
Abstract
Global climate change related to anthropogenic CO2 emissions is one of the most significant challenges for the future of human life on Earth. There are many potential options for reducing or even eliminating atmospheric CO2 emissions including underground sequestration, carbon mineralization and ocean storage. One of the most promising materials for carbon mineralization is Mg(OH)2 which is highly reactive and capable of forming stable carbonates. Here we show a novel low-carbon method of producing Mg(OH)2, from globally abundant olivine-rich silicate rocks. A combination of acid digestion and electrolysis of olivine were used to produce Mg(OH)2 in a fully recoverable system. The use of Mg(OH)2 from olivine provides a viable pathway for significant industrial scale reductions in global anthropogenic greenhouse gas emissions.
Similar content being viewed by others
Introduction
The effects of unchecked CO2 emissions on global climate change are being increasingly seen and felt across the world. Examples of issues include increased land and sea temperatures, glacier and sea ice loss, and sea-level rise1. Urgent action is needed to limit future emissions and sequester existing atmospheric CO2 in order to circumvent issues related to global warming2. In 2018, ~37 billion tonnes of CO2 was released into the atmosphere3. Point source emissions such as power generation and industrial production account for ~60% of the total CO2, but this is expected to decrease to 50% by 20504.
Methods for long-term storage of CO2 can be classified as either underground sequestration or carbon mineralization, of which underground sequestration in sedimentary formation is considered the most mature technology5. Carbon mineralization can be further divided into three approaches: (1) ex situ, where the material is transport to the site and reacted with CO2 typically at elevated temperatures and pressures, (2) surficial, using dilute or concentrated CO2, and (3) in situ, where the CO2 is transported to site with suitable geological formations, typically containing serpentine or olivine-bearing basalts5. Ocean storage has also been proposed as a potential means of CO2 sequestration but comes with a number of environmental consequences4.
A number of materials have been proposed for carbon mineralization, including serpentine, olivine, wollastonite, magnesium oxide, and magnesium hydroxide6,7,8,9,10. Of the various materials which may be suited for transport to the emissions source, Mg(OH)2 is one of the most reactive for carbonization5. Aqueous carbonation of Mg(OH)2 can result in the formation of a hydrated Mg-carbonate such as nesquehonite (MgCO3·3H20), dypingite (Mg5(CO3)4(OH)2.5H2O), and hydromagnesite (Mg5(CO3)4(OH)2·4H2O) under atmospheric or near-atmospheric conditions11. The formation of magnesite (MgCO3) is also possible but this typically requires higher temperatures (>100 °C) and pressures above 100 bar. Seeding Mg(OH)2 slurries with magnesite at elevated pressures and temperatures has been shown to produce a stable anhydrous magnesite12. One of the benefits of Mg(OH)2 with regard to overall conversion rates and transportation is that it does not contain significant quantities of unreactive material, such as silica or iron which are typically found in olivine or serpentine.
MgO and Mg(OH)2 are usually sourced from either the calcination of magnesite or precipitated from Mg-rich brines and seawater13. Recovery of Mg(OH)2 from seawater has the potential to provide virtually limitless supplies of the material; however, it’s commercial production typically requires the use of lime, which itself has significant embodied CO2, resulting in little overall environmental benefit. The high-temperature pyrohydrolysis process of using MgCl2·6H2O to form MgO and HCl14 could potentially provide a low-carbon Mg(OH)2 alternative; however, concentrations of Mg-chloride in seawater are relatively low which limits the efficiency of the process unless highly concentrated brines are used.
Magnesium-rich silicate minerals, such as olivine ((Mg,Fe)2SiO4), in ultramafic and mafic rocks have the potential to produce reactive Mg(OH)2 with zero CO2 emissions. Until now, Mg(OH)2 recovery from olivine has not been used primarily because of the slow rate of hydration (i.e., serpentinization)15,16 or ultramafic mineral extraction efforts have been focused on silica recovery17 rather than Mg(OH)2 production. Furthermore, a satisfactory energy-efficient industrial process has not been developed yet18.
Here, we extract Mg(OH)2 directly from olivine using a combination of acid digestion and electrolysis. The acid used in the digestion process is completely recoverable and provides a source of usable energy. Recovered Mg(OH)2 was found to have a similar level of reactivity compared to commercially available Mg(OH)2. A slurry of recovered Mg(OH)2 and water was shown to effectively sequester CO2 forming a hydrated Mg-carbonate. In addition to Mg(OH)2, amorphous silica, which has the potential for use as a partial cement replacement, was also recovered from the olivine.
Methods: synthesis of Mg(OH)2
The synthesis of Mg(OH)2 uses olivine from ultramafic rocks and/or olivine-rich basalts as the raw material and source of magnesium (Fig. 1). Olivine is sparingly soluble under standard state conditions with a total Mg concentration of less than 6 mg L−1 resulting in excessive energy demands for the recovery process. As the pH of the reaction solution decreases, the solubility of Mg increases considerably. In our investigation, 100 g of finely ground forsteritic ((Mg0.9Fe0.1)2SiO4) olivine19, with a mean particle size of 28 µm, was combined with 500 ml of 2 M HCl, resulting in the formation of a solution containing MgCl2 and FeCl2 and SiO2. The silica is produced through a process of hydrolysis and subsequent polymerization and condensation of silicic acid (Si(OH)4). Strong acids such as HCl are known to accelerate the hydrolysis process, whereas, a high pH favors the polymerization and condensation process that is supported by the addition of Mg(OH)2 in this study. Here, the pH of the solution increased rapidly from less than 0 to ~3 after 4 h of mixing at 20 °C.

Energy requirements are shown where E1 = 0.32, E2 = 6.6 and E3 = −0.64 GJ tonne−1 of Mg(OH)2 produced. Energy consumption details are provided in Supplementary Methods. Note: Direct solid-gas carbon mineralization is also possible and the most likely approach for point source CO2 sequestration. *Note: The carbonation product is likely to be hydrated Mg-carbonate such as nesquehonite, dypingite, or hydromagnesite, but MgCO3 formation is also possible depending on reaction conditions27.
The concentration of Mg2+ in solution was determined by complexometric titration used to measure the total hardness of water. In total, 1 ml of sample solution was added in a conical flask containing 10 ml of 0.05 M ethylenediaminetetraacetic acid (EDTA) solution and 10 ml of ammonia buffer solution. Eriochrome black T was used as an indicator. The solution was titrated using a 0.025 M magnesium chloride solution until its color changed from blue to pink. The concentration of complexed EDTA solution was used to determine the magnesium ion concentration. Since the concentration of calcium ions in the sample solution was negligible, it was assumed that all the ions complexed with EDTA were magnesium. The Mg concentration was found to be ~24 g L−1, which is more than 19 times the concentration of Mg in seawater and over three orders of magnitude greater than the concentration resulting from serpentinization reactions involving pure water. The potential Mg extraction efficiency, therefore, is considerably higher using an acid digestion solution than it is from seawater.
Once the initial digestion phase was completed the solution was allowed to settle for 1 h after which it was decanted to separate the solution containing the Mg, Fe, and Si ions from any unreacted olivine (Fig. 1). Silica was precipitated from the solution by increasing the pH to >3.5 through the addition of 1 g of Mg(OH)2. The pH of the remaining solution was further increased to almost 7 to precipitate the dissolved iron using 0.32 g of NaOH. The silica and iron were separated from the solution using a centrifuge in this proof-of-principal testing, whereas, an industrial filtration system would be used in practice. It should be noted that the formation of gel-like products during the precipitation of silica and iron can cause challenges to any filtration system. Regular flushing and cleaning of the membranes would be necessary and likely require a number of crossflow filters, such as those used for silica removal from geothermal waters, to be run in parallel to maintain operations. An alternative approach and potentially more efficient method for the removal and recovery of the silica and iron is by precipitate flotation.
The MgCl2 solution was electrolyzed in an H-cell with a carbon anode and platinum cathode. A DC power supply was used to generate a current of approximately 100 mA resulting in the formation of Cl2 gas at the anode and H2 gas at the cathode. Mg(OH)2 was formed at the cathode where the pH of the solution increased rapidly to ~9.5. The recovered Mg(OH)2 was placed in a drying oven at 105 °C for 1 day after which it was analyzed by scanning electron microscope (SEM), thermogravimetric analysis (TGA) and X-ray powder diffraction (XRD). For the commercial production of Mg(OH)2, H2 and Cl2 would be combined to produce HCl for re-use and as an energy resource, as is typically done in the chemical process and manufacturing industry.
Results and discussion
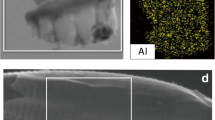
From this process, a total of 35 g of Mg(OH)2 was produced from 100 g of olivine, with 1 g of Mg(OH)2 added in the silica precipitation stage. Approximately 5 g of iron oxide was precipitated with the addition of 0.32 g of NaOH, which represented <1% of the total Mg(OH)2 recovered. In total, 35 g of amorphous silica was also recovered from the precipitation stage. The SEM image and TGA graph provided in Fig. 2a, b show the material recovered after electrolysis was primarily Mg(OH)2. XRD results provided in Fig. 2c show the recovered silica was predominantly amorphous SiO2 with some residual unreacted olivine and confirm the material recovered after electrolysis was predominately Mg(OH)2. The composition of the raw olivine sand, recovered Mg(OH)2, and recovered silica from olivine, determined by X-ray fluorescence (XRF), are provided in Table 1 and are consistent with the other material characterization analysis.

a SEM image of recovered Mg(OH)2 from the electrolysis of olivine digestion solution, b TGA of recovered Mg(OH)2 from olivine, c XRD of recovered Mg(OH)2 and of recovered silica (B: brucite, L: lizardite, F: forsterite).
In addition to the primary recovered Mg(OH)2, secondary materials of iron hydroxide and silica may also be of use. The iron hydroxide for instance could be used as a high purity raw material for iron production or as an absorbent while the amorphous silica can be used as a partial replacement for Portland cement, in the production of concrete, which is a significant industrial contributor to global CO2 emissions20. Our approach provides an almost completely closed system for the production of Mg(OH)2 with the only additions being olivine, as the source of Mg, and minor amounts of NaOH.
CO2 and energy implications of Mg-hydroxide extraction
Overall, conversion of olivine into Mg(OH)2 produces no direct CO2 emissions and the HCl used for digestion was completely recoverable. The large-scale and industrial use of strong acids such as HCl have serious consequences if released into the environment. However, this is only an issue if there is a loss of containment from the process. The large-scale handling of HCl without loss of containment is well established in the chemical process industries due to its use in commodity materials such as the manufacture of PVC and pickling of steel. What is most important is that neither HCl nor chlorine leaves the process described in our work, as they are recycled within the process.
The total energy required, including quarrying and grinding, to produce Mg(OH)2 from olivine was calculated to be 6.28 GJ tonne−1 (see Fig. 1 and Supplementary Methods for energy determinations and calculations). Further refinement of the process may allow greater use of recovered Mg(OH)2 for pH control, a decrease in the total energy required, and a reduction in NaOH. The Mg(OH)2 could be further processed into MgO but for CO2 sequestration; however, Mg(OH)2 is known to be a faster reactant than MgO10.
For every tonne of CO2 sequestered as a Mg-carbonate, including nesquehonite and hydromagnesite, 1.3 tonnes of Mg(OH)2 is required, resulting in an energy consumption 8.17 GJ tonne−1 of CO2 (Fig. 1, see Supplementary Methods for calculations). Using the method outlined in this paper would result in a net negative CO2 emission, even if coal was used to provide the necessary energy, though at ~25% sequestration efficiency. The overall carbon sequestration benefits would improve substantially if a low-carbon energy sources, or even a mixed electrical supply, was used. For example, in California where roughly 50% of the electricity is from non-fossil fuels, ~500 kg of CO2 would be emitted for every 1000 kg of CO2 captured and turned into Mg-carbonate.
To confirm the ability of the recovered Mg(OH)2 to sequester CO2, a Mg(OH)2 water slurry was pressurized to 4 bar with concentrated CO2. Over a 48-h period, >50% of the Mg(OH)2 was converted to a hydrated Mg-carbonate, demonstrating the potential conversion of CO2 into a solid. The reactivity of the recovered Mg(OH)2 was also confirmed by the rapid increase in pH to ~10.5 when it was added to de-ionized water. As previously noted, there are a number of other methods for sequestering CO2 with Mg(OH)2 using either aqueous12,21 or direct solid-gas reaction methods10. Our proposed process has the potential to provide a substantial source of energy-efficient, low-carbon Mg(OH)2 for use in various carbon sequestration techniques currently being developed by other investigators.
Olivine resources and feasibility
Ultramafic rocks, enriched in olivine, constitute ~1% of Earth’s terrestrial landscape, a high proportion of oceanic crust, and >50 % of the upper mantle22. Olivine-rich deposits (Fig. 3) are primarily present within populated areas of the Circum-Pacific and Mediterranean regions23,24. Olivine is present in many rocks such as basalt and other ultramafics. We would like to note that although olivine was assessed and used in this study, its metamorphic equivalent, serpentine (Mg3Si2O5(OH)4) with its high reactivity in HCl and worldwide deposits, could expand the scope and implications of this study. Our preliminary investigations have shown Mg can also be extracted from basalt; however, we will primarily focus our discussion on two enriched and accessible olivine deposits: the Semail ophiolite (Oman) and the Red Hills Ultramafic Complex (New Zealand) which conservatively contain 1.4 × 105 and 871 billion tonnes of olivine, respectively (see Supplementary Methods for sources and estimate calculations).

The general distribution of ultramafic rocks (including peridotites and serpentinites) is shown worldwide. Ultramafic rock distributions are based on location data from Oze et al.24 and compared to Real and Vishal23. Please note that more ultramafic rock deposits are present than shown and that the squares do not represent particular sites. The billions of tonnes of olivine per year needed to sequester all anthropogenic CO2, and reduce global atmospheric CO2 as well as the lifetime supply of olivine from the Red Hills (New Zealand) and Semail Ophiolite (Oman) is shown and based on calculations provided in Supplementary Methods.
Using Mg(OH)2 to remove and sequester anthropogenic CO2 estimated for 2020 (40 billion tonnes) in a Mg-carbonate would require ~105 billion tonnes of olivine. Reducing global atmospheric CO2 levels by an additional 10 billion tonnes would necessitate a further 26 billion tonnes of olivine. The sum of olivine required to do all of this would be 131 billion tonnes; 0.1% of the Oman ophiolite or 16% of Red Hills, New Zealand (Fig. 3). To sequester all anthropogenic CO2, these two deposits would last on the order of nearly a decade (Red Hills) to 1000 years (Semail), assuming the deposits contained at least 60% olivine. The degree of serpentinization of each deposit and the abundance of other minerals such as ortho- and clinopyroxene would affect the overall extraction efficiency. Smaller deposits, compared to the Semail ophiolite or Red Hills, around the world could significantly aid the global reduction of anthropogenic CO2.
Implications
The concept of using ultramafic rocks and its metamorphic equivalents as sources of Mg for CO2 sequestration has been considered for at least two decades. However, one limitation with regards to previous and current approaches is related to the distance between the CO2 gas source and the ultramafic site. Currently, concentrated CO2 is being transported/piped to ultramafic sites to be directly injected into the subsurface. Although ultramafic sites are around the world, many are not sufficiently close to areas of high CO2-producing industries. Our approach allows bulk Mg(OH)2 to be transported more efficiently to locations/industries where it can be used on-site for point source emissions control. For example, transporting enough Mg(OH)2 to sequester 1 tonne of CO2, from its production location to a point source such as a cement plant 1000 km away by rail, would result in 37 kg of CO2 emissions25.
In addition, slight modifications to this approach through the further refinement of Mg(OH)2 to MgO has the potential to produce construction materials such as Mg-masonry blocks26 rather than simply burying the CO2. There is considerable interest in the potential use of MgO as an alternative to traditional Portland cement13; however, there are a number of issues that must be addressed, including the low pH of the pore solution, which makes its use in steel-reinforced structures challenging, and high water demand which makes handling more difficult. One of the major environmental issues associated Mg-based cements is the vast majority of MgO is produced from MgCO3 which results in the release of CO2 at a similar proportion to that of Portland cement. The alternative of recovering MgO from seawater, as previously noted, also has considerable embodied CO2 due to the use of CaO, sourced from carbonates, in the recovery process. If MgO is to be used as a construction material then a low carbon mineral extraction approach will be needed. The scale and scope to mitigate climate change using Mg-bearing minerals from ultramafic and mafic rocks are massive, but our experiments and others support that this is feasible.
Scaling up and improving efficiencies with regards to the processes described and proven here will require significant development and industrialization efforts. We anticipate that the engineering challenges to produce Mg(OH)2 for use in CO2 sequestration, as shown in Fig. 1, are comparable to other well known industrial processes. One of the great challenges, however, is how to use the billions of tonnes of carbonate produced to offset anthropogenic CO2 emissions and this requires further circumspection. A more realistic and efficient approach to addressing global warming is to reduce CO2 emissions. Until then, we have demonstrated a process that is feasible and globally communal where many countries and industries can participate. Overall, the recovery of magnesium hydroxide from olivine-rich (and potentially serpentine-rich) rocks provides the basis for a direct route to reduce global anthropogenic CO2 and its associated climate change impacts.
Data availability
Data files containing XRD and TGA results of the recovered materials from this investigation are available from the figshare dataset repository: DOI 10.6084/m9.figshare.13543091.
References
Stocker, T. F. et al. Technical summary. in Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (eds Stocker, T.F. et al.) pp. 1535 (Cambridge University Press, 2013).
IPCC, 2018: Summary for policymakers. in Global Warming of 1.5 °C. An IPCC Special Report on the Impacts of Global Warming of 1.5°C Above Pre-industrial Levels and Related Global Greenhouse Gas Emission Pathways, in the Context of Strengthening the Global Response to the Threat of Climate Change, Sustainable Development, and Efforts to Eradicate Poverty (eds Masson-Delmotte, V. et al.). pp. 3–24 (Intergovernmental Panel on Climate Changepages, 2019).
Tollefson, J. Global industrial carbon emissions to reach all-time high in 2018. Nature, 3, 4 (2018).
IPCC, IPCC Special Report on Carbon Dioxide Capture and Storage. Prepared by Working Group III of the Intergovernmental Panel on Climate Change (eds Metz, B., O. Davidson, H. C. de Coninck, M. Loos, and L. A. Meyer). pp. 442 (Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2005).
Kelemen, P., Benson, S. M., Pilorge, H., Psarras, P. & Wilcox, J. An overview of the status and challenges of CO2 storage in minerals and geological formations, Front. Clim. 1, 9 (2019).
Prigiobbe, V., Hänchen, M., Werner, M., Baciocchi, R. & Mazzotti, M. Mineral carbonation process for CO2 sequestration. Energy Procedia 1, 4885–4890 (2009).
Olsson, J. et al. Olivine reactivity with CO2 and H2O on a microscale: Implications for carbon sequestration. Geochimica et Cosmochimica Acta 77, 86–97 (2012).
Munz, I. A. et al. A continuous process for manufacture of magnesite and silica from olivine, CO2 and H2O. Energy Procedia 1, 4891–4898 (2009).
Goff, F. et al. Evaluation of ultramafic deposits in the Eastern United States and Puerto Rico as sources of magnesium for carbon dioxide sequestration (LA-13694-MS, Los Alamos National Lab., NM (US). https://doi.org/10.2172/754045 (2000).
Fagerlund, J. & Zevenhoven, R. An experimental study of Mg(OH)2 carbonation. Int. J. Greenhouse Gas Control 5, 1406–1412 (2011).
Zarandi, A., Larachi, F., Beaudoin, G., Plante, B. & Sciortino, M. Nesquehonite as a carbon sink in ambient mineral carbonation of ultramafic mining wastes. Chem. Eng. J. 314, 160–168 (2017).
Swanson, E., Fricker, K., Sun, M. & Park, A. Directed precipitation of hydrated and anhydrous magnesium carbonates for carbon storage. Phys. Chem. Chem. Phys. 16, 23440–23450 (2014).
Walling, S. A. & Provis, J. L. Magnesia-based cements: a journey of 150 years, and cements for the future? Chem. Rev. 116, 4170–4204 (2016).
Shand, M. A. The Chemistry and Technology of Magnesia (John Wiley & Sons, 2006).
Neubeck, A. et al. Olivine alteration and H2 production in carbonate-rich, low temperature aqueous environments. Planet. Space Sci. 96, 51–61 (2014).
McCollom, T. M. & Bach, W. Thermodynamic constraints on hydrogen generation during serpentinization of ultramafic rocks. Geochimica et Cosmochimica Acta 73, 856–875 (2009).
Justnes, H. & Ostnor, T. Pozzolanic, amorphous silica produced from the mineral Olivine. Proceedings of the 7th CANMET/ACI. Am. Concrete Institute Special Publication 199, 769–782 (2001).
Gartner, E. & Sui, T. Alternative cement clinkers. Cement Concrete Res. 114, 27–39 (2018).
Pawson, J. F. Abiotic Methane Formation at the Dun Mountain Ophiolite, New Zealand. Msc thesis, University of Canterbury. (2015).
Scrivener, K. L., John, V. M. & Gartner, E. M. Eco-efficient cements: potential economically viable solutions for a low-CO2 cement-based materials industry. Cement Concrete Res. 114, 2–26 (2018).
Zhao, L., Sang, L., Chen, J., Ji, J. & Teng, H. H. Aqueous carbonation of natural brucite: relevance to CO2 sequestration. Environ. Sci. Technology. 44, 406–411 (2010).
Winter, J. D. Principles of Igneous and Metamorphic Petrology (Pearson Education, 2013).
García del Real, P. Vishal, V. in Geologic Carbon Sequestration: Understanding Reservoir Behavior. (eds Vishal, V. & Singh, T. N.) 213–229 (Springer International Publishing, 2016).
Oze, C., Bird, D. K. & Fendorf, S. Genesis of hexavalent chromium from natural sources in soil and groundwater. Proc. Natl Acad. Sci. USA 104, 6544–6549 (2007).
Kirschstein, T. Meisel, F. GHG-emission models for assessing the eco-friendliness of road and rail freight transports, Transport. Res. Part B 73, 13–33 (2015).
Vlasopoulos, N. & Cheeseman, C. R. Binder composition. https://patents.google.com/patent/US8496751/en (2013).
Rausisa, K., Ćwika, A. & Casanova, I. Phase evolution during accelerated CO2 mineralization of brucite under concentrated CO2 and simulated flue gas conditions. J. CO2 Utiliz. 37, 122–133 (2020).
Acknowledgements
The authors wish to acknowledge the contribution of Anthony Fairbanks and Travis Wallaby Horton for aid and discussions related to this research. The authors would also like to acknowledge the financial support provided by the Ministry of Business, Innovation, and Employment, New Zealand (1708) to this project.
Author information
Authors and Affiliations
Contributions
A.S.: conceptualization, methodology, resources, formal analysis, funding acquisition, and writing—original draft; V.S.: conceptualization; methodology, formal analysis, investigation, and writing—review; C.O.: conceptualization and writing—review and editing; B.S.: investigation and methodology; C.C.: writing—review and editing; N.Y.: investigation and methodology; M.W.: conceptualization and writing—review and editing; A.M.: conceptualization and writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Primary handling editor: Teresa Ortner
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scott, A., Oze, C., Shah, V. et al. Transformation of abundant magnesium silicate minerals for enhanced CO2 sequestration. Commun Earth Environ 2, 25 (2021). https://doi.org/10.1038/s43247-021-00099-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-021-00099-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
