. As, Cd, Pb and Se) deplete faster from Kīlauea’s volcanic plume than more widely studied species such as sulphur (Fig. 6). We therefore suggest that, in future studies of the atmospheric dispersal of volcanic gas and particulate emissions, both at Kīlauea and at other volcanoes worldwide, metal and metalloid pollutants should be studied independently. These metal/metalloid pollutants may potentially cause environmental build-up and chronic exposure in different areas to those identified as polluted based other commonly monitored volcanic pollutants.
There are tens, and potentially hundreds of actively degassing basaltic volcanoes worldwide, some with communities living very close to their degassing vents (e.g. ~2 km at Masaya in Nicaragua, one of the highest volcanic emitters of metal pollutants; Fig. 1). In recent years, links have been made between exposure to volcanogenic metal pollutants and a high incidence of certain diseases in communities living around volcanoes, such as thyroid cancer76 and multiple sclerosis77. The potential environmental and health impacts of rapid near-vent deposition of metal pollutants should be investigated in more detail, both as a matter of urgency and as part of routine monitoring, in areas surrounding persistently degassing volcanoes, particularly in communities that rely on rain catchment systems for household water supplies, as is the case on many volcanic islands like Hawai’i.
Methods
M1. Field campaign design
During the eruption campaign in July 2018, we sampled gas and PM near-source (as reported in Mason et al.26) in the plume of the main active vent of Kīlauea (‘Fissure 8’); and at a network of far-field stations (Table 1 and Fig. 3). The sampling was repeated in June–July 2019 at the same sites, using the same instrumentation allowing characterisation of the background atmospheric composition and non-volcanic pollution sources, during a period of no eruptive activity and negligible volcanic emissions. In 2019, we also collected a sample at the ocean shore of Mackenzie State Recreation Area (19.438703; −154.863279; 10 m a.s.l.), as sea-spray is one of the key non-volcanic PM sources on the Island of Hawai’i.
Near-source sampling set-up and instrumentation is described in Mason et al.26 The far-field sampling locations (latitudes and longitudes in Table 1) were co-located with:
-
Hawaii State Department of Health air quality stations in Kailua-Kona, Ocean View, and Pāhala;
-
a pre-existing SO2 monitoring station in Volcano village owned by University of Hawaii;
-
an eruption-response Environment Protection Agency SO2 monitoring station in a non-evacuated part of Leilani Estates (Alapai Street South); and
-
at Mauna Loa NOAA observatory.
The stations were visited, and sample collectors changed every 2–3 days in 2018 to give ~48–72 h time resolution. In Volcano village, in addition to the 48 h samples, 2 short-interval samples (1 and 4 h duration, respectively) were collected when the volcanic plume was being advected directly to this location (as detected by the SO2 sensor at this location; Table 1, Supplementary Fig. 2). The populated areas selected as sampling sites are small communities (1000–12,000 people) with low population densities of 20–100 residents km−2 38 and assumed relatively low levels of anthropogenic pollution.
During the non-eruptive period in 2019, the atmospheric concentrations of aerosol and gas were significantly lower (as indicated by the Hawaii State Department of Health air quality monitoring of PM2.5 and SO2). The sampling time was increased to 4 days in order to maximise the sampled mass and the chance of exceeding the analytical detection limits for trace elements. At Mauna Loa, the sampling time was increased to 9 days due to the very low local concentrations78 (Table 1).
M2. Filter packs
Filter pack samplers were used to collect simultaneous samples of gas and bulk PM. We followed a well-established technique79 previously used in multiple volcanological studies (for example3,19). The filter packs comprised of one particle filter followed by 2–4 base-treated gas filters in an all-Teflon cartridge.
Gas filters (Whatman Quantitative Filter Papers, Ashless, Grade 41, 55 mm diameter) were pre-soaked with a 0.1 M K2CO3 (+glycerol) and dried ~1 week before use in the field. This base treatment of the gas filters captures acidic gases (e.g. SO2, HF and HCl). For some samples, the last gas filter in the filter pack contained >10% of the total captured gas concentration—this is evidence that the gas filters had become saturated. The saturated samples are identified in Supplementary Data 5. Gas concentrations in the saturated samples represented a minimum value and should not be used for calculating gas-to-PM ratios. Note that concentrations in the PM phase are not affected (cannot be saturated) and therefore these samples are still valid for analysing the PM phase. The SO2 concentrations in non-saturated samples generally agreed well (<10% difference) with that measured by the co-located Department of Health reference-grade air quality instruments.
The particle filter collects bulk (non size-resolved) PM. The particle filter used was Whatman PTFE 47 mm diameter, pore size 0.8 µm. The filters were pre-washed with UPA grade nitric acid before use on the 2019 campaign. The filters were not pre-washed before the 2018 campaign due to a rapid mobilisation. Field and lab filter blanks were used to quantify the level of contamination due to the absence of acid wash. The contamination was found to be negligible in most samples due to the high sampled concentrations in the eruption-affected atmosphere in 2018. Air flow through the filter pack was generated using an external 12 V pump (Charles Austin Capex) running at ~20 l min−1. The flow rate was measured at the start and end of each sampling period. The uncertainty introduced by variations in the flow rate, and by the accuracy of the flow meter are 10%. Immediately after sampling, the filter pack was closed by Parafilm to prevent particle loss and contamination. Filters were later transferred into individual polypropylene bags (2 layers) using metal-free tweezers and gloves.
M3. Cascade impactors
Cascade impactors size and collect particles through inertial impaction onto a series of stages. A filter is placed onto each stage to collect the PM. We used a 5-stage SKC Inc. Sioutas impactor with Whatman and Zefluor PTFE filters (25 mm diameter on stages 1–4 and 37 mm diameter on stage 5, 0.2 µm pore size). Filters were acid washed following the same procedure as described above for filter packs. The Sioutas impactor resolves 5 size fractions between >2.5 µm and >0.25 µm at a flow rate of 9 l min−1. Air flow is created by an external pump with inbuilt battery (SKC Leland Legacy). The pumps had been calibrated prior to both campaigns by the manufacturer, with reported accuracy in flow rate of 5%.
M4. Sample extraction procedures
2018: Pre-sampling treatment and post-sampling extraction of gas and PTFE filters was carried out in class-10000 clean labs in the Department of Earth Sciences at the University of Cambridge, UK. Drying down of extraction solutions was made in a class-100 laminar flow cabinet.
2019: Pre-sampling treatment and post-sampling extraction of gas and PTFE filters was carried out in class-10000 clean labs, under class-100 laminar hood in the School of Earth and Environment at the University of Leeds, UK.
All filters were cut into quarters using acid-cleaned metal-free scissors and tweezers prior to the extraction process. Laboratory procedural blanks were analysed for each sample treatment and extraction stage. A total of 28 field and lab blanks representing all variations of sampling and extraction method were subjected to the same procedure as the samples.
Gas filters were transferred to acid-cleaned 50 ml centrifuge tubes using acid-cleaned metal-free tweezers and exposed to a solution of ultrapure Milli-Q water (>18.2 MΩ, MQ, 20 ml) and peroxide (250 µl). The tubes were then shaken for 30 min. 2 × 2 ml were pipetted from these solutions for measurements of pH and anion concentrations by ion chromatography (IC), respectively. Blank filters and field blanks (i.e. filters transported into the field and inserted into the instruments in an identical manner to samples but not attached to a pump) were also analysed and found to be negligible compared to the measured values.
A two-stage sequential leaching method was employed to assess the solubility of particle phases on the PM filters using previously reported procedures19. Steps 1–2 below are designed to extract the water-soluble fraction of the particle matter which typically include sulphate and halide salts in volcanic particle matter. Steps 3–6 are designed to extract the poorly soluble and water-insoluble fractions, which typically includes sulfides, carbonates, oxides and silicates. As with the gas filters, blank filters and field blanks were also analysed and found to be negligible compared to the measured values.
-
1.
PTFE filters were transferred to acid-cleaned 50 ml metal-free centrifuge tubes using acid-cleaned metal-free tweezers. 20 ml of MQ and trace element grade propanol (to alleviate PTFE hydrophobicity) were added to all tubes. Samples were then shaken for 30 min and centrifuged at 3000 rpm for 15 min to separate the water-insoluble particles. 2 × 2 ml were pipetted from these solutions for measurements of pH and anion concentrations by IC, respectively. Every 10th sample was analysed twice on the IC.
-
2.
10 ml of remaining MQ solution was pipetted to acid-cleaned 15 ml metal-free centrifuge tube for analysis by inductively coupled plasma mass spectrometry (ICP-MS)/optical emission spectroscopy (ICP-OES). The sample was stored in a fridge between extraction and analysis.
-
3.
Remaining MQ solution and filters were transferred to an acid-cleaned 22 ml PFA (Savillex) beaker. The filters were handled with acid-cleaned metal-free tweezers. 5 ml UPA grade concentrated HNO3 added to the PFA beaker and samples dried on a hot plate at 90 °C.
-
4.
1 ml concentrated distilled HNO3 and 1 ml UPA grade concentrated HF was pipetted to the solid residue and refluxed for 3 h at 120 °C with lids on the PFA beakers. The lids were then removed, and samples dried at 90 °C.
-
5.
10 ml UPA grade concentrated HNO3 was pipetted to the solid residue and refluxed on a hot plate at 120 °C for 3 h. Filters were then removed from the beakers using acid-cleaned metal-free tweezers and stored in original 50 ml centrifuge tubes. The solution was then dried down at 90 °C.
-
6.
0.5 ml UPA grade concentrated HNO3 was pipetted to the final solid residue and refluxed at 120 °C for 1 h to get the sample back into solution. 10 ml MQ pipetted into the PFA beakers and then transferred to an acid-cleaned sample tube for analysis by ICP-MS/ICP-OES (final sample volume 10.5 ml, 3.5% HNO3 solution).
M5. Compositional analysis
All samples (gas and PTFE filters) were analysed for anions (SO42–, F–, Cl–) on Thermo Dionex ion chromatograph system in School of Geography, University of Leeds, UK.
2018 PTFE filter samples and PTFE blanks were analysed for major and trace elements by ICP-MS (Thermo iCAP Qc ICP-MS) and/or ICP-OES (Thermo iCAP 7400 ICP-OES) in School of Earth and Environment, University of Leeds, UK. All major elements were measured by ICPMS, with the exception of Mg, Ca, K, Ba, Sr, Al, P, Na, S, Fe, which were measured by ICP-OES.
2019 PTFE filter samples and PTFE blanks were analysed for major and trace elements by ICP-MS (Agilent 8800 ICP-MS/MS) in School of Environment, Earth and Ecosystem Sciences at the Open University, UK. Bi and In were not analysed in 2019 due to methodology limitations (they are used as internal standards). Prior to the 2019 analysis, a subset of volcanic PM samples was analysed on both University of Leeds and Open University instruments to ensure that the 2018 and 2019 results were comparable.
Uncertainties in the compositional data were calculated using error propagation26 and are included in Supplementary Data 3.
M6. Back-trajectory plume dispersion simulations
A time-reversed model was used to estimate the age and travel-distance of the plume at each site where direct samples were collected 18–31 July 2018. Full results are tabulated in Supplementary Data 4.
The Hybrid Single-Particle Lagrangian Integrated Trajectory model (HYSPLIT80) was used to create the back-trajectories necessary for these estimates. As a Lagrangian particle dispersion model, HYSPLIT simulates paths that parcels of air traverse. HYSPLIT uses meteorological inputs from a numerical weather prediction to simulate atmospheric transport processes. A random component simulates the stochastic effects of turbulence.
The back-trajectories rely on accurate meteorological simulations of the wind field. Complex terrain in a tropical, marine environment means a variety of mesoscale meteorological processes substantially impact the weather over the Hawaiian islands81. It is necessary to simulate these processes in any attempt to estimate the plume age and cumulative distance travelled.
To characterise the important meteorological processes that impact the Hawaiian Islands, a customised version of the Weather Research and Forecasting – Advanced Research (WRF-ARW) model was used. The WRF-ARW creates the driving meteorological fields used for the HYSPLIT back-trajectory runs. Details of the configuration of WRF-ARW and HYSPLIT known collectively as the ‘vog model’ are described in previous works82,83. These runs were made at 900 m resolution. These runs differ from the vog model run in real-time at the University of Hawai’i in only one respect. Instead of being run in forecast mode, the vog model is run in reverse-time mode to create back-trajectories.
These back-trajectories were initiated from the direct sampling sites of Kailua-Kona, Ocean View, Pāhala, and Volcano village every 6 h and ran backwards for 24 h (Supplementary Fig. 4). This duration was selected based on the amount of time it took Lagrangian particles to reach the furthest site (Kailua-Kona) when the vog model was run in forecast mode. The results were analysed for whether the Lagrangian particles could originate from two emission sources: the erupting and highly gas-rich Fissure 8 vent; and Kīlauea summit crater which produced no significant gas but periodic small ash explosions. Trajectories that originated within a 3 km radius of the emission source were used to calculate the average plume age for each sample. The cumulative distance each Lagrangian particle travelled from source to sample is averaged to estimate the travel-distance (Supplementary Data 4).
M7. Calculation of elements’ plume lifetimes
The objective of this study was to investigate the source-to-far field evolution of the volcanic plume with a focus on element in-plume lifetimes. For calculations of sulphur conversion rate, and elements-specific depletion rates, we selected a subset of samples from 2018 which were significantly impacted by the volcanic plume, as described below. Samples from 2019 were used for subtracting the non-volcanic background composition from all of the selected 2018 samples.
Results for all near-source samples (n = 5) are reported by Mason et al.26,43. Selection of representative near-source samples was done according to the following considerations. Two of the ground-based filter pack samples and one UAS filter pack sample became saturated with respect to the base-treated gas filters and were therefore unusable for determining the total concentration of elements found both in the gas and PM (however, PM concentrations can still be used). There was a good agreement in element mass ratios between the filter pack samples (agreement within an order of magnitude or less)26. The mass concentrations in the UAS sample were higher than in the ground-based samples by a factor of 9 ± 5. For calculating depletion rates, we consider the UAS sample to be the most representative of the at-source plume composition as it is the least diluted sample.
Selection of representative far-field samples was done according to the following considerations. Typical trade wind conditions persisted during the period of data collection in 2018. This meant that Pāhala, Ocean View and Kailua-Kona were almost always impacted by the grounding volcanic plume with little variation between individual samples collected during our campaign (Supplementary Data 1); this was typical for the whole 2018 eruption period as confirmed using back-trajectory plume dispersion analysis (M4) and Hawaii State Department of Health operational monitoring of SO2 and PM2.5 co-located with our sampling sites38. All collected filter pack samples were used for the analysis (6 samples from each location). At these three sites, the concentrations of all analysed elements/ions were significantly higher in 2018 compared to 2019 (Fig. 3).
Volcano village received the plume periodically, confirmed by the same dispersion simulations and operational monitoring. The sample collected on 23 July 2018 in Volcano village represented an event where a highly concentrated plume being advected directly from the eruption site (Supplementary Fig. 2) and was selected as a representative sample for this analysis. For the remainder of the field campaign, Volcano village atmosphere was dominated by the background atmosphere (including no or minor plume component); these samples were excluded from depletion rate calculations.
Leilani sampling site was located several blocks from the active volcanic vent but was out of the plume’s prevalent path and there was no significant difference between 2018 and 2019 levels (Fig. 3); this site is excluded from depletion rate calculations.
The plume path distance to Mauna Loa sampling site was highly irregular (either reaching the site directly in a few hours, or after circumnavigating the island in over 24 h, M4). The relatively coarse time resolution of our sampling (48 h) did not allow us to discriminate between the composition of a relatively young versus a relatively mature plume composition in the Mauna Loa samples; they are therefore excluded from depletion rate calculations.
The elements for which plume depletion rates were calculated are those which had concentrations above detection limits and above atmospheric background levels in the samples collected at-source and at least 3 far-field sampling sites.
The depletion of elements’ mass concentration was observed to proceed at two distinct rates: (i) initial and very rapid depletion between the source (Fissure 8) and the first downwind sampling site (Volcano village, average plume age ~3 h); (ii) a slower depletion between Volcano village and the subsequent downwind sampling sites, Pāhala, Ocean View and Kailua-Kona (average plume age ~3–19 h).
-
(i)
The initial and rapid depletion rate of each element in the first 3 h after emission was expressed as the mass ratio between the source and Volcano village. As we only have two sampling locations, it was not justifiable to fit a depletion rate curve.
-
(ii)
The subsequent and slower depletion rate between 3 and 19 h (4 sampling sites) was modelled by an exponential curve: Ct = C0*ekt.
where C0 is the initial concentration at-source (time = 0), Ct is the concentration at time t (expressed here in seconds), and k is the depletion rate constant. Time t is the average plume age for each sampling location, calculated as per M4. The exponential fit was weighted for each location based on the number of samples collected there. Error margins were set as 95% confidence intervals of the exponential fit.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and in Supplementary Data 1–5. Observational data of gas and PM composition (Supplementary Data 1, 2 and 5) have also been deposited in The Centre for Environmental Data Analysis (CEDA)43 with the https://doi.org/10.5285/656204c257144708a641507c78187aaa. Other data (Supplementary Data 3 and 4) were calculated using the methods outlined in this paper.
Change history
01 June 2021
A Correction to this paper has been published: https://doi.org/10.1038/s43247-021-00190-y
References
Brown, S. K., Sparks, R. S. J. & Jenkins, S. F. Global distribution of volcanic threat. in Global Volcanic Hazards and Risk (eds Vye-Brown, C., Brown, S. K., Sparks, S., Loughlin, S. C. & Jenkins, S. F.) 359–370 (Cambridge University Press, 2015). https://doi.org/10.1017/CBO9781316276273.025.
Gauthier, P.-J., Sigmarsson, O., Gouhier, M., Haddadi, B. & Moune, S. Elevated gas flux and trace metal degassing from the 2014–2015 fissure eruption at the Bárðarbunga volcanic system, Iceland. J. Geophys. Res. Solid Earth 121, 1610–1630 (2016).
Ilyinskaya, E. et al. Understanding the environmental impacts of large fissure eruptions: Aerosol and gas emissions from the 2014–2015 Holuhraun eruption (Iceland). Earth Planet. Sci. Lett. 472, 309–322 (2017).
Neal, C. A. et al. The 2018 rift eruption and summit collapse of Kīlauea Volcano. Science 363, 367–374 (2019).
Kern, C. et al. Quantifying gas emissions associated with the 2018 rift eruption of Kīlauea Volcano using ground-based DOAS measurements. Bull. Volcanol. 82, 55 (2020).
Moune, S., Gauthier, P.-J. & Delmelle, P. Trace elements in the particulate phase of the plume of Masaya Volcano, Nicaragua. J. Volcanol. Geotherm. Res. 193, 232–244 (2010).
Wright, T. L. & Klein, F. W. Two hundred years of magma transport and storage at Kīlauea Volcano, Hawai’i, 1790-2008. Two hundred years of magma transport and storage at Kīlauea Volcano, Hawai’i, 1790-2008 vol. 1806 258 http://pubs.er.usgs.gov/publication/pp1806 (2014).
Self, S., Schmidt, A. & Mather, T. A. Emplacement characteristics, time scales, and volcanic gas release rates of continental flood basalt eruptions on Earth. Geol. Soc. Am. Spec. Pap. 505, 319–337 (2014).
Oppenheimer, C., Pyle, D. M. & Barclay, J. Volcanic Degassing. (Geological Society of London, 2003).
World Health Organisation. Health risks of heavy metals from long-range transboundary air pollution. (World Health Organisation, 2007).
Department for Environment Food and Rural Affairs (DEFRA). Heavy Metals Network- Defra, UK. https://uk-air.defra.gov.uk/networks/network-info?view=metals.
US EPA, O. Metals. US EPA https://www.epa.gov/caddis-vol2/metals (2015).
Jaishankar, M., Tseten, T., Anbalagan, N., Mathew, B. B. & Beeregowda, K. N. Toxicity, mechanism and health effects of some heavy metals. Interdiscip. Toxicol. 7, 60–72 (2014).
Orr, S. E. & Bridges, C. C. Chronic Kidney Disease and Exposure to Nephrotoxic Metals. Int. J. Mol. Sci. 18, 1039 (2017).
Al osman, M., Yang, F. & Massey, I. Y. Exposure routes and health effects of heavy metals on children. BioMetals 32, 563–573 (2019).
Edmonds, M., Mather, T. A. & Liu, E. J. A distinct metal fingerprint in arc volcanic emissions. Nat. Geosci. 11, 790 (2018).
Rubin, K. Degassing of metals and metalloids from erupting seamount and mid-ocean ridge volcanoes: Observations and predictions. Geochim. Cosmochim. Acta 61, 3525–3542 (1997).
Symonds, R. B., Reed, M. H. & Rose, W. I. Origin, speciation, and fluxes of trace-element gases at Augustine volcano, Alaska: Insights into magma degassing and fumarolic processes. Geochim. Cosmochim. Acta 56, 633–657 (1992).
Mather, T. A. et al. Halogens and trace metal emissions from the ongoing 2008 summit eruption of Kīlauea volcano, Hawai’i. Geochim. Cosmochim. Acta 83, 292–323 (2012).
Zelenski, M. E. et al. Trace elements in the gas emissions from the Erta Ale volcano, Afar, Ethiopia. Chem. Geol. 357, 95–116 (2013).
Mandon, C. L., Christenson, B. W., Schipper, C. I., Seward, T. M. & Garaebiti, E. Metal transport in volcanic plumes: a case study at White Island and Yasur volcanoes. J. Volcanol. Geotherm. Res. 369, 155–171 (2019).
Allard, P. et al. Prodigious emission rates and magma degassing budget of major, trace and radioactive volatile species from Ambrym basaltic volcano, Vanuatu island Arc. J. Volcanol. Geotherm. Res. 322, 119–143 (2016).
Allard, P. et al. Acid gas and metal emission rates during long‐lived basalt degassing at Stromboli Volcano. Geophys. Res. Lett. 27, 1207–1210 (2000).
Aiuppa, A., Dongarrà, G., Valenza, M., Federico, C. & Pecoraino, G. Degassing of Trace Volatile Metals During the 2001 Eruption of Etna. Volcanism Earths Atmosphere 41–54 (2004), https://doi.org/10.1029/139GM03.
Lambert, G., Le Cloarec, M. F., Ardouin, B. & Le Roulley, J. C. Volcanic emission of radionuclides and magma dynamics. Earth Planet. Sci. Lett. 76, 185–192 (1985).
Mason, E. et al. Volatile metal emissions from volcanic degassing and lava–seawater interactions at Kīlauea Volcano, Hawai’i. Commun Earth Environ. 2, 79, https://doi.org/10.1038/s43247-021-00145-3 (2021).
Symonds, R. B. (Michigan T. U. & Reed, M. H. (Univ of O. Calculation of multicomponent chemical equilibria in gas-solid-liquid systems: Calculation methods, thermochemical data, and applications to studies of high-temperature volcanic gases with examples from Mt. St. Helens. Am. J. Sci. U. S. 293:8 (1993).
Ayris, P. M. et al. HCl uptake by volcanic ash in the high temperature eruption plume: Mechanistic insights. Geochim. Cosmochim. Acta 144, 188–201 (2014).
Calabrese, S. et al. Characterization of the Etna volcanic emissions through an active biomonitoring technique (moss-bags): Part 1 – Major and trace element composition. Chemosphere 119, 1447–1455 (2015).
Martin, R. S. et al. Major and trace element distributions around active volcanic vents determined by analyses of grasses: implications for element cycling and bio-monitoring. Bull. Volcanol. 72, 1009–1020 (2010).
Moune, S., Gauthier, P.-J., Gislason, S. R. & Sigmarsson, O. Trace element degassing and enrichment in the eruptive plume of the 2000 eruption of Hekla volcano, Iceland. Geochim. Cosmochim. Acta 70, 461–479 (2006).
Liotta, M. et al. Mobility of plume-derived volcanogenic elements in meteoric water at Nyiragongo volcano (Congo) inferred from the chemical composition of single rainfall events. Geochim. Cosmochim. Acta 217, 254–272 (2017).
Lettino, A. et al. Eyjafjallajökull volcanic ash in southern Italy. Atmos. Environ. 48, 97–103 (2012).
Armienta, M. A., De la Cruz-Reyna, S., Morton, O., Cruz, O. & Ceniceros, N. Chemical variations of tephra-fall deposit leachates for three eruptions from Popocatépetl volcano. J. Volcanol. Geotherm. Res. 113, 61–80 (2002).
Schmidt, A. et al. Satellite detection, long-range transport, and air quality impacts of volcanic sulfur dioxide from the 2014–2015 flood lava eruption at Bárðarbunga (Iceland). J. Geophys. Res. Atmospheres 120, 9739–9757 (2015).
Schmidt, A. et al. Excess mortality in Europe following a future Laki-style Icelandic eruption. Proc. Natl. Acad. Sci. 108, 15710–15715 (2011).
Reikard, G. Volcanic emissions and air pollution: forecasts from time series models. Atmospheric Environ. X 1, 100001 (2019).
Whitty, R. C. W. et al. Spatial and Temporal Variations in SO2 and PM2.5 Levels from 2007-2018 Kīlauea Volcano, Hawai’i. Front. Earth Sci. 8, 36 (2020).
Elias, T. & Sutton, A. J. Volcanic air pollution hazards in Hawaii. Volcanic air pollution hazards in Hawaii vols 2017–3017 http://pubs.er.usgs.gov/publication/fs20173017 (2017).
Tam, E. et al. Volcanic air pollution over the Island of Hawai’i: Emissions, dispersal, and composition. Association with respiratory symptoms and lung function in Hawai’i Island school children. Environ. Int. 92–93, 543–552 (2016).
Longo, B. M., Rossignol, A. & Green, J. B. Cardiorespiratory health effects associated with sulphurous volcanic air pollution. Public Health 122, 809–820 (2008).
Giambelluca, T. W. et al. Online Rainfall Atlas of Hawai’i. Bull. Am. Meteorol. Soc. 94, 313–316 (2013).
Ilyinskaya, E., Mason, E., Wieser, P., Liu, E. & Whitty, R. Filter pack and cascade impactor samples of gas and aerosol particulate matter on the Island of Hawai’i (2018, 2019). (2021), https://doi.org/10.5285/656204C257144708A641507C78187AAA.
Vernier, J.-P. et al. VolKilau: Volcano rapid response balloon campaign during the 2018 Kilauea eruption. Bull. Am. Meteorol. Soc. (2020), https://doi.org/10.1175/BAMS-D-19-0011.1.
Hinkley, T. K. Distribution of metals between particulate and gaseous forms in a volcanic plume. Bull. Volcanol. 53, 395–400 (1991).
Oppenheimer, C. et al. Influence of eruptive style on volcanic gas emission chemistry and temperature. Nat. Geosci. 11, 678–681 (2018).
Kroll, J. H. et al. Atmospheric Evolution of Sulfur Emissions from Kı̅lauea: Real-Time Measurements of Oxidation, Dilution, and Neutralization within a Volcanic Plume. Environ. Sci. Technol. 49, 4129–4137 (2015).
Eatough, D. J., Caka, F. M. & Farber, R. J. The Conversion of SO2 to Sulfate in the Atmosphere. Isr. J. Chem. 34, 301–314 (1994).
Oppenheimer, C., Francis, P. & Stix, J. Depletion rates of sulfur dioxide in tropospheric volcanic plumes. Geophys. Res. Lett. 25, 2671–2674 (1998).
Pattantyus, A. K., Businger, S. & Howell, S. G. Review of sulfur dioxide to sulfate aerosol chemistry at Kīlauea Volcano, Hawai’i. Atmos. Environ. 185, 262–271 (2018).
Beirle, S. et al. Estimating the volcanic emission rate and atmospheric lifetime of SO2 from space: a case study for Kīlauea volcano, Hawai’i. Atmospheric Chem. Phys. 14, 8309–8322 (2014).
Edmonds, M., Gerlach, T. M. & Herd, R. A. Halogen degassing during ascent and eruption of water-poor basaltic magma. Chem. Geol. 263, 122–130 (2009).
Bureau, U. C. City and Town Population Totals: 2010-2019. The United States Census Bureau https://www.census.gov/data/tables/time-series/demo/popest/2010s-total-cities-and-towns.html.
Improve – Interagency Monitoring of Protected Visual Environments. http://vista.cira.colostate.edu/Improve/.
Mather, T. A., Pyle, D. M. & Oppenheimer, C. Tropospheric Volcanic Aerosol. in Volcanism and the Earth’s Atmosphere 189–212 (American Geophysical Union (AGU), 2013), https://doi.org/10.1029/139GM12.
Desboeufs, K. V., Sofikitis, A., Losno, R., Colin, J. L. & Ausset, P. Dissolution and solubility of trace metals from natural and anthropogenic aerosol particulate matter. Chemosphere 58, 195–203 (2005).
US EPA, O. Lead (Pb) Air Pollution. US EPA https://www.epa.gov/lead-air-pollution (2016).
Whitby, K. T. The Physical Characteristics of Sulfur Aerosols. in Sulfur in the Atmosphere (eds Husar, R. B., Lodge, J. P. & Moore, D. J.) 135–159 (Pergamon, 1978), https://doi.org/10.1016/B978-0-08-022932-4.50018-5.
Fujita, S., Sakurai, T. & Matsuda, K. Wet and dry deposition of sulfur associated with the eruption of Miyakejima volcano, Japan. J. Geophys. Res. Atmospheres 108, (2003) https://doi.org/10.1029/2002JD00306462.
Webster H. N. and Thomson D. J., 2017, A particle size dependent wet deposition scheme for NAME, Forecasting Research Technical Report No: 624, (2017).
Oelkers, E. H. & Gislason, S. R. The mechanism, rates and consequences of basaltic glass dissolution: I. An experimental study of the dissolution rates of basaltic glass as a function of aqueous Al, Si and oxalic acid concentration at 25 °C and pH = 3 and 11. Geochim. Cosmochim. Acta 65, 3671–3681 (2001).
Martin, R. S. et al. Composition-resolved size distributions of volcanic aerosols in the Mt. Etna plumes. J. Geophys. Res. Atmospheres 113, (2008) https://doi.org/10.1029/2007JD00964863.
Martin, R. S. et al. Size distributions of fine silicate and other particles in Masaya’s volcanic plume. J. Geophys. Res. Atmospheres 114, (2009) https://doi.org/10.1029/2007JD00964865.
Sparks, R. S. J. The dimensions and dynamics of volcanic eruption columns. Bull. Volcanol. 48, 3–15 (1986).
Ilyinskaya, E., Martin, R. S. & Oppenheimer, C. Aerosol formation in basaltic lava fountaining: Eyjafjallajökull volcano, Iceland. J. Geophys. Res. Atmospheres 117 (2012) https://doi.org/10.1029/2011JD01681166.
Patrick, M. R. et al. Cyclic lava effusion during the 2018 eruption of Kīlauea Volcano. Science 366, (2019) https://doi.org/10.1126/science.aay907069.
Toutain, J. P. et al. Vapor deposition of trace elements from degassed basaltic lava, Piton de la Fournaise volcano, Reunion Island. J. Volcanol. Geotherm. Res. 40, 257–268 (1990).
Quisefit, J. P. et al. Evolution versus cooling of gaseous volcanic emissions from Momotombo Volcano, Nicaragua: Thermochemical model and observations. Geochim. Cosmochim. Acta 53, 2591–2608 (1989).
Martin, R. S., Mather, T. A. & Pyle, D. M. High-temperature mixtures of magmatic and atmospheric gases. Geochem. Geophys. Geosystems 7 (2006) https://doi.org/10.1029/2005GC001186.
Mather, T. A., Allen, A. G., Oppenheimer, C., Pyle, D. M. & McGonigle, A. J. S. Size-Resolved Characterisation of Soluble Ions in the Particles in the Tropospheric Plume of Masaya Volcano, Nicaragua: origins and Plume Processing. J. Atmospheric Chem. 46, 207–237 (2003).
Jones, M. T. & Gislason, S. R. Rapid releases of metal salts and nutrients following the deposition of volcanic ash into aqueous environments. Geochim. Cosmochim. Acta 72, 3661–3680 (2008).
Stier, P. et al. The aerosol-climate model ECHAM5-HAM. Atmospheric Chem. Phys. 5, 1125–1156 (2005).
Waldman, J. M. & Hoffmann, M. R. Depositional Aspects of Pollutant Behavior in Fog and Intercepted Clouds. in Sources and Fates of Aquatic Pollutants vol. 216 79–129 (American Chemical Society, 1987).
Li, W. & Shao, L. Characterization of mineral particles in winter fog of Beijing analyzed by TEM and SEM. Environ. Monit. Assess. 161, 565–573 (2010).
Gerlach, T. M. & Graeber, E. J. Volatile budget of Kilauea volcano. Nature 313, 273 (1985).
Vigneri, R., Malandrino, P., Gianì, F., Russo, M. & Vigneri, P. Heavy metals in the volcanic environment and thyroid cancer. Mol. Cell. Endocrinol. 457, 73–80 (2017).
Nicoletti, A. et al. Incidence of multiple sclerosis in the province of Catania. A geo-epidemiological study. Environ. Res. 182, 109022 (2020).
Hyslop, N. P., Trzepla, K., Wallis, C. D., Matzoll, A. K. & White, W. H. Technical note: a 23-year record of twice-weekly aerosol composition measurements at Mauna Loa Observatory. Atmos. Environ. 80, 259–263 (2013).
Allen, A. G., Baxter, P. J. & Ottley, C. J. Gas and particle emissions from Soufrière Hills Volcano, Montserrat, West Indies: characterization and health hazard assessment. Bull. Volcanol. 62, 8–19 (2000).
Stein, A. F. et al. NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System. Bull. Am. Meteorol. Soc. 96, 2059–2077 (2015).
Smith, R. B. & Grubišić, V. Aerial Observations of Hawaii’s Wake. J. Atmospheric Sci. 50, 3728–3750 (1993).
Cherubini, T., Businger, S., Lyman, R. & Chun, M. Modeling Optical Turbulence and Seeing over Mauna Kea. J. Appl. Meteorol. Climatol. 47, 1140–1155 (2008).
Businger, S. et al. Observing and Forecasting Vog Dispersion from Kīlauea Volcano, Hawaii. Bull. Am. Meteorol. Soc. 96, 1667–1686 (2015).
National emissions reported to the Convention on Long-range Transboundary Air Pollution (LRTAP Convention) — European Environment Agency. https://www.eea.europa.eu/data-and-maps/data/national-emissions-reported-to-the-convention-on-long-range-transboundary-air-pollution-lrtap-convention-14.
Tian, H. Z. et al. Quantitative assessment of atmospheric emissions of toxic heavy metals from anthropogenic sources in China: historical trend, spatial distribution, uncertainties, and control policies. Atmospheric Chem. Phys. 15, 10127–10147 (2015).
Acknowledgements
The fieldwork was supported by the United States Geological Survey, Hawaiian Volcano Observatory, State of Hawaii Department of Health, and NOAA Global Monitoring Division – in particular by Tina Neal, Jim Ciszewski, Darryl Kuniyuki, Rich Thurau, Bobby Eisele, Geoff DeBenedetto, Frederic (Bill) Simonds, and Frank Trusdell. AirMonitors, SKC West, Prof Claire Horwell and Prof Francis Pope are acknowledged for providing field equipment. Volcano Arts Center is thanked for hosting our equipment. Residents of Leilani Estates and Lower Puna, in particular Steven Jacquier, David Moletti, Chris Brace, and Kris Burmeister are thanked for their support. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government. EI: NERC Centre for Observation and Modelling of Earthquakes, Volcanoes and Tectonics (COMET), a partnership between UK Universities and the British Geological Survey. UNRESP (NE/R009465/1, NE/P015271/1) and V-PLUS (NE/S00436X/1). M.E.: The Alfred P. Sloan Foundation via the Deep Carbon Observatory. P.W.: Leave to Work Away Research Fund. E.M.: EPSRC-CASE studentship. E.L.: Leverhulme Trust Early Career Fellowship. R.C.W.W.: NERC DTP-CASE studentship. C.O.: NERC NE/N009312/1.
Author information
Authors and Affiliations
Contributions
E.I., T.A.M., M.E., T.E., C.K. and P.N. initiated the project idea and field campaign planning. E.I. led the 2018 and 2019 field campaigns with significant contributions from E.M., P.W., E.L., and R.C.W.W. T.A.M., M.E., S.A., J.M.Q. and C.O. participated in campaign planning and/or execution. T.E., P.N., D.S., J.C., C.K. and D.D. provided local expertise and support for planning and execution of the field campaigns. E.M. led the methodology development and execution of the laboratory analysis of 2018 data with participation of P.W., E.I. and E.L.; E.I. performed the 2019 lab analysis. L.H. performed the plume dispersion modelling and back-trajectory calculations. Results analysis was jointly led by E.I. and E.M. with significant contributions from P.W., T.A.M., M.E. and E.L. Paper writing was led by E.I. All authors participated in the discussion of the results and contributed to the paper.
Corresponding author
Ethics declarations
Competing interests
Dr. E.L. is an Editorial Board Member for Communications Earth & Environment, but was not involved in the editorial review of, or the decision to publish this paper. All other authors declare they have no competing interests.
Additional information
Peer review information Primary handling editor: Joe Aslin
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ilyinskaya, E., Mason, E., Wieser, P.E. et al. Rapid metal pollutant deposition from the volcanic plume of Kīlauea, Hawai’i. Commun Earth Environ 2, 78 (2021). https://doi.org/10.1038/s43247-021-00146-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-021-00146-2
This article is cited by
-
Potentially toxic elements contamination in groundwater and human health risk assessment in the Mayo Bocki watershed, North Cameroon
Arabian Journal of Geosciences (2023)
-
A golden era for volcanic gas geochemistry?
Bulletin of Volcanology (2022)
-
Volcanic air pollution and human health: recent advances and future directions
Bulletin of Volcanology (2022)
-
Volatile metal emissions from volcanic degassing and lava–seawater interactions at Kīlauea Volcano, Hawai’i
Communications Earth & Environment (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
