
Abstract
Volcanoes represent one of the largest natural sources of metals to the Earth’s surface. Emissions of these metals can have important impacts on the biosphere as pollutants or nutrients. Here we use ground- and drone-based direct measurements to compare the gas and particulate chemistry of the magmatic and lava–seawater interaction (laze) plumes from the 2018 eruption of Kīlauea, Hawai’i. We find that the magmatic plume contains abundant volatile metals and metalloids whereas the laze plume is further enriched in copper and seawater components, like chlorine, with volatile metals also elevated above seawater concentrations. Speciation modelling of magmatic gas mixtures highlights the importance of the S2− ligand in highly volatile metal/metalloid degassing at the magmatic vent. In contrast, volatile metal enrichments in the laze plume can be explained by affinity for chloride complexation during late-stage degassing of distal lavas, which is potentially facilitated by the HCl gas formed as seawater boils.
Similar content being viewed by others

Introduction
Volcanoes have played a key role in shaping the composition of Earth’s atmosphere over geological time, influencing the planet’s habitability through the outgassing of hydrogen-, carbon- and sulphur-bearing species1,2,3,4. Volcanoes also supply significant amounts of volatile trace elements, including metals and metalloids, in the gas phase or as non-silicate particulate matter (PM, also called aerosol)5,6,7,8,9,10,11,12. Volcanogenic metal emissions have been sampled and studied at many volcanoes worldwide (e.g., refs. 6,7,8,9,10,11,12,13), and early studies date back to the 1960s and 70s (e.g., refs. 14,15). During periods of intense unrest or eruption, volcanic emission rates of metals such as cadmium (Cd), copper (Cu) and zinc (Zn) can be equal to or, in some cases, orders of magnitude greater than daily anthropogenic emissions from entire regions (e.g., the Mediterranean basin14) or countries16, and volcanoes are one of the largest natural sources of many metals17. Some trace metals present in volcanic plumes can act as nutrients for living organisms at low levels, yet at higher concentrations are categorised by environmental agencies as pollutants known to be harmful to health18,19. Basaltic volcanoes typically release relatively ash-poor plumes into the troposphere9, exposing both proximal populations (up to a few tens of kilometres away, e.g., Kīlauea, Hawai’i, 1983–201820; Masaya, Nicaragua, 1993–present21) and distal populations thousands of km downwind (e.g., Laki, Iceland22) to sustained high levels of volcanogenic gases (e.g., SO2) and trace metal-bearing PM. Understanding volcanic metal emissions also provides important constraints on the environmental impact of large, prehistoric basaltic eruptions, including flood basalts23,24. However, metal abundance and speciation in volcanic gas and aerosol emissions remain poorly understood.
Basaltic ocean island volcanoes, such as those found on the Island of Hawai’i, can produce an additional source of gas and PM emissions as lava flows reach coastlines and rapidly boil and evaporate seawater, with significant implications for the marine biosphere25. Lava–seawater interactions, which produce acidic ‘laze’ plumes, have occurred throughout Earth’s history, often associated with some of the most dramatic manifestations of volcanism, such as: when lava flows from continental flood basalts reached coastlines (e.g., Columbia River Basalt group26, Deccan Traps27); in periods of sub-aerial eruption during ocean plateau basalt emplacement (e.g., the Kerguelen28, Ontong Java29,30,31, and Manihiki32,33 Plateaus); and during the early stages of continental rifting34 (e.g., the North Atlantic Igneous Province35). Fragments of silicate material can be generated during lava–seawater interactions through a fuel–coolant-type interaction36 as seawater invades lava tubes, and during the collapse of lava deltas37. A major component of laze plumes after water (~98–99 mol% of the gas phase) is hydrogen chloride (HCl) gas38,39, and it has been suggested that HCl emission rates from laze plumes at Kīlauea Volcano are comparable to or may exceed that from coal-fired power plants in the United States38. However, despite the hazards and potential impacts of lava ocean entry plumes on the biosphere, only a few detailed studies have been carried out (e.g., refs. 40,41,42) and relatively little is known about their chemistry compared to magmatic plumes.
The 2018 eruption of Kīlauea—a basaltic shield volcano located in the south–east of the Island of Hawai’i (Fig. 1a)—presented a rare opportunity to study the emissions of volatile trace metals in substantial, genetically related, magmatic and laze plumes. Before 30 April 2018, the eruptive activity at Kīlauea was relatively stable at two locations: a lava lake within the Halema’uma’u summit crater (active since 2008), and the Pu′u ′Ō′ō cone and other vents in the East Rift Zone (ERZ), as part of the long-lived Pu′u ′Ō′ō̄-Kupianaha eruption of Kīlauea (1983–2018)43. On 30 April 2018 the Pu′u ′Ō′ō vent collapsed, and magma propagated down the lower East Rift Zone (LERZ) of Kīlauea44. On 3 May 2018, the first of 24 fissures opened in the Leilani Estates subdivision on the LERZ44, and by the end of May, activity had largely focused at a single vent known as Fissure 8 (19.4627 °N, 154.9091 °W, ~220-m a.s.l., Fig. 1a, b). Lava flows from the 2018 LERZ eruption reached the coast on 23 May 2018, creating a laze plume (Fig. 1c) as still-molten lava boiled and evaporated seawater44. Peak SO2 emission rates of more than 200 kt day−1 were recorded from the LERZ in June and early July45, exceeding Kīlauea’s 2014 to 2017 average SO2 emission rate (combined summit and ERZ46) of 5.1 ± 0.3 kt day−1 SO2, by two orders of magnitude. Population exposure to poor air quality (SO2 and PM) during the 2018 LERZ eruption surpassed that observed during the 1983 to early 2018 eruption episodes at Kīlauea20,47. Further, in terms of both eruption rates (50–500 m3 s−1, dense rock equivalent)44,48 and erupted volume (~1.5 km3 in 94 days)49, the 2018 eruption was 1–2 orders of magnitude larger than any other in the preceding 180 years of activity in the LERZ49. The lava effusion and SO2 emissions from Fissure 8 declined dramatically on the 4 August 201844.

a Map of the final extent of the 2018 lower East Rift Zone (LERZ) eruption lava flows (red area, including extension of pre-LERZ coastline, adapted from Neal et al.44) and plume sampling locations during this study. The yellow star marks the location of the Leilani Community Association, where UAS (Unoccupied Aircraft System) flights into the magmatic plume were launched. White triangles mark the locations from which UAS flights to sample the laze plume were launched (Mackenzie State Recreation Area is marked by a smaller triangle as the sample launched here (9_1) was more distal and saturated, and was therefore excluded from the discussion—see Supplementary Note 1). During the July–August 2018 campaign, we sampled the laze plume, which at that time was being released from the Isaac Hale Park area. Inset map of Island of Hawai’i with locations of Kīlauea summit (S), Pu′u ′Ō′ō̄ (P) and Fissure 8 (F8). b Aerial image (U.S. Geological Survey: USGS) of Fissure 8 and the magmatic plume, taken on 24 July 2018 during a sampling flight, looking ~SE. UAS sampling of the magmatic plume was carried out ~300 m above the active vent. The cylindrical filter pack symbol (Fig. S1) marks the approximate location of ground-based sampling. c Aerial image (USGS) of the ocean entry and laze plume taken during a flight from Mackenzie State Recreation Area on 24 July 2018, looking ~NE. UAS sampling of the laze plume was carried out ~100 m above the ocean entry.
During the 2018 LERZ eruption of Kīlauea, we used ground-based and Unoccupied Aircraft System (UAS) platforms (Supplementary Note 1; ‘Methods’, Figs. S3, S4) to quantify the major and trace element compositions of gas and size-segregated PM emitted in the magmatic plume from the main active vent (Fissure 8, Fig. 1b, Supplementary Data 1 Tables S7–S9), and the laze plume generated at the ocean entry (Fig. 1c, Supplementary Data 1 Tables S11–S13). Here, we quantify element volatility in the magmatic plume, and demonstrate the consistency of Kīlauea’s volatile metal fingerprint across different eruptive periods (Supplementary Data 1 Table S10). We also model speciation during the oxidation and cooling of Kīlauea’s magmatic plume close to the lava–air interface (Supplementary Data 2 Tables S20–S27) and use these results to decipher the origin of the lava–seawater interaction plume.
Results and discussion
Element volatility at Kīlauea Volcano
Measured concentrations of elements in the magmatic plume (Supplementary Data 1 Tables S7–S9) can be used to estimate the relative volatility of each element during degassing (i.e., to what degree elements degas from magmas50). Emanation coefficients describe the degree to which an element is lost during syn-eruptive degassing at the vent according to:
where \({[X]}_{i}\) is the concentration of \(X\) in the melt just prior to syn-eruptive degassing, and \({[X]}_{f}\) is the final degassed concentration of element \(X\) in the melt, respectively (originally defined by Lambert et al.51). Emanation coefficients can be estimated in several ways, including: (1) using enrichment factors and the assumed constant emanation coefficient of Pb from molten basalt52 and; (2) comparing undegassed and degassed melts (e.g., ref. 53).
In this study, \({[X]}_{i}\) is calculated by adding the concentration of a degassed element in the magmatic plume (using X/S ratios) to a degassed matrix glass composition, i.e., \({[X]}_{f}\) (e.g., ref. 50; data sources in Supplementary Data 1 Table S14, ‘Methods’). We use the calculated ε values to group elements into volatile (ε > 0.001%) and refractory elements (ε < 0.001%; Fig. 2a, b). Within these groupings, and particularly within the volatile group, there remains a large amount of variability in ε values. These groupings were chosen because elements with ε > 0.001% have weighted ash fractions (WAFs) in our plume samples of 5% or less (i.e., they are predominantly released as gases or non-silicate particles in the plume; ‘Methods’, Supplementary Data 1 Table S10). Further, the two groups display contrasting behaviour during transport in and deposition from the volcanic plume16. For elements with a calculated ε above zero (i.e., they degas), their order of increasing volatility is: ε < 0.001%: Gd, La, Sm, Tb, Nd, Pr, Ce, Fe, Eu, Al, Ti, Mg, Ta; ε > 0.001% Ba, Na, Cu; ε > 0.01%: Zn, Ag; ε > 0.1%: In, Sn, Pb; then at ε > 1%: As; and ε > 10% Bi, Cd, Cl, Se, Re, Te, S (as calculated for non-saturated samples; ‘Methods’, Supplementary Note 1).

Note the different emanation coefficient scales on a and b. a Refractory (ε < 0.001%) elements in the magmatic plume, with emanation coefficients (ε; triangle and circle markers with white fill) calculated for the magmatic plume for the two non-saturated ground-based samples also shown. ε error bars represent the propagated errors (see Supplementary Note 7; ε data in Supplementary Data 1 Table S10). b Volatile elements (ε > 0.001%) in the magmatic plume, with emanation coefficients. c Refractory elements in the laze plume. d Volatile elements in the laze plume. All elements were measured by ICP-MS or ICP-OES except where indicated; IC ion chromatography, D cutoff diameter as defined in methods, BDL below detection limits (of either the instrument, or below filter blanks). Elements are ordered by average emanation coefficient, increasing from left to right.
Size-segregated concentrations of particles in the magmatic plume provide information on the mechanisms of particle formation, and can be used to corroborate ε calculations. Previous work has ascribed broad formation processes to particle diameter (D) ranges: (1) D < 0.1 µm: the ‘nucleation’ mode, associated with cooling, oxidation and quenching of magmatic gases as they mix with the atmosphere; (2) 0.1 < D < 2 µm: the ‘accumulation’ mode, formed from the condensation of low volatility vapours and from agglomeration (‘sticking together’) and growth of particles in the nucleation mode; (3) D > 2 µm: the ‘coarse particle’ mode, composed largely of mechanically generated particles (e.g., ash)54.
In the magmatic plume, simultaneous gas and PM measurements (using filter packs, ‘Methods’) showed that PM accounted for <1% of the total mass of S and Cl, i.e., these elements are present in the near-source plume almost entirely as gases (Fig. 3). For the PM, S is concentrated in the smallest size fraction (~91% collected with diameter, D ≤ 0.25 µm; ~0.5% collected with D ≥ 2.5 µm; Fig. 2b), while Cl concentrations are bimodal with the majority (~60%) found in D ≤ 0.25 µm, and ~27% found in D ≥ 2.5 µm. The presence of Cl and to a much lesser extent S, in the coarse fraction could indicate adsorption of HCl and SO2 gases onto ash in the plume55,56. Alternatively, it could represent halogen uptake by sulphuric acid that formed by homogeneous nucleation (potentially catalysed by metals, e.g., Cu, Fe, Mn57) during rapid cooling58; however, further investigation into this process at Kīlauea is beyond the scope of this study.

Concentrations (mol/m3) of S (measured as SO42−) and Cl (measured as Cl−) in the gas and PM phases of the magmatic (left, two unsaturated ground samples) and laze plumes (right, unsaturated Isaac Hale Park sample). Percentages indicate the proportion of S and Cl measured as gas and PM for each element. S/Cl ratios are total (gas + PM) molar ratios. Cl in PM at Fissure 8 is comparable to background levels measured in 201916 (Supplementary Data 1 Table S7). Errors are ±33% for Cl− and ±35% SO42−. Data and propagated errors can be found in Supplementary Data 1 Tables S7 and S11 for the magmatic and laze plumes, respectively.
Elements measured in this study can be subdivided into a refractory coarse fraction present as silicates (i.e., ash; Fig. 2a) and a volatile fine fraction present as non-silicate PM (Fig. 2b). Refractory elements (ε < 0.001%) in the magmatic plume are generally found in coarse size fractions: 10–50% (most REEs, Zr, Fe, Mn, Al, Ba), or 50–100% (P, Ca, Eu, Ti) of their concentration in PM is found within the D > 2.5-µm size fraction. These elements are likely derived from ash generated during lava fountaining and fine fragmentation at the Fissure 8 vent. Average WAFs calculated for these elements in the magmatic plume (ash correction in ‘Methods’, Supplementary Data 1 Table S10) support this conclusion: for all of the refractory elements in Fig. 2a apart from Al, Fe, Ti and Mg, >90% of their concentrations can be accounted for by ash (WAFs: Al = 89%; Fe = 81%; Ti = 72%; Mg = 71%). In contrast, volatile elements (ε > 0.001%) are predominantly found at D < 0.25 µm, consistent with typical diameters of the ‘nucleation mode’54, and have WAFs < 0.3%, except Ba (55%), Cu (4%), Na (4%) and Zn (1%). The size-segregated concentrations of elements observed in Kīlauea’s 2018 magmatic plume match broadly with those made at Kīlauea’s summit in 20089; however, concentrations of volatile trace elements are more evenly distributed across the size bins in 2008 than in 2018 (Supplementary Note 8 and Fig. S14).
Kīlauea’s trace metal compositional ‘fingerprint’
While volatile trace element emission rates at Kīlauea in 2018 are the highest recorded at the volcano45, the relative abundances of volatile trace elements measured at Kīlauea agree closely between different eruptive periods (Fig. 4). To eliminate the effects of variable plume dilution, we compare the composition of Kīlauea’s 2018 magmatic plume to other basaltic volcanic plumes using trace element to SO2 mass ratios (X/SO2, Supplementary Data 1 Table S10 and Fig. 4). X/SO2 ratios at Kīlauea in 2018 agree within an order of magnitude with previous measurements made at Kīlauea’s summit in 20089. It is important to note that the differences in relative abundances of volatile metals and metalloids between arc and intraplate settings59 are only consistent for mafic magma compositions (i.e., basaltic to basaltic andesite). More evolved or peralkaline intraplate volcanoes (e.g., Nyiragongo60, Erebus61,62) can produce higher volatile trace element emissions than those at mafic intraplate volcanoes—this has been attributed to the higher halogen contents of gases emitted at these volcanoes13.

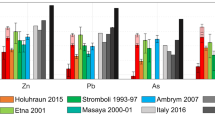
Trace element concentrations, X, ratioed to SO2 gas (SO2) concentration to account for variable plume dilution, normalised to the X/SO2 ratios in the Kīlauea 2008 eruptive plume9. Data are shown as shaded regions, the bounds of which represent the standard deviation of the samples used to calculate the X/SO2 mass ratio. Data sources: Holuhraun 201411; Etna 20017,112; Stromboli 1993–19976; Masaya 2000–20018; Ambrym 2007–0812; Yasur 201613. Elements are ordered by the average arc X/SO2 ratio for the volcanoes shown. Kīlauea 2018 data are an average of non-saturated samples 8_5 and 8_6 and error bars represent the maximum and minimum values from these two samples.
Emanation coefficients calculated for Kīlauea in 2018 (‘Methods’) also agree, to within an order of magnitude, with petrological estimates obtained through the comparison of variably degassed Kīlauea melt inclusions and matrix glasses53 (Fig. 5). However, we note that all methods of ε calculation are approximations, relying on assumptions that lead to relatively large uncertainties (including the method used here; ‘Methods’). For example, we note that the melt inclusion-matrix glass comparisons used in petrological estimates are hindered by the complex process of sulphide resorption into the degassing melt upon ascent53. Further, the composition of co-erupted olivine-hosted melt inclusions are unlikely to have a simple relationship with their carrier melt (i.e., the matrix glass), due to crystal scavenging from deep mush piles at Kīlauea63.

Emanation coefficients at a selection of basaltic to basaltic-andesite intraplate and arc volcanoes, ordered to their magnitude in Kīlauea 2018 (average of two ground samples). All emanation coefficients are calculated using X/SO2 ratios as outlined in ‘Methods’, with the exception of Holuhraun, for which the values are those given in the paper, calculated using \({{\boldsymbol{\varepsilon }}}_{{\boldsymbol{Pb}}}\). Data sources are as in Fig. 4, in addition to a study of degassed and undegassed melts53, and a compilation (Rubin) of ε at a mixture of tectonic settings72. Data for the figure can be found in Supplementary Data 1 Table S16 and a description of ε calculations can be found in ‘Methods’. Error bars represent the emanation coefficients calculated for the maximum and minimum X/SO2 ratio measured during each measurement campaign. Errors are not available for the Rubin compilation72 or the Holuhraun 2014–2015 data11.
The order of element volatility at Kīlauea matches that derived from measurements made at Erta ′Ale volcano, an intraplate (rift) basaltic volcano in Ethiopia10 (Fig. 5). However, X/SO2 ratios and emanation coefficients at Holuhraun, a hotspot-related, basaltic volcano, are 1–2 orders of magnitude lower than those measured at Kīlauea and Erta ′Ale (Figs. 4 and 5). This may be due to the low chlorine contents in the Holuhraun plume, reducing the availability of ligands11 (total, gas + PM, S/Cl mass ratio at Holuhraun = 40–5211; at Kīlauea = 32–35, this study; at Erta ‘Ale = 6–1510).
Speciation of gases in Kīlauea’s magmatic plume
The speciation of volatile trace elements during degassing provides key initial conditions for the subsequent atmospheric transport, solubility, deposition and resulting hazard from these elements16. Major elements—such as S, O, H and Cl (and other halogens)—are the main ligand-forming elements for volatile trace metals and metalloids in volcanic gas emissions (e.g., as sulphates (\({{\rm{SO}}}_{4}^{2-}\)), sulphides (\({{\rm{S}}}^{2-}\)), chlorides (\({{\rm{Cl}}}^{-}\)), bromides (\({{\rm{Br}}}^{-}\)), fluorides (\({{\rm{F}}}^{-}\)), oxides (\({{\rm{O}}}^{2-}\)) and hydroxides (\({{\rm{OH}}}^{-}\))10,64). The magmatic plume emitted from Fissure 8 was S-rich relative to halogens, with a molar gas ratio for SO2/HCl of 32–35. This ratio is similar to those measured from previous persistent emissions at the Pu‘u ‘Ō‘ō vent in 2004–2005 (SO2/HCl ~23)65, and from the summit in 2008–2009 (SO2/HCl ~20–28)9. However, due to the preferential (or fractional) loss of sulphur during early degassing, residual halogen concentrations in lava flows that travel towards the ocean entry will increase relative to sulphur (decreasing SO2/HCl ratios). For example, gas composition measurements made at Kīlauea in 1983–1984 indicated that while ~78% of S was lost from the melt during initial degassing, this corresponded to a loss of only ~10% of Cl66 (this process has also been observed at other basaltic volcanoes, e.g., Holuhraun67, Etna68, Piton de la Fournaise69).
The speciation of volatile trace elements at thermodynamic equilibrium during volcanic degassing has been modelled at several arc volcanoes (e.g., refs. 13,64) but not, to our knowledge, at any intraplate volcanoes such as Kīlauea. Here we model the simultaneous oxidation (by atmospheric mixing) and cooling of the magmatic gas mixture emitted from the Fissure 8 vent. Atmospheric mixing rapidly oxidises magmatic gas mixtures close to the lava–air interface, leading to a compositional discontinuity70,71 (Supplementary Note 6). This compositional discontinuity corresponds to the near-complete oxidation of reduced gas species such as H2S and S2 (Fig. 6a), and a shift in the speciation, to a greater or lesser degree, of all trace elements. This oxidised gas mixture will eventually ‘quench’ (i.e., cool to a largely non-reactive state71), and then undergo further chemical changes through low temperature reactions during atmospheric transport.

a The effect of atmospheric mixing (+78% N2, 21% O2, 1% Ar) on the speciation of major sulphur-bearing gases in the volcanic plume, with the compositional discontinuity (CD) at ~14% air. b The speciation (gas phase only) of elements in the magmatic plume at 0% percent air (\({V}_{A}/{V}_{M}\) = 0; no atmospheric mixing). c The speciation after the compositional discontinuity, at 25% air (\({V}_{A}/{V}_{M}\) = 0.33; the proportions of different species do not change significantly between 20 and 25% air). Elements are ordered according to their average emanation coefficient (Supplementary Data 1 Table S10), increasing upwards. The model inputs can be found in Supplementary Data 2 Table S20 and the outputs can be found in Supplementary Data 2 Table S21.
Factors that control the speciation of elements in volcanic gas emissions at the point of degassing include the composition of the volatiles released from the melt, as well as temperature, pressure and the composition of the magma from which degassing occurs (i.e., its oxidation state)59,64,70,72. Kinetic effects also play an important role in determining speciation73, and are not accounted for in the model presented here. Therefore, speciation modelling in this study is relevant only to the high-temperature mixture of atmospheric and magmatic gases generated close to the lava–air interface, where gases are assumed to attain equilibrium rapidly, and kinetic effects are assumed to be negligible70,74. Mixing between a magmatic plume and the background atmosphere has been previously described using a ratio \({V}_{A}/{V}_{M}\), where \({V}_{A}\) is the volume of air in the mixture, and \({V}_{M}\) is the volume of undiluted magmatic plume in the mixture (mixing can also be described as a mol% of air in the mixture).
We modelled the equilibrium gas phase speciation in the magmatic plume using the Gibbs Energy Minimisation module of HSC Chemistry (version 9.9.2, Outotec Research Oy, Finland; ‘Methods’). From an initial magmatic gas equilibrium temperature of 1145 °C (from MgO content of Fissure 8 lavas; ‘Methods’), we model speciation during early mixing of magmatic gases with the atmosphere (\({0} < ({V_A}/{V_M}) < {0.33}\)); atmosphere is added as a 78:21:1 mixture of N2:O2:Ar in increments of \({V}_{A}/{V}_{M}=0.01\). To determine the temperature decrease during mixing, we use a simple model of temperature change during mixing of two gases of different temperatures. We use N2, O2 and Ar gases to represent the atmosphere at 25 °C and H2O vapour at 1145 °C to represent the magmatic gases (following Ilyinskaya et al.75; ‘Methods’, Fig. S11). This generates a temperature decrease from 1145 °C to 1016 °C between \({V}_{A}/{V}_{M}=\) 0 and \({V}_{A}/{V}_{M}=\) 0.33.
Broadly, at the point of emission with no atmospheric mixing, \({V}_{A}/{V}_{M}\) = 0, volatile trace elements can be grouped into those complexed as gaseous species with S2− (TeS, SeS, BiS, AsS, PbS, SnS) and those complexed with Cl− (InCl, AgCl, CuCl), and/or elemental gases (Cd, Bi, Ag, Zn, Cu; Fig. 6b). The speciation observed at \({V}_{A}/{V}_{M}\) = 0 highlights the critical role that the oxidation state of magmas (which determines the availability of certain ligands, e.g., S2−), as well as their S and Cl contents, play in determining degassing of volatile trace metals59. At \({V}_{A}/{V}_{M}\) = 0.33 (i.e., after the compositional discontinuity), sulphide-complexing elements (e.g., Se, Te, As) are now present predominantly as oxides, and those that were present in elemental forms are now present as chlorides, hydroxides or oxides (Fig. 6c). The concentration of chloride complexes increases after the compositional discontinuity (Fig. 6c), consistent with previous work70,74 that has shown that the oxidation of magmatic gases creates more reactive halide species, thus increasing the proportion of elements present as chloride compounds. Elements that do not complex with chloride between \({V}_{A}/{V}_{M}\) = 0 and \({V}_{A}/{V}_{M}\) = 0.33 (e.g., Se, Te, As) generally will not form chlorides unless the HCl gas content of the plume is increased by between 100 and 1000 times (\({V}_{A}/{V}_{M}\) = 0, Fig. S13; \({V}_{A}/{V}_{M}\) = 0.33, Fig. 7). In the case of Se, even 1000 times the original HCl gas concentration does not cause Se to complex with chloride.

Elements are shown in order of increasing emanation coefficient (Supplementary Data 1 Table S10) upwards, as in Fig. 6. From left to right, the input concentration (mol) of HCl(g) in the original model is increased by an order of magnitude per column, and each column represents a discrete step of the model. The model uses the same input composition as in Fig. 6 (Supplementary Data 2 Table S20) and data outputs from the altered HCl(g) models can be found in Supplementary Data 2 Tables S21–S27. Results are shown at \({V}_{A}/{V}_{M}\) = 0.33 or 25% mixing with ambient atmosphere. Results at \({V}_{A}/{V}_{M}\) = 0 or 0% air are shown in Fig. S13.
Several previous studies have demonstrated or inferred the importance of chloride for trace metal degassing, from both natural sampling (at arcs13,59,64,76,77,78 and other hotspot volcanoes11,79) and experimental work80. Thermodynamic speciation modelling of magmatic gases released at arc volcanoes, which typically have a higher Cl content and more oxidised magmas than intraplate volcanoes, has shown that volatile trace metals and metalloids are speciated predominantly as chloride gases during high-temperature degassing13,64. For example, in the magmatic plume of Yasur, a basaltic-andesitic volcano, at magmatic gas temperatures of 1100 °C and no atmospheric mixing (i.e., \({V}_{A}/{V}_{M}\) = 0) speciation modelling using HSC Chemistry indicates that Pb is mainly speciated as PbCl(g)13. This is in contrast to the Kīlauea plume modelled in this study, in which Pb is present predominantly as PbS(g) at \({V}_{A}/{V}_{M}\) = 0 and 1145 °C (modelling of the Kīlauea plume at 1100 °C does not change the dominant speciation, i.e., differences between Yasur and Kīlauea are not simply temperature effects). Similarly, BiS(g) is considerably less abundant in the Yasur plume speciation model compared to the Kīlauea model, with elemental Bi(g) and BiCl(g) dominant instead. Similar results arose from thermodynamic models of the magmatic plume of Mount Augustine, Alaska64.
Volcanic plumes from basaltic volcanoes in arc settings display characteristic differences in the relative abundances of volatile trace elements when compared to intraplate settings59 (although this does not apply to all magma compositions, particularly halogen-rich intraplate magma compositions13). X/SO2 ratios (Fig. 4) and emanation coefficients (Fig. 5) for some elements (e.g., In, Cu) are up to 2–4 orders of magnitude higher in arc plumes, while others (e.g., Te, Se) differ little between different tectonic settings. These differences have been explained by more oxidized magmas, and higher concentrations of chlorine (mean arc molar SO2/HCl ~1.974, versus SO2/HCl = 32–35 at Kīlauea in 2018) and water at arc settings, which promote metal partitioning directly into aqueous/saline aqueous fluid59. Speciation modelling in this study (Figs. 6 and 7) supports these conclusions—it shows that the elements most enriched in arc emissions are present as chloride gases (e.g., InCl(g), CuCl(g), Fig. 6b) at the point of emission. In contrast, elements that behave similarly between volcanoes at different tectonic settings are emitted as sulphide gases at Kīlauea (e.g., TeS(g), SeS(g), SnS(g), Fig. 6b).
The origin of the laze plume and late-stage degassing at the ocean entry
The composition of the laze plume differs significantly from the magmatic plume. As has been shown in previous work38, the laze plume is relatively Cl-rich and S-poor, which reflects the very large contribution of boiled seawater to the laze plume. The total (gas and PM) molar ratio for S/Cl is 0.1 (versus ~32–35 in the magmatic plume, Fig. 3). In the laze plume, a larger fraction of S and Cl concentrations is measured in the PM (~49% S, ~50 Cl), while the magmatic plume is gas-dominated at source for these elements (<1% of S and Cl in PM; Fig. 3). S and Cl concentrations are distributed uniformly across the five size fractions (Fig. 2d and Supplementary Data 1 Table S13).
Concentrations of volatile trace elements (ε > 0.001%) are generally lower in the laze plume than in the magmatic plume (with the exception of Cu and Ag, Fig. 8b and Supplementary Data 1 Tables S7, S11). As in the magmatic plume, volatile elements are found predominantly in the smallest size fraction: for all but Te, >40% of the concentration of volatile elements is found in the D ≤ 0.25-μm fraction, consistent with particles formed by gas-to-particle conversion post degassing54 (Fig. 2d and Supplementary Data 1 Table S13). Note that Te concentrations are close to detection limits (Supplementary Data 1 Table S7; ‘Methods’). In contrast to the magmatic plume, the size distribution of particulates containing refractory elements (ε < 0.001%) in the laze plume is strongly bimodal, with the concentrations of many elements only above detection limits in the smallest (D ≤ 0.25 μm) and largest (D ≥ 2.5 μm) size fractions (Fig. 2c and Supplementary Data 1 Table S13).

a The laze plume composition presented as X/Cl mass ratios in the plume compared to X/Cl mass ratios in a compilation of seawater compositions (data sources in Supplementary Data 1 Table S16) and discriminated using symbol colours according to emanation coefficients (‘Methods’; Supplementary Data 1 Table S10). Ash-uncorrected compositions are shown as circles (unfilled when the silicate correction can account for ~100% of their concentration in the laze plume; ‘Methods’). Corrected compositions are shown as either: (1) a small square when concentrations are reduced after correction, but still elevated above seawater; (2) a triangle when concentrations are concordant with seawater and unaffected by correction; and (3) star symbols when concentrations are elevated above seawater, and do not change position after correction. It should be noted that challenges with measuring Al in seawater113 can lead to high (several orders of magnitude) variability in measured concentrations, therefore these values should be treated with a degree of caution. WAF weighted ash fraction. b Laze and magmatic plume compositions compared using element-to-Mn ratios. Mn was chosen as a reference element as it is assumed to behave similarly in both plumes, i.e., in both plumes it is predominantly sourced from silicate material.
The similarity of the laze plume composition to seawater is assessed by comparing element/chlorine ratios (X/Cl) measured in the plume to X/Cl ratios in seawater (Fig. 8a). X/Cl ratios may be fractionated during the rapid evaporation of seawater, particularly for those elements that form salts38. However, such fractionations are currently unconstrained and are not considered further here. For elements abundant in seawater (Ca, Mg, Na, K), the laze plume composition is close to the compiled seawater composition (data and sources in Supplementary Data 1 Table S15). However, for Al, Ti, Fe, the REEs and some volatile metals (Cd, Bi, Cu, Ag, Zn, Re) the laze plume is enriched relative to seawater by 1–6 orders of magnitude.
Correcting the laze plume composition for a silicate contribution (‘Methods’) can fully account for elevated concentrations of some refractory elements in the laze plume (Ti, Gd, Tb; Fig. 8a). This may come from rapidly quenched silicate fragments generated by explosive hydro-magmatic fragmentation of lava as it interacts with seawater, or from congruent dissolution of basalt by seawater41. However, several refractory elements remain elevated above seawater after this silicate correction (Al, Fe, Nd, Sm, Pr, Eu, Ce, La). Results from lava–seawater interaction experiments may provide an explanation for this. In these experiments41, some refractory elements (e.g., La and Al) display contrasting behaviour when the water is seawater versus distilled water (DIW). This has been linked to the high particle reactivity of these elements41 (i.e., they are rapidly scavenged by particles in seawater but not in DIW), and also affected the concentration of Cu. Refractory elements that have adsorbed to particles may become enriched in the laze plume relative to seawater if these particles are then lofted into the laze plume.
Silicate-corrected concentrations of Cd, Zn, Ag, Cu, Bi, Re also remain elevated in the laze plume above seawater (Fig. 8a). Cu and Cd enrichments were also observed in seawater close to Kīlauea’s ocean entry in July 201840. Due to the moderate-to-high volatility of all these elements (ε > 0.001%), it is likely they are, to some extent, degassing from ocean entry lavas (as previously proposed for Cu and Cd41). Critical factors that could explain the enhanced degassing of these elements include the oxidation state of the lavas/gases and the availability of ligands, such as Cl and S. Basaltic melts have been shown to become more reduced during S degassing81,82, therefore lava flow melts at the ocean entry might be more reduced that those emitted at the source vent. However, we note that a study focused on the 2018 eruption of Kīlauea83 found that melts (matrix glass and melt inclusions) sampled from distal lava flows were more oxidised than those at the source, potentially due to atmospheric interaction during sub-aerial transport.
We have investigated the effect of increasing/decreasing the Cl content at seven different initial HCl gas concentrations (above and below that measured in the magmatic plume; ‘Methods’) on the speciation of trace elements in a gas mixture, at comparatively reduced (\({V}_{A}/{V}_{M}\) = 0), and oxidised (\({V}_{A}/{V}_{M}\) = 0.33) conditions (‘Methods’). We use these endmembers as an approximation of the speciation within the gas mixture due to changes in the oxidation state of the melt. While this simplified approach is not exactly the same as changing the initial melt oxidation state, and therefore the exact gas speciation may differ somewhat from that presented here, it provides a useful route to exploring the effects of oxidation on gas speciation.
The S/Cl ratio of the laze plume is comparable to the model run with ~100 times the amount of HCl gas in the magmatic plume (data in Supplementary Data 1 Tables S7, S11; Fig. 7). Focusing on those moderately to highly volatile elements elevated above seawater in the laze plume: Cu, Ag and Zn complex with chloride, at both \({V}_{A}/{V}_{M}\) = 0 and 0.33 (Fig. 6), and their degree of association with chloride increases markedly when the HCl gas content of the plume is increased (Figs. 7 and S13). At \({V}_{A}/{V}_{M}\) = 0.33, and at 100 times the initial HCl gas content, ~100% of Cu, Ag, Zn, Cd and Bi are present as chloride gaseous species (Fig. 7). At \({V}_{A}/{V}_{M}\) = 0, Cu, Zn and Ag are present almost exclusively as chloride gaseous species, while only a small proportion of Bi and Cd are complexed with chloride (Fig. S13). In contrast, Re is present as an chloride compound at \({V}_{A}/{V}_{M}\) = 0 and as an oxide compound at \({V}_{A}/{V}_{M}\) = 0.33. The true oxidation state of degassing from ocean entry lavas is likely to be somewhere in between these two model endmembers, which might allow almost all elements to degas as chlorides, to at least some extent. The high volatility of Re and the fact that it does not degas complexed with S (the majority of which degasses at the vent83,84) might combine to allow small amount of Re to degas at the ocean entry (the measured Re concentration is only 1.25 times detection limits). From the results of the speciation model, we might also expect other chloride-complexing elements to be elevated in the laze plume, such as Pb and In. These elements were not above detection limits in both filter pack samples (‘Methods’); however, In was above detection limits in the size-segregated sampling (Fig. 2d), and Pb was in a previous study of a laze plume at Kīlauea41.
Our speciation results show that the moderately (Cu, Zn, Ag) to highly volatile (Cd, Bi) elements found in the laze plume complex predominantly with chloride, especially at the high chloride concentrations of the laze. The concentration and emission rate of Cu in the laze plume may even be greater than that of the magmatic plume (Fig. 8b and Supplementary Note 9).
In contrast, volatile elements that complex with sulphide on degassing (Se, Te and As, Fig. 6b) appear to be absent from, or at very low concentrations in the laze plume. Previous studies at Kīlauea have shown that 50–90% of the sulphur in the melt is degassed during ascent to and extrusion at the surface (Fig. 9a)83,84, while significantly less Cl (~10%) is lost during this initial degassing66. We suggest that there is insufficient sulphur remaining in the melt in distal ocean entry lavas to facilitate significant degassing of Se, As and Te, and/or Se, As and Te concentrations in the lava reaching the coast are themselves extremely low, due to extensive degassing at the active vent (Fissure 8) and along the lava flows.

Occurring in a the magmatic plume and b the laze plume. Speciation changes before and after the compositional discontinuity are shown for the magmatic plume. For the laze plume, those elements below detection limits are crossed out with a solid line, and those that were not detected in our filter pack samples, but were in our impactor samples, or have been in other studies, are crossed out with a dashed line (see main text for full description). Molar S/Cl ratios are shown for the magmatic and laze plumes, and for seawater. Molar Na/Cl ratios are compared for the laze plume and seawater.
The degassing of chloride-complexing elements may be enhanced relative to sulphide-complexing elements at the ocean entry for several reasons (Fig. 9b). First, the early degassing of sulphur described above will decrease the S/Cl ratio of the remaining undegassed volatiles67. The higher solubility of Cl in silicate melts means that Cl is more likely to degas during lava flow emplacement than at the vent, while the majority of the sulphur will degas at the vent83,84. Second, the Cl-rich environment created at the ocean entry due to the extensive boiling of seawater may facilitate the degassing of Cl-complexes. This second mechanism is a much greater potential source of Cl than magmatic degassing alone. Alternatively, high-temperature Cl-rich seawater (heated through interaction with lavas) at the ocean entry may preferentially dissolve chloride-complexing elements (e.g., Cu) over refractory elements (e.g., REEs). However, lava–water interaction experiments41 showed that seawater produced no preferential dissolution of chloride-complexing elements compared to DIW. Our observations of the critical role of chloride from speciation modelling are supported by experimental work80, which has shown that increasing the level of chloride available during degassing can increase the amount of Cu hosted in the resulting sublimates80.
The mechanisms by which (1) volatiles degas from rapidly quenched lavas at the ocean entry, and (2) Cl from seawater enhances the degassing of elements with Cl-affinity, remain speculative. We suggest that the high levels of Cl at the ocean entry may act to stabilise metal chlorides at the high temperatures of lava–seawater interaction.
Synthesis
In this study, we demonstrate the strongly contrasting compositions of the genetically related magmatic and laze plumes associated with the 2018 eruption of Kīlauea Volcano.
The chemistry of these plumes at basaltic intraplate volcanoes such as Kīlauea (and other hotspot volcanoes in e.g., Iceland) provide valuable analogues for the gas and PM emissions during periods of very heightened volcanism in Earth’s history such as flood basalt eruptions (forming large igneous provinces, LIPs). Understanding LIP emissions is important due to their correlation with periods of relatively rapid global change within the geological record. Scaled up to estimates of the degassed magma volumes of LIPs, present-day plume compositions can be used to estimate the magnitude of volatile trace element emission rates that we might expect during flood basalt events. It should be noted, however, that X/SO2 values may differ by two orders of magnitude or more between different intraplate settings. The exact durations of pulses of activity during the eruption of LIPs such as the Deccan Traps are not known. However it is thought that a single eruption could produce up to several 1000 km3 of lava, and that 1000 km3 of lava would release ~3.6–5.4 × 103 Tg SO285.
Using X/SO2 ratios from Kīlauea in 2018, over a single eruption of 1000 km3 of lava, the Deccan Traps could have released huge amounts of trace metals and metalloids. For a duration of between 10 and 100 years, this yields an average emission rate of 300–7800-kg Cu day−1 (Kīlauea 2018 Cu/SO2 = 2.9 × 10−6–5.3 × 10−6) and 3100–72000-kg Se day−1 (Kīlauea 2018 Se/SO2 = 3.1 × 10−5–4.9 × 10−5). Using Cl emissions and X/Cl ratios yields emission rates of the same order of magnitude. These emission rates are extremely high; they are up to an order of magnitude higher than emission rates measured at Kīlauea in 2018 (with a duration of only 3 months rather than several years/decades). Further, Se emission rates from the Deccan Traps could have exceeded present-day anthropogenic emission rates from entire European countries (e.g., UK, Germany, Italy) by up to three orders of magnitude, and may have been comparable to anthropogenic emission rates from large industrialised countries such as China16.
Further, our results highlight the unique metal signature of lava–seawater interaction plumes, which would have occurred throughout Earth’s history during: oceanic plateau basalt eruptions28,29,30,31,32,33,34,35, continental flood basalt eruptions that reached coastlines26,27 and the early stages of continental rifting34,35. Degassing of trace metals from late-stage lava flows at chlorine-rich ocean entries, where melts are enriched in Cl over S due to fractional degassing66,67,68,69, produces a fundamentally different fingerprint of trace metals to magmatic plumes. Our results suggest that laze plumes have the potential to produce higher emission rates of Cu than even large magmatic plumes. Laze plumes can transfer elements directly to the marine biosphere25,40,41, where elements such as Cu can act as both pollutants (e.g., ref. 86) and important nutrients for microorganisms87. During large basaltic eruptions, distal ocean entry environments may have been important sites where volcanogenic metals are transferred from magmas into the environment and the biosphere, potentially hundreds of km27 from magmatic source vents.
Methods
Multi-GAS sampling
During the ground-based measurements of the magmatic plume emitted from Fissure 8, local wind conditions and thermal upwelling of the plume over the vent meant that the plume was only directed towards our instruments periodically. To account for this, a mobile multi-component Gas Analyser System (Multi-GAS88,89) was used to simultaneously measure concentrations of CO2, SO2, H2S (and H2O where possible) as well as pressure, temperature and relative humidity (RH). Air was sampled through a 1.0-μm particle filter exposed to ambient air, at a flow rate of 1.0 l min−1. SO2 and H2S electrochemical sensors (T3ST/F-TD2G-1A and T3H-TC4E-1A, both City Technology) were calibrated for 0–200 and 0–50 ppmv, respectively, with an accuracy of ±2% and a resolution of 0.1 ppmv. A non-dispersive infra-red spectrometer (Gascard EDI030102NG, Edinburgh Instruments) was calibrated for 0–3000-ppmv CO2 with an accuracy of ±1.5% and a resolution of 1 ppmv. Pressure, temperature and RH were measured at 1 Hz (KVM3/5 Galltec-Mela T/Rh sensor). The multi-GAS was calibrated at the University of Palermo prior to and following the field campaign; no sensor drift was identified. CO2/SO2 molar ratios were calculated using Ratiocalc 3.290 (Supplementary Note 2). A stable, background CO2 concentration of 370 ppm (when SO2 = 0) was subtracted from the measured signal (we note that this is lower than the expected background but this does not affect the derived CO2/SO2 ratios, which are based on relative changes in concentration). H2O concentrations were calculated from records of temperature and RH, using an ambient pressure of 1003.5 mbar (according to the Arden Buck equations relating the pressure of vapour saturation to temperature for moist air)91. Multi-GAS SO2 concentrations (Figs. S6, S7 and Supplementary Data 1 Tables S17, S18) were used to determine the time in plume (and therefore the volume of plume sampled) for ground-based sampling, which gives a time-averaged concentration. The SO2 concentration at which the instruments were inferred to be exposed to the grounding magmatic plume (rather than local fumarolic emissions) was set at 1-ppm SO2, which corresponds to the concentration baseline between SO2 peaks (Fig. S6b).
Filter pack and cascade impactor sampling
Filter packs were used to collect simultaneous samples of gas and bulk particulate material in both the magmatic and the laze plumes (Fig. S1), following a well-established technique92 previously used in multiple volcanological studies (e.g., refs. 9,93). The filter packs contained one particle filter (Whatman™ WTP polytetrafluoroethylene (PTFE) 47-mm diameter, pore size 1.0 μm) followed by 3-4 alkali-impregnated gas filters (Whatman™ Quantitative Filter Papers, Ashless, Grade 41, 55-mm diameter). Gas filters were washed, impregnated with a 5% K2CO3 + 1% glycerol mixture and dried in a clean-lab environment prior to use in the field. Base treatment of gas filters captures acidic gases (e.g., SO2 and HCl). PTFE filters were not pre-washed. Airflow through the filter pack was generated using an external 12-V pump (Charles Austin Capex) running at ~20 l min−1, if ground-based, and using an SKC Leland Legacy pump running at ~9 l min−1 if UAS-based (specific flow rates in Supplementary Note 1). The uncertainty introduced by variations in the flow rate, and by the accuracy of the flow meter are 10%. Immediately after sampling, the filter pack was closed by Parafilm to prevent particle loss and contamination. Filters were later transferred into individual polypropylene bags using metal-free tweezers and gloves.
We note that, due to limitations of access to the hazardous sampling area close to Fissure 8 and saturation of some samples, we were only able to acquire two non-saturated ground-based samples (FP_8_5 and FP_8_6 on 31/07/18). We assess the saturation of samples in the Supplementary Note 1 and individual gas filter concentrations are given in Supplementary Data 1 Tables S7 and S11. Filter pack samples can show broad compositional variations in element to SO2 ratios (X/SO2; sometimes several orders of magnitude; e.g., refs. 7,13) between sampling periods, even when these are only a few hours apart. However, we note that differences in X/SO2 ratios between samples in this study are comparatively small (and always well within 1 order of magnitude).
Cascade impactors consist of a series of stages between apertures of decreasing diameter. Larger particles are aerodynamically deposited on earlier collection stages, while finer particles are only deposited after passing through later narrower apertures, once they achieve a sufficiently high flow velocity to impinge on a collection stage. The cutoff diameter is conventionally reported as \({D}_{p50}\)—the particle diameter retained with 50% efficiency on the given stage, and is dependent on Stokes number parameters94. The Sioutas cascade impactor (SKC Inc., Fig. S2) was used to collect size-segregated particles in five size bins. Cutoff diameters are 2.5, 1.0, 0.50 and 0.25 μm, with all particles smaller than 0.25-μm collected on the final ‘after-stage’95,96. A flow rate of 9 l min−1 was maintained for PM collection by a Leland Legacy (SKC Inc.) dual diaphragm pump and PTFE filters were used on each stage of the impactor (Zefluor™, 25-mm diameter, 0.5 μm), including the after-stage (SKC Inc., 37-mm diameter, 2.0 μm).
A UAS (DJI Matrice 600 Pro, Fig. S3), owned and operated by the United States Geological Survey, was used to sample the magmatic and laze plumes, ~300 and ~100 m above the ground, respectively. The filter pack and cascade impactor were flown on separate flights due to the UAS payload capacity. However, instruments were flown in succession to obtain samples under as similar conditions as possible. Two sampling flights were launched from the Leilani Community Association (19.4660° N, 154.9156° W, 230-m a.s.l.) and flown into the magmatic plume, one each for the filter pack and cascade impactor. For all UAS flights, an approximate ‘in-plume’ duration was estimated using visual identification of the plume in footage acquired by the UAS-mounted camera during flight. It should also be noted that rotor turbulence (or thermal updrafts) may affect the ingestion of airborne PM during UAS sampling (e.g., by altering the ‘true’ particle size distribution), compared to traditional ground-based sampling, and this effect is not yet constrained. Even if these effects are subtle, due to the low concentrations of some of the elements considered, they might cause significant differences in measured values.
Aerial measurements were also complemented by ground-based sampling near to Fissure 8. The Fissure 8 plume was sampled by placing the sampling equipment in a specially designed drop-and-run backpack frame (Fig. S4) with the instruments attached ~50 cm off the ground to minimize contamination from windblown silicate material in the tephra field. The drop-and-run frame was designed to minimize the time spent setting up the equipment near the active volcanic vent. Ground-based sampling was carried out near the south-western edge of Fissure 8 by walking in through the evacuated section of Leilani Estates (Kupono Street) where the plume grounded periodically (approximate location shown in Fig. 1a, ~30 m away from the vent rim, which was ~10-m high at the time of sampling). Two filter packs were run simultaneously (Supplementary Note 1) alongside a multi-GAS. Only filter pack samples, where flow rates were measured successfully at the start, and end of sampling are presented here. Some filter packs showed evidence of saturation of the gas filters (Supplementary Note 1 and Fig. S5)—only data from the PM collected on the first filter should be interpreted in this case (Supplementary Data 1 Table S7).
The laze plume was sampled using solely aerial techniques, as the plume was lofting over the sea and therefore inaccessible to ground-based sampling. The UAS was flown from Isaac Hale Park (19.4595, −154.8423, 12-m a.s.l.), and Mackenzie State Recreation Area (19.4390, −154.8642, 37-m a.s.l.), both on the southeast coast. At Mackenzie State Recreation Area, one filter pack flight was undertaken. At Isaac Hale Park, one filter pack sampling flight was undertaken, followed by three consecutive cascade impactor sampling flights. The same impactor was flown on each flight to ensure a total exposure time sufficient to collect concentrations above detection limits for trace elements.
Background atmospheric measurements were made in 2019, after the eruption had ended and emissions of volcanic gases and particulates at Kīlauea were almost non-existent. These data are published in a separate study16, and comparison data are included in Supplementary Data 1 Table S8.
Sample extraction and analysis
All filter extractions were carried out in a class-10000 clean-lab environment at the University of Cambridge. PM on PTFE filters was first extracted in Milli-Q (MQ) water + propan-2-ol (to reduce the hydrophobicity of the PTFE filters), shaken and left for 24 h. After centrifugation, the water-soluble supernatant was extracted for analysis by ion chromatography (IC) and solution inductively coupled plasma-mass spectrometry and/or inductively coupled plasma-optical emission spectroscopy (ICP-MS and ICP-OES, respectively). The remaining solution and filters were transferred to acid-cleaned perfluoroalkoxy vials and refluxed at 120 °C for 3 h with concentrated distilled nitric acid (HNO3) + hydrofluoric acid (HF), followed by concentrated distilled HNO3 that was subsequently diluted with MQ to a 5% HNO3 solution for analysis. Solutions were dried down in between reflux stages at 90 °C in a class-100 laminar flow cabinet. Volatile species collected on gas filters were extracted in MQ water (20 ml) + H2O2 (250 μl; used to oxidise), shaken and left for 24 h (following97,98). Solution was then extracted for pH and IC measurements. SO2 and HCl, collected on the gas filters were measured (as SO42− and Cl−, respectively) by IC at the University of Leeds (on Thermo Dionex ion chromatograph system). These major anions were also measured by IC on the PTFE PM filters. Major and trace elements were measured in extraction solutions by ICP-MS (Thermo iCAP Qc ICP-MS) and/or ICP-OES (Thermo iCAP 7400). All major elements were measured by ICP-MS, with the exception of Mg, Ca, K, Ba, Sr, Al, P, Na, S, Fe, which were measured by ICP-OES. For filter pack measurements, Ba, Sr, Be, B, V, Cr, Co, Ni, Mo, Cs, Lu, U were measured but were below detection limits of the ICP-MS and/or ICP-OES instruments. For cascade impactor measurements, these elements were also below detection limits, in addition to Li, K, Sc and Ta. The presence of more elements below detection limits for impactor measurements reflects the fact that these instruments segregate PM by size onto five separate filters, whereas filter packs deposit all particulate material on a single filter. All elements and species are at least one order of magnitude above background levels measured in 2019 in the Leilani Estates16, except for Sc, Ta and Tb; these elements have not been considered further (Supplementary Data 1 Table S7). Chloride measured in the PM at Fissure 8 is also below background levels measured in 2019. Propagated errors are provided alongside data in the supplement and error propagation is detailed in Supplementary Note 7. Errors for all elements measured by ICP-MS or ICP-OES were ±10–18%. The H2O2 used to oxidize captured acidic gases during sample extraction is highly reactive, and can cause baseline effects during IC analysis that may lead to inaccurate quantification, especially for anions with short retention times (e.g., F−)99. Excess H2O2 was not removed (e.g., by reaction with MnO299) before analysis. Glycerol used to pre-treat filters before sampling can also interfere with the F− signal. Therefore, we do not present F− data here. Based on experimental results99, excess H2O2 can introduce errors for SO42− concentrations of ~30% (however we note that this may vary depending on the concentration of excess H2O2 in the solution). This source of error is incorporated into the propagated errors for IC measurements to give ±33% for Cl− and ±35% SO42−. Blank filters were also measured, with concentrations negligible (with the exception of W and P) compared to in-plume samples (Supplementary Data 1 Table S19).
SO2 and multi-element emission rates in the magmatic plume
SO2 emission rates of the magmatic plume from Fissure 8 were measured on 31 July 2018 (the same day as ground-based filter pack sampling) using a PiSpec instrument, a low-cost, 3D-printed UV spectrometer100,101(Supplementary Note 3). Typical traverse-mode differential optical absorption spectroscopy (DOAS) retrievals were employed102, traversing beneath the plume on state highway Route 130 (Fig. 1a) with the PiSpec looking upwards. A total of four traverses were performed, between 15:20 and 16:40 local time. Due to extremely high column densities of SO2 (at times exceeding 10,000 ppm m), the spectrometer retrievals were corrected for non-linearity following a H2O saturation correction (which can also be applied to DOAS retrievals of other gas species)103,104, using a modelled PiSpec response to account for the saturation effect. Relatively long wavelength fitting windows, typically 322–332 nm, were used to exploit weaker absorption bands where the saturation effect is less pronounced; due to the wide range of column densities found during traverses, each spectrum was processed manually with an optimised fitting window. Wind speed was taken from the National Oceanic and Atmospheric Administration Global Data Assimilation System, which provides 1˚ latitudinal/longitudinal resolution. The speed was estimated to be 6.5 m s−1 during the times of the traverses.
Trace element fluxes cannot be measured directly, and are estimated based on the measured X/SO2 ratio in the plume and the independently measured SO2 emission rate, as
The weighted mean of SO2 emission rates measured during our fieldwork were 39 ± 11 kt day−1 (where the error is the standard deviation of three measurements and the full range is 27–54 kt day−1, Supplementary Note 3). Another highly detailed study found SO2 emission rates of 71 ± 31 kt day−1 on the same day of sampling45—these are within 1 SD of this study’s average. Earlier in the eruption, during June and early July 2018, a higher emission rate of >200 kt day−1 SO2 was reported45. If X/SO2 ratios remain constant with changing SO2 emission rate, the metal emission rates earlier in the eruption may have been up to four times higher than those presented here.
Silicate (ash) correction
To determine the concentration of elements in the non-silicate aerosol phase, the contribution from a silicate (ash) component must be removed. Here, for our ash composition, we used a matrix glass composition (Supplementary Data 1 Table S16). Ideally, this composition would be measured in a melt that is closely temporally related to the melt from which the measured plume emissions are degassed. However, this is often not possible as it requires sampling in hazardous or inaccessible environments (e.g., active lava flows or recent tephra falls). Here, where data availability and quality allowed for the elements of interest, we use an average composition of basaltic matrix glass from two 2018 samples (one overflow sample, one lava channel sample) collected close to the time and location at which the magmatic plume was sampled105. The elements in these matrix glass samples were measured by electron microprobe and LA-ICP-MS (laser ablation—ICP-MS)105. Where elements were not measured in these 2018 samples, either a 1971 summit composition53 or preferred BHVO values were used (Supplementary Data 1 Table S14).
For the ash correction, we consider the concentration of element A on the filter to be derived from two volcanic components, silicate ash and non-silicate aerosol
Where \({X}_{{\mathrm{ash}}}\) is the proportion of element A on the filter that is present in the ash phase. If A is a lithophile/refractory element (e.g., REEs, Sc Th or some combination of lithophiles), we assume that the concentration of this element in the aerosol phase is zero (\({\left[A\right]}_{{\mathrm{aerosol}}}\) = 0), allowing the equation to be simplified to
In the ash corrections performed in this study, we use a combination of major and trace refractory elements to calculate \({X}_{{\mathrm{ash}}}\): Fe, Al, Ti, Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb (other combinations of refractory elements are compared in Fig. S8). Light REEs La and Ce were excluded as they displayed evidence of volatile behaviour (Fig. 2). The higher concentrations of major elements (compared to REEs) in silicate material, allow smaller ash contributions to be resolved, and are thus more sensitive to the small amounts of ash in the Fissure 8 or laze plumes. WAFs can then be calculated for each element in each sample
Regardless of how ash-poor a plume appears during the sampling period, small amounts of ash can have a disproportionately large effect on the concentrations measured on the filter, and therefore the fluxes and measures of volatility calculated from the data, due to the high concentrations of many elements in silicate material. For example, a contribution of only 0.001% ash by weight to the concentration on the particulate filter, would result in a measured ash-derived concentration of 85-ppm Fe and 71-ppm Al.
Measures of volatility
We calculate emanation coefficients (\({\varepsilon }_{x}\)) using the ratio of element concentration to sulphur (\(X/S\)) in the volcanic plume, combined with an estimate of the total S degassed during sub-aerial eruption
where \({X}_{{\mathrm{aerosol}}}\) is the concentration of element \(X\) measured on particulate filters (corrected for ash, as above); \({S}_{{\mathrm{total}}}\) is the total S measured on filter pack particulate and gas filters and \({X}_{{MG}}\) is the concentration of element \(X\) remaining in the degassed matrix glass (same composition as used in the ash correction, described above). We note that this method of emanation coefficient calculation relies on the assumption that metal degassing scales linearly with increasing S degassing, which may not fully take into account those metals that might leave the magma as halides.
Ideally, \({S}_{{\mathrm{degassed}}}\) is the concentration of S (in ppm) degassed from the melt from which the measured emissions are released, measured by comparing degassed and undegassed glass compositions. However, this data is not yet available for the Fissure 8 lavas. Recent work53 suggests that during the 1971 summit eruption as much as ~1200–1300 ppm of S was degassed, with degassed amounts of up to ~1465 ppm possible in the most primitive eruptions of Kīlauea. Therefore to cover the full range of possible \({S}_{{\mathrm{degassed}}}\) values, we use a \({S}_{{\mathrm{degassed}}}\) value of 1250 ± 300 ppm to calculate \({\varepsilon }_{x}\). As we calculate emanation coefficients using silicate-corrected plume composition data, our emanation coefficients are relative, i.e., during the ash-correction elements used to calculate \({X}_{{\mathrm{ash}}}\) are assumed to be only present in a silicate phase, and therefore these elements have emanation coefficients of zero, but may degas in the gas phase to a limited extent.
Equilibrium chemistry modelling
The Gibbs free energy minimisation (GEM) module of HSC Chemistry (version 9.9.2, Outotec Research Oy, Finland) was used to model gas phase speciation in the Fissure 8 plume. Detailed descriptions of the model are provided in previous works70,74,106. Model inputs are major and trace species gas concentrations (kmol), pressure, temperature and the phases expected in the plume. Using the method from Helz and Thornber107, the MgO content (6.50 ± 0.13 wt%105) of Fissure 8 lavas give an equilibrium temperature of the magmatic gases emitted at the time of plume sampling of ~\(1145{\pm }_{3.6}^{1.6}\) °C. Further cooling of gases may occur during adiabatic expansion of bubbles; the extent of this cooling depends on the size of the bubbles, with bubbles of radius 2 m producing a cooling of ~100 °C108. Based of footage of degassing taken during sampling, we assess that this is a reasonable upper limit for bubble size during degassing at Fissure 8. Therefore an additional lower uncertainty of 100 °C is considered but did not have a significant effect on the dominant speciation of volatile trace elements. We determine the temperature of the gas mixture at the range of mixing ratios considered (\({0} < ({V_A}/{V_M}) < 0.33\)) using a simple fluid mixing model, defined as follows:
where \({T}_{{\mathrm{mixture}}}\) is the temperature of the mixture of magmatic gases; \({T}_{n}\) is the input temperature of the gases (i.e., 25 °C for air; 1145 °C for magmatic gas); \({m}_{n}\) is the mass of gas (in kg) present in the mixture and \({c}_{n}\) is the specific heat of the gas (in kJ/(kg*K)) at the relevant \({T}_{n}\). Specific heat values for atmospheric N2, O2 and Ar gases at 25 °C are 1.0, 0.9 and 0.5 kJ/(kg*K), respectively; the specific heat for magmatic H2O vapour at 1145 °C is 2.5 kJ/(kg*K)). The mixing model produces an approximately linear relationship between temperature (from 1145 to 1016 °C) and \({V}_{A}/{V}_{M}\) (0–0.33; Fig. S11), which is then used to parameterise the HSC Chemistry GEM model. Input concentrations of major gas species (e.g., H2O, CO2, SO2, H2S etc.) are from Gerlach109, and trace metal concentrations are from the filter pack samples (ash-corrected average of samples 8_5 and 8_6; sample summary in Supplementary Note 1; data in Supplementary Data 1 Table S8) presented in this study. While a more recent gas composition for major species would be favourable, the speciation model is very sensitive to the concentrations of reduced species and this dataset provides the most complete composition of the Kīlauea plume and allows comparison with previous work70. The data presented in Gerlach109 display a similar CO2/SO2 ratio (~0.3) to that measured by the multi-Gas in this study (0.38; Supplementary Note 2) and others110. Approximate HBr and HF concentrations for the plume were calculated using ratios from 2008 gas measurements at Kīlauea’s summit9. Trace metal data were corrected for the dilution that occurs over the distance between emission at source and sampling location by multiplication of element ratios to total sulphur as follows:
where \({X}_{{\mathrm{source}}}\) is the concentration of the element of interest at the point of emission, \({S}_{T}\) is the total sulphur concentration (mol) measured on all filters of the filter pack, \({X}_{{FP}}\) is the ash-corrected concentration (mol) of element \(X\) measured on the particle filter of our filter packs and \({S}_{{\mathrm{source}}}\) is the concentration (mol) of S in the plume at the point of emission. In this case \({S}_{{\mathrm{source}}}\) is the total sulphur concentration (measured as SO2, H2S, S2 and COS gases) as determined by Gerlach109. An assumption of the modelling is that the elemental composition of the plume is not fractionated (i.e., not changed by any depositional processes) between the point of emission at the lava–air interface and the point of sampling, which we consider reasonable given that the travel time for gases and particulates to reach the sampling instruments—either on the ground or to the UAS—is likely to be on the order of a few minutes or less. The speciation of the magmatic plume was also modelled at 0.001, 0.01, 0.1, 10, 100 and 1000 times the actual measured initial HCl gas concentration to assess the effect of changing Cl concentrations in the plume on speciation. All input conditions, including the simple temperature model, were the same as in the main model (Supplementary Data 2 Table S20).
Data availability
All data used and generated in this study can be found in the Supplementary Data Files: Supplementary Data 1—contains Supplementary Tables S7–S19—raw measured compositions and calculated values (Tables S1–S6 are in the Supplementary Information). Supplementary Data 2—contains Supplementary Tables S20–S27—chemical speciation model inputs and outputs. Measured data (gas and PM compositional data from filter pack and cascade impactor measurements) have also been deposited in the Natural Environment Research Council (NERC) Centre for Environmental Data Analysis (CEDA) database (doi: 10.5285/656204c257144708a641507c78187aaa)111.
Change history
01 June 2021
A Correction to this paper has been published: https://doi.org/10.1038/s43247-021-00189-5
References
Carn, S. A., Fioletov, V. E., McLinden, C. A., Li, C. & Krotkov, N. A. A decade of global volcanic SO2 emissions measured from space. Sci. Rep. 7, 1–12 (2017).
Martin, R. S., Mather, T. A. & Pyle, D. M. Volcanic emissions and the early Earth atmosphere. Geochim. Cosmochim. Acta 71, 3673–3685 (2007).
Lee, C., Jiang, H., Dasgupta, R. & Torres, M. A framework for understanding whole-Earth carbon cycling. in Deep Carbon: Past to Present (eds Orcutt, B., Daniel, I. & Dasgupta, R.) 313–357 (Cambridge University Press, 2019).
von Glasow, R., Bobrowski, N. & Kern, C. The effects of volcanic eruptions on atmospheric chemistry. Chem. Geol. 263, 131–142 (2009).
Mather, T. A., Pyle, D. M. & Oppenheimer, C. Tropospheric volcanic aerosol. in Volcanism and the Earth’s Atmosphere 189–212 (American Geophysical Union (AGU), 2013), https://doi.org/10.1029/139GM12.
Allard, P. et al. Acid gas and metal emission rates during long‐lived basalt degassing at Stromboli Volcano. Geophys. Res. Lett. 27, 1207–1210 (2000).
Aiuppa, A., Dongarrà, G., Valenza, M., Federico, C. & Pecoraino, G. Degassing of trace volatile metals during the 2001 eruption of Etna. Washington DC American Geophysical Union Geophysical Monograph Series 139, 41–54 (2003).
Moune, S., Gauthier, P.-J. & Delmelle, P. Trace elements in the particulate phase of the plume of Masaya Volcano, Nicaragua. J. Volcanol. Geotherm. Res. 193, 232–244 (2010).
Mather, T. A. et al. Halogens and trace metal emissions from the ongoing 2008 summit eruption of Kīlauea volcano, Hawai’i. Geochim. Cosmochim. Acta 83, 292–323 (2012).
Zelenski, M. E. et al. Trace elements in the gas emissions from the Erta Ale volcano, Afar, Ethiopia. Chem. Geol. 357, 95–116 (2013).
Gauthier, P., Sigmarsson, O., Gouhier, M., Haddadi, B. & Moune, S. Elevated gas flux and trace metal degassing from the 2014–2015 fissure eruption at the Bárðarbunga volcanic system, Iceland. J. Geophys. Res. Solid Earth 121, 1610–1630 (2016).
Allard, P. et al. Prodigious emission rates and magma degassing budget of major, trace and radioactive volatile species from Ambrym basaltic volcano, Vanuatu island Arc. J. Volcanol. Geotherm. Res. 322, 119–143 (2016).
Mandon, C. L., Christenson, B. W., Schipper, C. I., Seward, T. M. & Garaebiti, E. Metal transport in volcanic plumes: a case study at White Island and Yasur volcanoes. J. Volcanol. Geotherm. Res. 369, 155–171 (2019).
Buat‐Ménard, P. & Arnold, M. The heavy metal chemistry of atmospheric particulate matter emitted by Mount Etna Volcano. Geophys. Res. Lett. 5, 245–248 (1978).
Murata, K. J. Occurrence of CuCl emission in volcanic flames [Hawaii]. Am. J. Sci. 258, 769–772 (1960).
Ilyinskaya, E. et al. Rapid metal pollutant deposition from the volcanic plume of Kīlauea, Hawai’i. Commun Earth Environ. 2, 78, https://doi.org/10.1038/s43247-021-00146-2 (2021).
Nriagu, J. O. A global assessment of natural sources of atmospheric trace metals. Nature 338, 47–49 (1989).
Sabra, S., Malmqvist, E., Saborit, A., Gratacós, E. & Gomez Roig, M. D. Heavy metals exposure levels and their correlation with different clinical forms of fetal growth restriction. PLoS ONE 12, https://doi.org/10.1371/journal.pone.0185645 (2017).
Rehman, K., Fatima, F., Waheed, I. & Akash, M. S. H. Prevalence of exposure of heavy metals and their impact on health consequences. J. Cell. Biochem. 119, 157–184 (2018).
Whitty, R. C. W. Spatial and Temporal Variations in SO2 and PM2. 5 Levels Around Kīlauea Volcano, Hawai'i During 2007–2018. Frontiers in Earth Science 8, 36, https://doi.org/10.3389/feart.2020.00036 (2020).
Delmelle, P., Stix, J., Baxter, P., Garcia-Alvarez, J. & Barquero, J. Atmospheric dispersion, environmental effects and potential health hazard associated with the low-altitude gas plume of Masaya volcano, Nicaragua. Bull. Volcanol. 64, 423–434 (2002).
Schmidt, A. et al. Excess mortality in Europe following a future Laki-style Icelandic eruption. Proc. Natl. Acad. Sci. 108, 15710–15715 (2011).
Percival, L. M. E. et al. Mercury evidence for pulsed volcanism during the end-Triassic mass extinction. Proc. Natl. Acad. Sci. 114, 7929–7934 (2017).
Broadley, M. W., Barry, P. H., Ballentine, C. J., Taylor, L. A. & Burgess, R. End-Permian extinction amplified by plume-induced release of recycled lithospheric volatiles. Nat. Geosci. 11, 682–687 (2018).
Wilson, S. T. et al. Kīlauea lava fuels phytoplankton bloom in the North Pacific Ocean. Science 365, 1040–1044 (2019).
Wells, R. E., Niem, A. R., Evarts, R. C. & Hagstrum, J. T. The Columbia River Basalt Group—from the gorge to the sea. GSA Field Guides, 15, 737–774, https://doi.org/10.1130/2009.fld015 (2009).
Self, S., Jay, A. E., Widdowson, M. & Keszthelyi, L. P. Correlation of the Deccan and Rajahmundry Trap lavas: are these the longest and largest lava flows on Earth? J. Volcanol. Geotherm. Res. 172, 3–19 (2008).
Coffin, M. F. & Eldholm, O. Large igneous provinces: crustal structure, dimensions, and external consequences. Rev. Geophys. 32, 1–36 (1994).
Tarduno, J. A. et al. Rapid formation of Ontong Java Plateau by Aptian Mantle Plume volcanism. Science 254, 399–403 (1991).
Thordarson, T. Accretionary-lapilli-bearing pyroclastic rocks at ODP Leg 192 Site 1184: a record of subaerial phreatomagmatic eruptions on the Ontong Java Plateau. Geol. Soc. Lond. Spec. Publ 229, 275–306 (2004).
Chambers, L. M., Pringle, M. S. & Fitton, J. G. Phreatomagmatic eruptions on the Ontong Java Plateau: an Aptian 40Ar/39Ar age for volcaniclastic rocks at ODP Site 1184. Geol. Soc. Lond. Spec. Publ 229, 325–331 (2004).
Schlanger, S. O., Jenkyns, H. C. & Premoli-Silva, I. Volcanism and vertical tectonics in the Pacific Basin related to global Cretaceous transgressions. Earth Planet. Sci. Lett. 52, 435–449 (1981).
Beiersdorf, H., Bach, W., Duncan, R., Erzinger, J. & Weiss, W. New evidence for the production of EM-type ocean island basalts and large volumes of volcaniclastites during the early history of the Manihiki Plateau. Mar. Geol. 122, 181–205 (1995).
White, R. & McKenzie, D. Magmatism at rift zones: the generation of volcanic continental margins and flood basalts. J. Geophys. Res. Solid Earth 94, 7685–7729 (1989).
Stokke, E. W., Liu, E. J. & Jones, M. T. Evidence of explosive hydromagmatic eruptions during the emplacement of the North Atlantic Igneous Province. Volcanica 3, 227–250 (2020).
Büttner, R. & Zimanowski, B. Physics of thermohydraulic explosions. Phys. Rev. E 57, 5726–5729 (1998).
Mattox, T. N. & Mangan, M. T. Littoral hydrovolcanic explosions: a case study of lava–seawater interaction at Kilauea Volcano. J. Volcanol. Geotherm. Res. 75, 1–17 (1997).
Edmonds, M. & Gerlach, T. M. The airborne lava–seawater interaction plume at Kīlauea Volcano, Hawaiʻi. Earth Planet. Sci. Lett. 244, 83–96 (2006).
Gerlach, T. M., Krumhansl, J. L., Fournier, R. O. & Kjargaard, J. Acid rain from the heating and evaporation of seawater by molten lava: a new volcanic hazard. EOS 70, 1421–1422 (1989).
Hawco, N. J. et al. Metal isotope signatures from lava-seawater interaction during the 2018 eruption of Kīlauea. Geochim. Cosmochim. Acta https://doi.org/10.1016/j.gca.2020.05.005 (2020).
Resing, J. A. & Sansone, F. J. The chemistry of lava-seawater interactions II: the elemental signature. Geochim. Cosmochim. Acta 66, 1925–1941 (2002).
Resing, J. A. & Sansone, F. J. The chemistry of lava–seawater interactions: the generation of acidity. Geochim. Cosmochim. Acta 63, 2183–2198 (1999).
Wright, T. L. & Klein, F. W. Two Hundred Years of Magma Transport and Storage at Kīlauea Volcano, Hawai’i, 1790–2008. 258 http://pubs.er.usgs.gov/publication/pp1806 (2014).
Neal, C. A. et al. The 2018 rift eruption and summit collapse of Kīlauea Volcano. Science 363, 367–374 (2019).
Kern, C. et al. Quantifying gas emissions associated with the 2018 rift eruption of Kīlauea Volcano using ground-based DOAS measurements. Bull. Volcanol. 82, 55 (2020).
Elias, T., Kern, C., Horton, K. A., Sutton, A. J. & Garbeil, H. Measuring SO2 emission rates at Kīlauea Volcano, Hawaii, using an array of upward-looking UV spectrometers, 2014–2017. Front. Earth Sci. 6, 214, https://doi.org/10.3389/feart.2018.00214 (2018).
Tam, E. et al. Volcanic air pollution over the Island of Hawai’i: Emissions, dispersal, and composition. Association with respiratory symptoms and lung function in Hawai’i Island school children. Environ. Int. 92–93, 543–552 (2016).
Patrick, M. R. et al. Cyclic lava effusion during the 2018 eruption of Kīlauea Volcano. Science 366, eaay9070, https://doi.org/10.1126/science.aay9070 (2019).
Kauahikaua, J. P. & Trusdell, F. A. Have Humans Influenced Volcanic Activity on the Lower East Rift Zone of Kīlauea Volcano? A Publication Review. https://pubs.er.usgs.gov/publication/ofr20201017 (2020).
Mather, T. A. Volcanoes and the environment: lessons for understanding Earth’s past and future from studies of present-day volcanic emissions. J. Volcanol. Geotherm. Res. 304, 160–179 (2015).
Lambert, G., Le Cloarec, M. F., Ardouin, B. & Le Roulley, J. C. Volcanic emission of radionuclides and magma dynamics. Earth Planet. Sci. Lett. 76, 185–192 (1985).
Gauthier, P.-J. & Le Cloarec, M.-F. Variability of alkali and heavy metal fluxes released by Mt. Etna volcano, Sicily, between 1991 and 1995. J. Volcanol. Geotherm. Res. 81, 311–326 (1998).
Wieser, P. E., Jenner, F., Edmonds, M., Maclennan, J. & Kunz, B. E. Chalcophile elements track the fate of sulfur at Kīlauea Volcano, Hawai’i. Geochim. Cosmochim. Acta 282, 245–275 (2020).
Whitby, K. T. The physical characteristics of sulfur aerosols. in Sulfur in the Atmosphere (eds Husar, R. B., Lodge, J. P. & Moore, D. J.) 135–159 (Pergamon, 1978). https://doi.org/10.1016/B978-0-08-022932-4.50018-5.
Óskarsson, N. The interaction between volcanic gases and tephra: fluorine adhering to tephra of the 1970 hekla eruption. J. Volcanol. Geotherm. Res. 8, 251–266 (1980).
Delmelle, P., Wadsworth, F. B., Maters, E. C. & Ayris, P. M. High temperature reactions between gases and ash particles in volcanic eruption plumes. Rev. Mineral. Geochem. 84, 285–308 (2018).
Berresheim, H. & Jaeschke, W. Study of metal aerosol systems as a sink for atmospheric SO2. J. Atmospheric Chem. 4, 311–334 (1986).
Coffey, M. T. Observations of the impact of volcanic activity on stratospheric chemistry. J. Geophys. Res. Atmospheres 101, 6767–6780 (1996).
Edmonds, M., Mather, T. A. & Liu, E. J. A distinct metal fingerprint in arc volcanic emissions. Nat. Geosci. 11, 790–794 (2018).
Calabrese, S. et al. Passive degassing at Nyiragongo (D.R. Congo) and Etna (Italy) volcanoes. Ann. Geophys. 57, https://doi.org/10.4401/ag-6637 (2015).
Wardell, L. J., Kyle, P. R. & Counce, D. Volcanic emissions of metals and halogens from White Island (New Zealand) and Erebus volcano (Antarctica) determined with chemical traps. J. Volcanol. Geotherm. Res. 177, 734–742 (2008).
Zreda‐Gostynska, G., Kyle, P. R., Finnegan, D. & Prestbo, K. M. Volcanic gas emissions from Mount Erebus and their impact on the Antarctic environment. J. Geophys. Res. Solid Earth 102, 15039–15055 (1997).
Wieser, P. E., Edmonds, M., Maclennan, J., Jenner, F. E. & Kunz, B. E. Crystal scavenging from mush piles recorded by melt inclusions. Nat. Commun. 10, 5797 (2019).
Symonds, R. B., Reed, M. H. & Rose, W. I. Origin, speciation, and fluxes of trace-element gases at Augustine volcano, Alaska: Insights into magma degassing and fumarolic processes. Geochim. Cosmochim. Acta 56, 633–657 (1992).
Edmonds, M. & Gerlach, T. M. Vapor segregation and loss in basaltic melts. Geology 35, 751–754 (2007).
Greenland, L. P., Rose, W. I. & Stokes, J. B. An estimate of gas emissions and magmatic gas content from Kilauea volcano. Geochim. Cosmochim. Acta 49, 125–129 (1985).
Sigmarsson, O., Moune, S. & Gauthier, P.-J. Fractional degassing of S, Cl and F from basalt magma in the Bárðarbunga rift zone, Iceland. Bull. Volcanol. 82, 54 (2020).
Burton, M., Allard, P., Murè, F. & Oppenheimer, C. FTIR remote sensing of fractional magma degassing at Mount Etna, Sicily. Geol. Soc. Lond. Spec. Publ. 213, 281–293 (2003).
Toutain, J. P. et al. Vapor deposition of trace elements from degassed basaltic lava, Piton de la Fournaise volcano, Reunion Island. J. Volcanol. Geotherm. Res. 40, 257–268 (1990).
Martin, R. S., Mather, T. A. & Pyle, D. M. High-temperature mixtures of magmatic and atmospheric gases. Geochem. Geophys. Geosystems 7, Q04006 https://doi.org/10.1029/2005GC001186 (2006).
Gerlach, T. M. & Nordlie, B. E. The C-O-H-S gaseous system; part II, temperature, atomic composition, and molecular equilibria in volcanic gases. Am. J. Sci. 275, 377–394 (1975).
Rubin, K. Degassing of metals and metalloids from erupting seamount and mid-ocean ridge volcanoes: Observations and predictions. Geochim. Cosmochim. Acta 61, 3525–3542 (1997).
Roberts, T., Dayma, G. & Oppenheimer, C. Reaction rates control high-temperature chemistry of volcanic gases in air. Front. Earth Sci. 7, 154, https://doi.org/10.3389/feart.2019.00154 (2019).
Gerlach, T. M. Volcanic sources of tropospheric ozone-depleting trace gases. Geochem. Geophys. Geosystems 5, https://doi.org/10.1029/2004GC000747 (2004).
Ilyinskaya, E., Martin, R. S. & Oppenheimer, C. Aerosol formation in basaltic lava fountaining: Eyjafjallajökull volcano, Iceland. J. Geophys. Res. Atmospheres https://doi.org/10.1029/2011JD016811 (2018).
Wahrenberger, C., Seward, T. M. & Dietrich, V. Volatile trace-element transport in high-temperature gases from Kudriavy volcano (Iturup, Kurile Islands, Russia). Geochem. Soc. Spec. Publ 7, 307–327 (2002).
Henley, R. W. & Seward, T. M. Gas–solid reactions in arc volcanoes: ancient and modern. Rev. Mineral. Geochem. 84, 309–349 (2018).
Symonds, R. B., Rose, W. I., Reed, M. H., Lichte, F. E. & Finnegan, D. L. Volatilization, transport and sublimation of metallic and non-metallic elements in high temperature gases at Merapi Volcano, Indonesia. Geochim. Cosmochim. Acta 51, 2083–2101 (1987).
Renggli, C. J., King, P. L., Henley, R. W. & Norman, M. D. Volcanic gas composition, metal dispersion and deposition during explosive volcanic eruptions on the Moon. Geochim. Cosmochim. Acta 206, 296–311 (2017).
Scholtysik, R. & Canil, D. Investigation of the effect of Cl on the transport and sublimation of volatile trace metals in volcanic gases using benchtop fumarole experiments. J. Volcanol. Geotherm. Res. 395, 106838 (2020).
Moussallam, Y. et al. The impact of degassing on the oxidation state of basaltic magmas: a case study of Kīlauea volcano. Earth Planet. Sci. Lett. 450, 317–325 (2016).
Longpré, M.-A., Stix, J., Klügel, A. & Shimizu, N. Mantle to surface degassing of carbon- and sulphur-rich alkaline magma at El Hierro, Canary Islands. Earth Planet. Sci. Lett. 460, 268–280 (2017).
Lerner, A. The Depths and Locations of Magma Reservoirs and Their Consequences for the Behavior of Sulfur and Volcanic Degassing. (2020).
Cashman, K. V., Mangan, M. T. & Newman, S. Surface degassing and modifications to vesicle size distributions in active basalt flows. J. Volcanol. Geotherm. Res. 61, 45–68 (1994).
Self, S., Blake, S., Sharma, K., Widdowson, M. & Sephton, S. Sulfur and chlorine in late cretaceous deccan magmas and eruptive gas release. Science 319, 1654–1657 (2008).
Leal, P. P. et al. Copper pollution exacerbates the effects of ocean acidification and warming on kelp microscopic early life stages. Sci. Rep. 8, 14763 (2018).
Walsh, M. J., Goodnow, S. D., Vezeau, G. E., Richter, L. V. & Ahner, B. A. Cysteine enhances bioavailability of copper to marine phytoplankton. Environ. Sci. Technol. 49, 12145–12152 (2015).
Shinohara, H. A new technique to estimate volcanic gas composition: plume measurements with a portable multi-sensor system. J. Volcanol. Geotherm. Res. 143, 319–333 (2005).
Aiuppa, A. et al. Forecasting Etna eruptions by real-time observation of volcanic gas composition. Geology 35, 1115–1118 (2007).
Tamburello, G. Ratiocalc Software for processing data from multicomponent volcanic gas analyzers. Comput. Geosci. 82, 63–67 (2015).
Buck, A. L. New equations for computing vapor pressure and enhancement factor. J. Appl. Meteorol. Climatol. 20, 1527–1532 (1981).
Allen, A. G., Baxter, P. J. & Ottley, C. J. Gas and particle emissions from Soufrière Hills Volcano, Montserrat, West Indies: characterization and health hazard assessment. Bull. Volcanol. 62, 8–19 (2000).
Ilyinskaya, E. et al. Understanding the environmental impacts of large fissure eruptions: Aerosol and gas emissions from the 2014–2015 Holuhraun eruption (Iceland). Earth Planet. Sci. Lett. 472, 309–322 (2017).
Marple, V. A., Rubow, K. L. & Behm, S. M. A Microorifice Uniform Deposit Impactor (MOUDI): Description, calibration, and use. Aerosol Sci. Technol. 14, 434–446 (1991).
May, K. R. The cascade impactor: an instrument for sampling coarse aerosols. J. Sci. Instrum. 22, 187–195 (1945).
Marple, V. A. & Willeke, K. Impactor design. Atmospheric Environ. 1967 10, 891–896 (1976).
Witt, M. L. I. et al. Mercury and halogen emissions from Masaya and Telica volcanoes, Nicaragua. J. Geophys. Res. Solid Earth 113, https://doi.org/10.1029/2007JB005401 (2008).
Aiuppa, A. et al. Emission of bromine and iodine from Mount Etna volcano. Geochem. Geophys. Geosystems 6, https://doi.org/10.1029/2005GC000965 (2005).
Wittmer, J. et al. Active alkaline traps to determine acidic-gas ratios in volcanic plumes: sampling techniques and analytical methods. Geochem. Geophys. Geosystems 15, 2797–2820 (2014).
Wilkes, T. C., McGonigle, A. J. S., Willmott, J. R., Pering, T. D. & Cook, J. M. Low-cost 3D printed 1nm resolution smartphone sensor-based spectrometer: instrument design and application in ultraviolet spectroscopy. Opt. Lett. 42, 4323–4326 (2017).
Wilkes, T. C. et al. The PiSpec: a low-cost, 3D-printed spectrometer for measuring volcanic SO2 emission rates. Front. Earth Sci. 7, 65 https://doi.org/10.3389/feart.2019.00065 (2019).
Platt, U. & Stutz, J. Differential optical absorption spectroscopy (DOAS). Physics of Earth and Space Environments (Springer-Verlag, 2008).
Wagner, T., Heland, J., Zöger, M. & Platt, U. A fast H2O total column density product from GOME—validation with in-situ aircraft measurements. Atmospheric Chem. Phys. 3, 651–663 (2003).
Kern, C., Masias, P., Apaza, F., Reath, K. A. & Platt, U. Remote measurement of high preeruptive water vapor emissions at Sabancaya volcano by passive differential optical absorption spectroscopy. J. Geophys. Res. Solid Earth 122, 3540–3564 (2017).
Wieser, P. E. et al. Reconstructing magma storage depths for the 2018 Kı̄lauean eruption from melt inclusion CO2 contents: the importance of vapor bubbles. Geochem. Geophys. Geosystems 22, e2020GC009364, https://doi.org/10.1029/2020GC009364 (2021).
Symonds, R. B., & Reed, M. H. Calculation of multicomponent chemical equilibria in gas-solid-liquid systems: Calculation methods, thermochemical data, and applications to studies of high-temperature volcanic gases with examples from Mt. St. Helens. Am. J. Sci. U. S. 293, 8 (1993).
Helz, R. T. & Thornber, C. R. Geothermometry of Kilauea Iki lava lake, Hawaii. Bull. Volcanol. 49, 651–668 (1987).
Oppenheimer, C. et al. Influence of eruptive style on volcanic gas emission chemistry and temperature. Nat. Geosci. 11, 678–681 (2018).
Gerlach, T. M. Oxygen buffering of Kilauea volcanic gases and the oxygen fugacity of Kilauea basalt. Geochim. Cosmochim. Acta 57, 795–814 (1993).
Clor, L. E. et al. Gas composition monitoring with multi-GAS during the 2018 eruption of Kīlauea, Hawai’i. AGU Fall Meet. Abstr. 2018, V43J–V40271 (2018).
Ilyinskaya, E., Mason, E., Wieser, P. E., Liu, E. J. & Whitty, R. C. W. Filter pack and cascade impactor samples of gas and aerosol particulate matter on the Island of Hawai’i (2018, 2019). Cent. Environ. Data Anal. https://doi.org/10.5285/656204c257144708a641507c78187aaa (2021).
Aiuppa, A., Federico, C., Paonita, A., Pecoraino, G. & Valenza, M. S, Cl and F degassing as an indicator of volcanic dynamics: the 2001 eruption of Mount Etna. Geophys. Res. Lett. 29, 54-1–54-4 (2002).
Tria, J., Butler, E. C. V., Haddad, P. R. & Bowie, A. R. Determination of aluminium in natural water samples. Anal. Chim. Acta 588, 153–165 (2007).
Acknowledgements
This project received funding from the NERC Centre for Observation and Modelling of Earthquakes, Volcanoes and Tectonics (COMET), UNRESP (NE/R009465/1, NE/P015271/1) and V-PLUS (NE/S00436X/1). COMET is a partnership between UK Universities and the British Geological Survey. Thanks to Tina Neal, Frank Trusdell, the Hawaiian Volcano Observatory and Frederic ‘Bill’ Simonds for assistance in accessing sampling areas. Thanks also to the USGS team of UAS pilots for flying our instruments into the airborne plumes and to the helicopter pilot (David Okita) for access to the ocean entry plume. Finally, our thanks to all agencies that managed and monitored the impacts of 2018 LERZ eruption and supported our fieldwork during a very challenging period of Kīlauea’s eruptive history. E.M. is funded by an EPSRC-CASE studentship, P.E.W. by a NERC studentship, and R.C.W.W. by a NERC-CASE studentship in partnership with the Icelandic Meteorological Office. E.J.L. was supported by a Leverhulme Trust Early Career Fellowship. A.J.S.M. acknowledges support of the Rolex Institute. C.O. acknowledges funding from the NERC (Grant NE/N009312/1). The Multi-GAS was kindly contributed by S. Aiuppa, University of Palermo, through funding from the Alfred P. Sloan Foundation’s support of the Deep Carbon Observatory DECADE programme. Any use of trade, firm or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Author information
Authors and Affiliations
Contributions
E.M., P.E.W., E.J.L., E.I. and R.C.W.W. were the fieldwork team with local assistance from C.K., T.E., D.J.S. and P.A.N., and additional support in planning stages from C.O., M.E. and T.A.M. T.C.W. and A.J.S.M. collected SO2 flux data, with assistance from E.J.L., T.D.P. and F.M.M. E.M. carried out the majority of the clean-lab work, along with E.J.L., E.I. and P.E.W. P.E.W. carried out the EPMA and LAICPMS measurements of matrix glasses. E.M. created figures, processed data and wrote the manuscript with assistance from all co-authors.
Corresponding author
Ethics declarations
Competing interests
E.J.L. is an Editorial Board Member for Communications Earth & Environment, but was not involved in the editorial review of, or the decision to publish this article. The authors declare no competing interests.
Additional information
Peer review information Primary handling editor: Joe Aslin
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mason, E., Wieser, P.E., Liu, E.J. et al. Volatile metal emissions from volcanic degassing and lava–seawater interactions at Kīlauea Volcano, Hawai’i. Commun Earth Environ 2, 79 (2021). https://doi.org/10.1038/s43247-021-00145-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-021-00145-3
This article is cited by
-
Recent trends and sources of lead toxicity: a review of state-of-the-art nano-remediation strategies
Journal of Nanoparticle Research (2024)
-
Microbiomes in the Challenger Deep slope and bottom-axis sediments
Nature Communications (2022)
-
A golden era for volcanic gas geochemistry?
Bulletin of Volcanology (2022)
-
Volcanic air pollution and human health: recent advances and future directions
Bulletin of Volcanology (2022)
-
A look ahead to the next decade at US volcano observatories
Bulletin of Volcanology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
