
Abstract
Due to the SARS-CoV-2 pandemic renewed attention has been directed towards viral neutralization assays and neutralizing antibodies quantification, for vaccine pre-clinical trials and determining vaccine efficacy over time. The gold standard to assess antibody titer is the plaque reduction neutralization test, an end-point assay which evaluates the highest serum antibody dilution that neutralizes viral replication, by inspecting the cytopathic effect induced on cell cultures. Here, we use planar, PEDOT:PSS-based organic electrochemical transistors for real-time, remote-controlled, reliable and fast electrical monitoring of the cytopathic effect induced by SARS29 CoV-2 on Vero E6 cell lines, allowing the quantification of serum neutralizing titer. Our low-cost and scalable device has the potential to speed-up large-scale viral neutralization screening without the need for cancerous staining or highly specialized operators. Finally, the technology could be easily transferred to assess neutralizing antibody response towards different viruses in their permissive cell substrates.
Similar content being viewed by others

Introduction
In November 2019, cases of atypical pneumonia were detected in China. The etiological agent was quickly identified as a β-coronavirus (named SARS‐CoV‐2), which has since caused the current pandemic. Some coronaviruses are zoonotic being transmitted from animals to humans and vice versa1.
Several studies performed on severe acute respiratory syndrome (SARS) and the Middle East respiratory syndrome (MERS) have demonstrated the presence of specific neutralizing antibodies against these viruses in 80–100% of patients 2 weeks after the onset of symptoms2. Lau et al. showed that neutralizing antibody responses in SARS-CoV-2 infections is comparable to what is observed with SARS with neutralizing antibody maintained over the first year after mild or severe disease with higher antibody titers and longer duration of detectable antibody in those with severe disease3. Antibodies may suppress SARS-CoV-2 replication through viral neutralization, but they may also participate in COVID-19 pathogenesis and progression through a process termed antibody-dependent enhancement4,5.
For this reason, there is a significant and urgent need of assessing the presence of neutralizing antibodies, produced during SARS-CoV-2 infection, for prognostic purposes in clinical practice. Furthermore, to plan the next steps in COVID-19 vaccine development, it is vital to understand the relationship between the measured immunity and the clinical protection against SARS-CoV-26.
The immunological assays available to quantify neutralizing antibodies in patients are key to providing information and data on vaccine efficacy, in order to prioritize different vaccine candidates and to support regulatory submissions for vaccine licensure7. The harmonization of neutralizing antibody test results (both against live SARS-CoV-2 and pseudoviruses) is particularly important also in vaccine pre-clinical trials, involving non-human primates, to allow accurate comparisons with those obtained in clinical ones4.
There are several methods for determining the antibody titer, but serum neutralization (SN) is considered the gold standard for measuring the presence and levels of neutralizing antibodies in tested sera. In addition to the conventional SN, to obtain a precise quantification of the titer of neutralizing antibody for a virus, an assay called plaque reduction neutralization test (PRNT) was developed for studies of inactivation (or neutralization) of animal viruses and modified to measure serum antibody neutralization titer8,9. The PRNT assay has been already used to test the level of protection of the population in countries affected by other zoonotic coronaviruses, such as MERS10. PNRT involves significant economic costs, from the materials used to the highly specialized operators needed and toxic waste disposal, and requires long processing times (72 h post infection), hampering rapid reporting. Due to the high infectivity and pathogenicity of the SARS-CoV-2, the virus needs to be handled in biosafety level 3 (BSL-3) specific facilities. PRNT is thus technically demanding and difficult to automate, making it unsuitable for large-scale studies of serum samples, such as phase III human clinical vaccine trials7.
To avoid dealing with infectious CoV-2, several safe, biosafety level 2 (BSL2) pseudovirus-based systems have been developed11. Although they have been shown to be sensitive and reliable, they could be considered a proxy of the “real” neutralization assay, and often suffer from being farraginous, time-consuming, and expensive in procedural terms. To overcome some of these disadvantages also microneutralization assay (MNA) has been developed, The MNA offers the advantages, over the PRNT, of a reduced assay time, allowing increased throughput and reducing operator workload while remaining dependent upon the use of wild-type virus7.
Lately, electrical measurements are emerging as a viable alternative for quantitative investigations over in vitro cell cultures12,13,14, particularly employing Organic Electrochemical Transistors (OECTs). OECTs are three-terminal devices (source, drain, and gate), in which the current flowing in the semiconducting channel (source-drain current, Ids) is modulated by the potential applied to the gate electrode (Vgs) throughout an electrolyte solution. Any impediment positioned between the channel and the gate of the device will hinder the ion flux, slowing down the transistor response to gate potential switching. Thus, cell layers grown on top of OECTs will affect the device’s performance. It has been recently theoretically15,16 and experimentally17 demonstrated that poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (Pedot:Pss) OECTs provide a reliable technology to study different in vitro cell cultures18,19,20,21,22. In particular, planar devices23,24, having the semiconducting polymer employed for the channel and the gate of the device, allow for the direct cell growth on the OECT, resulting in fully biocompatible, permitting simultaneous optical and electrical monitoring25, and doubling the device sensitivity to the cell layer26. A fully automated integrated system (labeled Tissue Engineering Cell Holder for Organic Electrochemical Transistors, TECH-OECT26) has been recently designed and proposed by our group, employing planar, Pedot:Pss-based OECTs to carry out a real-time evaluation of the cell layer integrity over time, inside a humidified incubator. We have tested this technology on two different cell lines, stressed by a toxic agent (i.e., silver nanoparticles)27 or Trypsin26, obtaining fast, quantitative, reliable, and real-time analysis on cell layer formation and disruption/detachment.
Pizzorno et al. proved that high SARS-Cov-2 viral replication stresses cell layer integrity in Human Airway Epithelial (HAE), reducing cellular transepithelial resistance (TEER) values, thus facilitating the ionic flux passing through the cell layer28.
Here, we propose the use of Pedot:Pss-based OECTs to monitor the CPE induced by SARS-Cov-2 on Vero E6 cell line by investigating the ion flux differences recorded for healthy and viral-infected cells during growth. Our work aims at the realization of reliable electrically probed, real-time, and fast in vitro SN assays. In the present study, sera with known neutralization titers, obtained with the classical PRNT method, were evaluated by OECT devices, speeding up the reporting response times and increasing the information on the kinetics of infection.
Results
Planar Pedot:Pss-based OECTs were employed in this work, since we proved their high sensitivity to cell tissue disruption/stress, once incubated with a toxic agent (i.e., silver nanoparticles)27. The device schematic and electrical characterization are shown in Fig. 1a and Supplementary Fig. 1, respectively, while its geometry and fabrication steps are reported in the “Methods” section. VeroE6 were used as a cellular model since they have β-interferon response deficiency due to the absence of the β-interferon gene29 and are therefore highly permissive to many viral infections, including SARS-CoV-2, as explained in the “Methods” sections.

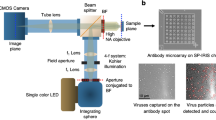
a Rendering of the planar Organic Electrochemical Transistor, consisting in a glass substrate with Cr/Au contacts and Pedot:Pss channels and gate, enclosed in a PDMS perforated cylinder to implement the culture well. b TECH-OECT with six devices inserted and electrically connected. Optical zooms highlight a viral-infected (red) and a healthy cell line (blu) after 48 h from seeding. Circle diameter of 500 μm. c Schematical graph (left) of the expected OECT responses discriminating a viral-infected culture and a healthy one, with the relative cross-section representations (right). Black arrows represent ionic fluxes across cell layers, which intensify upon layer destruction caused by viral proliferation.
Aiming at developing a fast, simple and reliable protocol for virus neutralization assays, cells, serum, and virus were mixed together (concentrations are reported in the “Methods” section) in an Eppendorf tube before introducing the mix directly onto the OECTs. The cultures were electrically monitored in real-time for 44–48 h, inside the incubator under constant physiological conditions (37 °C, 5% CO2), by means of our prototype, the TECH-OECTs integrated system (Fig. 1b). Supplementary Methods explains the schematic representation of the experimental setup and wiring connections reported in Supplementary Fig. 2a, while Supplementary Fig. 2b shows TECH-OECT during seeding at the beginning of an experiment.
Owing to its cytopathic effect, once SARS-CoV-2 enters the VeroE6 hosting cells, the formation of a cell layer is stressed and disrupted if the virus is free to replicate. This effect results in reduced barrier-like properties of the cell line towards the ion flux between their apical and basal poles (Fig. 1c). Figure 1b shows optical micrographs and the schematic cross-section of VeroE6 line seeded on top of a Pedot:Pss transistor, showing a healthy cell line in which the virus activity was stopped due to neutralizing antibodies (blue circles) and a stressed cell layer where the virus freely proliferated (red circles). Different cell morphology and spreading are evident in the micrographs, the former (blue) presenting a confluent and tight monolayer, while the latter (red) showing stretched and isolated cells. Accordingly, to our previous works26,27, the OECT is able to discriminate between the two growths, as reported in Fig. 1c, by measuring the different time response to current modulation upon a pulsed gating (i.e., ion flux into the Pedot:Pss thin film), which is dependent on the cell layer integrity and health (Fig. 1c).
We employed planar, Pedot:Pss OECTs to monitor in real-time VeroE6 growth infected by SARS-CoV-2 (300 TCID50/mL), with and without highly concentrated (dilution 1:10) neutralizing (positive serum = serum P) and non-neutralizing antibodies (negative serum = serum N). OECT time response results are outlined in Fig. 2a, while PCR tests (as described in the Materials and methods section), performed at the beginning (0 h) and at the end (48 h) of each experiment to confirm the expected virus proliferation/stop, are reported in Fig. 2b. It is worth noting that OECTs show an initial growth trend in common to all cell cultures in the 20–25 h following the seeding. After that time, viral replication starts to influence the cell layer integrity (red and magenta lines), as highlighted by a drop in the device time response, which is greater when neutralizing antibodies are absent in the cell medium (red line). The healthy growth, monitored when the neutralizing antibodies stop the viral proliferation (blue line), reaches a normalized time response close to 2 a.u. after 44 h, a significantly different value from the one recorded on viral-proliferating cultures (<1.2 a.u.). The behavior of the healthy growing layer well follows the control samples, in which cells were seeded with serum N (dark green line) and P (light green line) to confirm their non-toxic effect to the cell line. The time response of the three healthy cell cultures after 44 h differs only slightly, mostly due to the final distribution of the cell layer formed on the active areas of our devices. Kinks and fluctuations in the curves are likely induced by transitory biological events and/or external electrical noises/mechanical vibrations and are irrelevant for the assay outcome since the real-time monitoring allows for complete trend analyses, extracting the correct results. To validate our sensor response, optical images (in brightfield and using crystal violet staining) together with cytofluorimeter analysis were taken at the end of the experiments. Data of the viral-infected cultures and controls are shown in Fig. 2c and Supplementary Fig. 3, respectively. Cells were fixed and stained with Crystal Violet, as explained in the Methods Section. It is clearly visible that SARS-CoV-2 infected cells exhibit a lower percentage of cell mortality in presence of neutralizing antibodies (40%), than in their absence. Accordingly, a confluent and uniform cell layer is visible in the viral-neutralized cell culture and controls (with round-shaped VeroE6 tightly linked to one another); on the other hand, wide empty areas are shown in the infected ones, in which stressed/dead cells were easily detached during the washing steps of the staining (the few cells remaining displaying a stretched shape). Electrical controls (without cells) on the device response to serum P, serum N, and SARS-CoV-2 have also been carried out, underlining that no effects are present, and a stable OECT time response is maintained over the 44 h of the experiments (Supplementary Fig. 4).

a OECT real-time monitoring of SARS-Cov-2-infected cell lines (red), with neutralizing (blu) and non-neutralizing (magenta) antibodies and sera controls (light and dark green for neutralizing and non-neutralizing antibodies, respectively). The horizontal dashed black line is inserted as the threshold discriminating between viral-proliferating and healthy cell cultures. b Graph reporting qRT-PCR Allplex Seegene data normalized to the starting value at the beginning of each experiment. The orange dashed line is inserted to stress the difference between viral proliferating cultures, having reduced PRC value after 48 h, and viral-neutralized ones, with increasing PCR. ***p < 0.001 denote significant differences with respect to the “Virus” data (red column). c Cytofluorimeter analysis on cell population and optical micrographs on Crystal Violet stained cells of cell cultures for viral-infected cell lines (red), with neutralizing (blu) and non-neutralizing (magenta) antibodies. The black histogram corresponds to unstained healthy cells whereas the red histogram represents suffering cells characterized by membrane alterations that allow the DAPI to enter. White scale bars in the micrographs of 100 μm.
The reproducibility and reliability of our devices have been assessed by replicating the experiment three times (Fig. 3), using devices fabricated in different batches, and carrying out the assays in two different laboratories (see Materials and Methods). Figure 3a shows the average trends of three different devices, having comparable polymerase chain reaction (PCR) N-gene variation in the 44 h experiment (Fig. 3b), chosen since it is the specific gene for SARS-CoV-2. The shadows reported in the graph highlight the standard deviation from the average. The OECTs monitoring cell cultures in which the virus was neutralized (blue line) show a rising time response, with a final value between 1.7 a.u. and 2 a.u., very similar to the control (green line), in which the slight decrease in the last 12 h was ascribed to cell overpopulation (the overabundant serum probably boosted cell proliferation and growth30,31). On the contrary, OECTs studying cells infected with SARS-CoV-2 with (red line) or without (magenta line) negative serum show off final time responses below 1.2 a.u., confirming the previous results. Noteworthy, the recorded OECT trends closely follow the normalized PCR ones (Fig. 3b), taken at the beginning and at the end of the experiment, highlighting that our results are correlated to viral replication/inhibition in the cell culture.

a Average OECT normalized time response over three different devices with standard deviation (semi-transparent shadow) monitoring viral-infected cell lines (red), with neutralizing (blu) and non-neutralizing (magenta) antibodies, and Serum P on cells as a control (light green). b Graph reporting the average normalized values of qRT-PCR Allplex Seegene analysis of the selected device cultures at the beginning and at the end of the experiments. **p < 0.01 and ***p < 0.001 denote significant differences with respect to the “Virus” data (red column).
Finally, we employed the TECH-OECT system for an SN test, using a neutralizing serum with titer above 80, according to previous PRNT tests (as described in the Materials and methods section). The standard viral concentration was thus incubated with progressive positive serum dilution (1:20, 1:40, 1:80, and 1:160) and without serum: results are shown in Fig. 4a, with the corresponding PCR test reported in the histogram in Fig. 4b. OECTs prove that the positive serum has a neutralizing activity down to 1:80, in agreement with PRNT, while the next dilution (1:160) displays an uncertain response, slightly higher than the ideal threshold of 1.2 a.u. This response may be ascribed to the fact that 1:160 serum dilution represents a borderline antibodies concentration, which decelerates viral proliferation but still allows for strong, viral-induced cell stress highlighted by the low-increasing curve trend and final value. It is worth noting that PCR analysis for 1:160 positive serum dilution reports a “standard” viral stop after 48 h, hindering the identification of this borderline concentration, which may be interesting for more accurate analysis on the virus-cell interactions. Indeed, it is crucial to point out that OECT and PCR are not exactly comparable since the former provides measurements of cell layer integrity (thus evaluating potential viral replication and CPEs on the cell culture), while the latter detects the viral genome, independently on viral activity or replication.

a Serum neutralization assay was performed using the TECH-OECT system. A neutralizing serum (Sp), with titer above 80, was progressively diluted in cultures infected by SARS-CoV-2, real-time monitoring the cell line using the automatized OECT technology for 44 h. b qRT-PCR Allplex Seegene data normalized to the starting value at the beginning of each experiment. The orange dashed line is inserted to stress the difference between viral proliferating cultures, having reduced or similar PCR value after 48 h, and viral-neutralized ones, with increasing PCR value. **p < 0.01 and ***p < 0.001 denote significant differences with respect to the “Virus” data (red column).
Repeated neutralization test sessions have been carried out with the devices here described to test the possibility to reuse them multiple times. With this purpose, a revitalization protocol to detach the cell layer and clean the device surface at the end of the experiment has been developed, revitalizing the OECTs for a second test: briefly, after medium removal, the well was washed with a physiological solution and incubated with 300 μL of Trypsin for 15 min at 37 °C; Trypsin was discarded (together with the detached cells) and the devices were sterilized again under UV light for 20 min. The device characterization after the revitalization protocol is reported in Supplementary Fig. 5, displaying a reduction in the OECT conducibility but still confirming the device’s current modulation upon gating. We performed up to three consecutive experiments to prove the device reuse, switching the “incubation mix” at the seeding for the second and third tests among the devices to increase the randomness of the experiment. We assessed that, despite mild changes or signal reduction, the devices were still able to discriminate between viral-infected growth and viral-neutralized ones. Figure 5 reports the comparison of the first (Fig. 5a), second (Fig. 5b), and third (Fig. 5c) test. To compare the mild changes in the device response produced by the cleaning procedure, we outlined repeated measurements on devices having the same “incubation mix” in Supplementary Fig. 6. Clearly, revitalized OECTs seeded with cells infected with the proliferating virus show very repeatable results (Fig. 5a, b). Similarly, healthy cell growth is correctly monitored (Fig. 5c, d), only with slight reductions of the final normalized time response. For completeness, we report in Supplementary Fig. 7 repeated measurements on the same revitalized devices, using different seeding “mix” for the repeatability assessment. We can conclude that OECTs correctly identify the healthy and infected cultures after up to 2 revitalization protocols, even though their initial time response parameter mildly shifts towards a higher value after the second revitalization (Supplementary Fig. 8).

Device repeatability tests. OECTs have been employed up to three consecutive times (developing a cleaning procedure between the experiments) to monitor Sars-Cov-2-infected cell lines (red), with non-neutralizing (blue) and neutralizing (magenta) antibodies, together with neutralizing serum control (green). OECT first, second, and third uses are reported in (a)–(c), respectively.
Discussion
We have designed and implemented an accurate real-time technology for quantitative SN assay, based on OECTs. This is proof-of-concept research of a technology that could potentially replace (or work in parallel with) subjective optical evaluation or expensive laboratory equipment. The OECTs work in real-time, as in vitro, cell tissue integrity sensors, remotely monitoring the cell layer health during growth to discriminate between viral-infected and viral-neutralized cell cultures. It is worth noting that OECTs monitoring cell layers exhibit a normalized time response threshold of 1.2 a.u. after 44–46 h from seeding: lower values are attributed to unhealthy cell layers, where the viral effect disrupted the cell growth process, while higher values define healthy cell lines in which the viral activity has been neutralized (usually presenting normalized time response above 1.3 a.u.). We have assessed that an SN assay can be reliably carried out using these values and the trends recorded in real-time measurements. The here proposed technology allows also to highlight the borderline serum dilution (not detectable with standard PCR tests) which presents a final time response between 1.2 and 1.3 a.u. It is worth emphasizing that the normalized time response final outputs depend on OECT geometry, cell line, and viral activity thus they may be calibrated before clinical trials. Optimizing the device geometry for the target cell–virus coupling would allow increasing the sensitivity of the assay and preliminary tests on microfabricated devices have already been carried out, foreseeing the method scalability. Our technology, thus, introduces a robust and reliable method for SARS-CoV-2 neutralization tests that nowadays rely on cell culture staining and subjective optical investigation.
Furthermore, OECTs are able to distinguish the different behavior of virally infected cell lines with and without neutralizing antibodies, showing milder infection when the non-neutralizing antibodies are in the culture well. The normalized time response at the end of the experiment is lower than 1.2 a.u. in the presence or absence of non-neutralizing antibodies. PCR values, compared to validate OECTs response, also report different increasing viral load, well supporting the obtained results.
This behavior may be explained considering a slight specific activity of the antibodies that is not enough to stop viral activity but still mitigates the cell layer disruption/viral proliferation. Indeed, sera usually contain growth factors that boost cell proliferation and health32, which would also explain the average faster increase in the response trend of cells cultured only with neutralizing serum in Fig. 3a (green line).
A final consideration can be made on the cell culture distribution and device dimensions: despite the device’s ability to investigate cell layer integrity only in its active areas (Pedot:Pss gate and channels), the selected threshold of 1.2 a.u. correctly discriminates viral infected culture from viral neutralized ones. The curve shadows in Fig. 3a highlight the variable cell culture distribution on the OECTs, which decrease while approaching the 44–46 h from seeding and clearly not affecting the assessment of the cell culture state (infected or healthy).
The advantages of employing OECTs, as an in-vitro serum-neutralization assay technology, are manifold. Firstly, OECTs allow avoiding the use of formaldehyde and other toxic/expensive substances, since there is no need to fix or stain the cell culture. The serum-neutralization assay procedure does not require experienced lab technicians, thanks to the completely automatized TECH-OECT prototype that presently permits real-time analysis of up to six different cell cultures in parallel. Noteworthy, we have obtained robust and reproducible results in less than 48 h, thus faster than for the typical PRNT test, which requires a SARS-CoV-2 incubation of 72 h for a complete quantitative assay. Moreover, OECTs can be revitalized and re-used for up to three consecutive experiments reducing plastic waste and their effective cost/experiment. The here proposed OECT technology could be scaled down to multiple wells numbers and speed up the viral neutralization screening on larger scales with higher automated reproducibility, monitoring the population immunity after direct contact with the virus or vaccination, thus quantifying the neutralizing antibodies produced and their temporal decay. Khoury and collaborators (2021) have recently provided an evidence-based model of SARS-CoV-2 immune protection showing that the neutralization level is highly predictive of immune protection. For this reason, reliable and secure methodologies for the rapid quantification of the neutralizing response against the SARS-CoV-2 will be crucial for future vaccine strategies.
S. Inal’s group very recently developed an OECT-based sensor for detecting SARS-CoV-2 spike proteins on clinical nasopharyngeal swabs and saliva samples to extract viral loads33. Similarly, biosensors for the detection of SARS-CoV-2 viral RNA, antigens, and antibodies (e.g., type G Immunoglobulins) have been developed, exploiting the transistor geometry and surface functionalization for signal amplification and selectivity, respectively34,35,36. However, to the best of our knowledge, our system represents the first example of a viral neutralization test using remotely-controlled OECTs on SARS-CoV-2, which combine high quantitative reproducibility with the real-time investigation and fast output response. Furthermore, differently from the previous works, our assay is not SARS-Cov-2 specific but could be translated towards several viruses and cell lines, accordingly setting the health/infection threshold and normalized time response ranges. Examples of real-time electrical viral proliferation or Virus neutralization tests are present in scientific literature37,38,39, even using commercially available instruments40, but they rely on microelectrodes, which lack the signal amplification given by OECT architecture and may thus lead to difficulties in monitoring leaky-barrier or non-barrier cell lines. On the contrary, it has already been theoretically16 and experimentally26,27 demonstrated the ability of OECTs to evaluate non-barrier cell tissue integrity. Our OECT-based technology would therefore help in reducing operator error and enhance assay precision and it could also be easily adapted to lower biocontainment by using pseudotyped viruses.
Our electrical system is still at an early stage of development, and we just demonstrated it as a proof-of-concept. Scale-up of OECT devices, as well as of multiple reader and software analysis, is well known and could be at a low cost. Actual OECT dimensions are compatible with a well of 11 mm diameter corresponding to those of an existing 24 multiwell plate. However, using standard lithography fabrication, OECTs can be miniaturized, reducing costs and wall dimensions, and multiwell designs up to 48 wells are easily envisaged.
As stated above, the role of the neutralizing humoral response in disease progression has been demonstrated5, thus, our technology may also be used for prognostic purposes in clinical practice.
Moreover, the susceptibility of several animal species to SARS-CoV-2 has been shown during the pandemic41. Animals may play different epidemiological roles ranging from dead-end hosts to possible reservoir species42 allowing the reintroduction of the virus once its circulation has been reduced or stopped by mass vaccination in humans. To in-depth study and monitor the role of animals in the infection dynamic, serological tests need to be available for many different species, which is currently only feasible with virus neutralization tests43. Since our technology is not species-specific it can be also employed for epidemiological purposes, for detecting neutralizing antibodies in domestic and wildlife species to investigate their role in the ecology of the infection.
Finally, owing to the specific nature of the SN screening here proposed, our low-cost and scalable devices could be easily transferred to the study of other cytopathic viruses and animal cells, for extensive, real-time monitoring and operator-safe evaluations over several critical viral infections.
Methods
Cell line
Cell studies were conducted in two different BSL-3 virology laboratories, the laboratory at Unit of Microbiology, Great Romagna Hub Laboratory, Cesena, Italy and the Istituto Zooprofilattico Sperimentale della Lombardia e dell’Emilia-Romagna (IZSLER), Brescia, Italy.
Vero E6 cells (ATCC CRL-1586) were used since they are sensitive and permissive to SARS-CoV-2 infection, leading to high titer replication44,45. Vero E6 cells are used for studies of viruses that have an affinity for the ACE2 receptor, this being highly expressed in this cell model and coronaviruses enter cells via this receptor. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% penicillin–streptomycin (P/S), and 1% l-glutamine (l-Gln). Culture medium and supplements were all purchased from EuroClone (Milan, Italy). The cell line was cultured following the protocol of growth and maintenance of Vero cell lines46. Cells were seeded at a density of 15,000 cells/well on the OECTs and cultured in 500 µL of DMEM with 2% FBS, 1% penicillin–streptomycin (P/S), and 1% l-glutamine (L-Gln).
Sera samples and virus infection
This study has been performed by using anonymized leftover serum samples. The samples have been collected from healthy blood donors for the detection of the immune response against SARS-CoV-2 by chemiluminescence assay and by PRNT for the evaluation of serum neutralizing activity in vitro.
Our PRNT assay is modified in MNA according to our already published work47. It requires the formation of a virus-antibody (in an equal volume) complex in vitro during incubation for 30 min at 37 °C in thermo shaker and subsequent seeding on cells susceptible to viral infection. After incubation (typically 72 h), the CytoPathic Effect (CPE) is evaluated by fixing and staining the cells with formaldehyde in Crystal violet and, subsequently, reading the absorbance using the spectrophotometer at 560 nm. The highest serum dilution that neutralizes 90% of viral replication and thus its CPE is reported as the neutralizing titer. At the end of the diagnostic workflow, all the samples used in this study were totally anonymized according to the “ANONYMIZING PROCEDURE” of the Great Romagna Hub Laboratory (AVR PPC 09) that has been approved by the local Ethics Committee.
PRNT assays were performed at the BSL-3 laboratory at Unit of Microbiology, Great Romagna Hub Laboratory, Cesena, Italy. The sera were not disrupted at 56 °C for 30 min as no different from the PRNT test was detected in previous tests. The antibody titer against SARS-CoV-2 was detected by SARS-CoV S1/S2 IgG Chemiluminescent ImmunoAssay (LIAISON® SARS-CoV-2 S1/S2 IgG test) and the PRNT of periodic voluntary plasma donors. Neutralizing sera with titer above 80, as determined by standard PRNT evaluation, were used for this work. PRNT tests on the OECT device were carried out using progressive serum dilution of 1:20, 1:40, 1:80, and 1:160.
The SARS-CoV-2 (titred 106.5 TCID50/mL) virus employed for all SN tests was supplied by the Virology Laboratory of Policlinico San Matteo in Pavia (Italy) and is named VR PV10734. In order to mimic, as closely as possible, the conditions of use, a wild-type viral strain belonging to lineage B1 has been employed (GISAID code EPI_ISL_1908157). Viral strain was sequenced using CleanPlex SARS-CoV-2 Flex (Paragon Genomics Inc., Hayward, CA, USA) and Illumina MiSeq (Illumina Inc. San Diego, CA, USA). Viral titers, expressed as TCID50/mL, were calculated according to the Reed and Muench method based on eight replicates for dilution48,49 and Sperman–Karber50. OECT tests employed a dilution of 300 TCID/well, prepared in DMEM/2% FBS.
Cytopathic effect value
After 72 h from the infection, cells were fixed and stained using 300 µL of a 4% formaldehyde (Fisher Chemical, Milan, Italy) solution in Crystal violet (Delcon, Bergamo, Italy) and incubated for 30 min at room temperature. The dye stains only the living cells, fixed to the well, therefore it allows to discriminate the living cells from the dead ones, which are washed away in the next step. After the incubation time, the dye is washed off with tap water and the plate is dried in a hood. Optical micrographs were taken using an inverted optical microscope, with magnification 10× (Hund Wetzlar).
The highest serum dilution capable to neutralize 90% of the CPE is reported as a neutralizing titer. The sera control is taken as a 100% neutralization reference. Hence, it is possible to calculate the optical density which corresponds to the IC90.
OECT device fabrication
Glass substrates (25 × 25 mm2) were cleaned by sonication in distilled water/acetone/isopropanol baths. Afterward, 10 nm of chrome and 50 nm of gold were deposited by thermal evaporation. After that, substrates were treated with air plasma (20 W for 4 min), and then PEDOT:PSS solution was spin-coated (3000 rpm for 10 s) using a Teflon mask. The thin film thickness was 140 ± 10 nm. The solution was made of 94% PEDOT:PSS (Heraeus, Clevios PH1000) with 5% of ethylene glycol (EG) (Sigma-Aldrich), 1% of 3-glycidoxypropyltrimethoxysilane (GOPS), and 0.25% of 4-dodecylbenzenesulfonicacid (DBSA). This suspension was treated in an ultrasonic bath for 10 min and filtered using 1.2 μm cellulose acetate filters (Sartorius) before the deposition. The samples were subsequently baked at 120 °C for 1 h. Planar geometry OECTs were patterned, having two channels with a length (L) of 1 mm and width (W) of 0.75 mm and an inner gate electrode (L = 2 mm, W = 3 mm). The dual-channel configuration doubled up the device monitoring of the same well, thus taking into account possible cell layer dis-homogeneity after seeding, that may conceal the correct outcome. Then, devices were immersed in distilled H2O for 1 h and dried with a nitrogen flux. In the end, a Polydimethylsiloxane (PDMS) transparent, cylindrical well, having an inner diameter and height of 12 and 8 mm, respectively, was bound to the device to realize the culture well.
OECT integrated system and electrical characterization set-up
All measurements were performed in DMEM as the electrolyte solution. Experiments were performed using an integrated system, the TECH-OECTs, reported in Fig. 1 and in our previous work26. Cells were seeded inside cylindrical PDMS wells, having a diameter of 12 mm. Noteworthy, TECH-OECTs allows to carry out measurements inside the humidified incubator (constant temperature of 37 °C and a CO2 level of 5%), without direct interaction with the virus SARS-Cov-2, thus can be reused. A multiplexer system was used to measure sequentially 12 channels. We measured the source-drain current by means of a Keysight B2912A Source Measure Unit (SMU), while biasing the channel with Vds = –0.1 V and introducing a square wave potential on the gate electrode, from Vgs(OFF) = 0.0 V to Vgs(ON) = 0.3 V, with ton = 0.5 s and toff = 1.5 s. Keysight and the multiplexer were both controlled with customized PC software.
OECT data analysis
Output data were analyzed with a customized Matlab routine: each single channel response to a pulse on the gate was isolated, normalized, and fitted with the bi-exponential curve \({I}_{d}={a}\,{{\exp }}^{-\frac{x}{{\tau }_{1}}}+{{\exp }}^{-\frac{x}{{\tau }_{2}}}+{e}\), as reported in our previous work26. As described elsewhere15, labeling \({\tau }_{1}\) greater than \({\tau }_{2}\), \({\tau }_{1}\) represents the charging time of the PEDOT:PSS influenced by the ion-blocking properties of the cell layer, while \({\tau }_{2}\) relates to the charging time of the cell layer. Thus, we will focus and examine \({\tau }_{1}\) as the device time response to a gate potential pulse, averaging its value over five pulses on the same channel and then normalizing it, using the following equation: \({{{{{\rm{OECT time response}}}}}}({{{{\rm{a.u.}}}}})=\tau /{\tau }_{{{{{\rm{No}}}}}\; {{{{\rm{Cells}}}}}}\), with \({\tau }_{{{\rm{No}}}} {{{\rm{Cells}}}}\) is the response time of the device before cell seeding. We then choose 1.2 a.u as the threshold value at the end of the experiment to separate viral-infected cultures (< 1.2 a.u.) from healthy ones (>1.2 a.u.), since it was consistent with the averaged experimental trends carried out in two different laboratories, on three devices, and for the different “incubation mix”.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Viral load is monitored by sampling the cell culture at the end of the experiment and using qRT-PCR Allplex Seegene analysis.
Allplex SARS-CoV-2 Extraction-Free (Seegene Inc., Seoul, Korea) is a real-time qRT-PCR assay that does not require a preparatory RNA-extraction but rather relies on the thermal lysis taking place during the reverse transcription reaction in which the specimen is warmed up to 50 °C for 20 min (reverse transcription) and then to 95 °C for 15 min (polymerase activation). The assay enables the simultaneous detection of three target genes, namely the E gene (common to all Sarbecoviruses), RdRP/S gene, and N gene (both specific for SARS-CoV-2). Sample preparation, reaction setup, and analysis were performed accordingly to the manufacturer’s instructions. In brief, 15 µL of each sample were diluted 1:4 with 45 µL of RNase-free water in a 96-well PCR plate and hence 5 µL were transferred to another plate with 16 µL of PCR master mix, containing 5 µL of MOM (MuDT Oligo Mixture, containing dNTPs, oligos, primers, and Taq-Man 5’ fluorophore / 3’ Black Hole Quencher probes), 5 µL of enzymes, 5 µL of RNase-free water and 1 µL of internal control for every reaction. A positive and negative control were included. The assay was run on a CFX96 real-time thermal cycler (Bio-Rad, Feldkirchen, Germany). The amplification process relies on the first step for cDNA denaturation at 95 °C for 10 s, followed by primers annealing at 60 °C for 15 s and elongation at 72 °C for 10 s (44 cycles). Fluorescent signals were acquired after every amplification cycle for FAM (E gene), Cal Red 610 (RdRP/S gene), Quasar 670 (N gene), and HEX (internal control) fluorophores. In a real-time PCR assay, a positive reaction is detected by the accumulation of a fluorescent signal. The Ct (cycle threshold) is defined as the number of cycles required for the fluorescent signal to cross the threshold. Ct levels are inversely proportional to the amount of target nucleic acid in the sample (i.e., the lower the Ct level the greater the amount of target nucleic acid in the sample). The fold change reported in our graph represents the Ct level at the end of the experiment (T48h), normalized with respect to its initial value at the beginning of the experiment (T0h). Results analysis and target quantification were carried out employing a 2019-nCoV viewer from Seegene Inc.
Cell death evaluation
Cell death was detected by measuring the permeability of the plasma membrane to the normally impermeable fluorescent dye DAPI. At the end of the experiment, cells were washed with PBS and incubated with DAPI for 20 min at room temperature. Cells were then harvested, washed in PBS, and fixed in 4% paraformaldehyde (PFA), and DAPI uptake was quantified by FACS analysis (Cytoflex-S Beckman Coulter, California, USA).
Statistical analysis
Differences between qRT-PCR results were analyzed using a double-sided Student’s t-test. P-values <0.05 were considered statistically significant. Statistical analyses were performed using SPSS version 19.0 (IBM Corp., Armonk, NY).
Data availability
Data are available in a public repository at the following https://doi.org/10.5281/zenodo.5771725.
Code availability
The code generated for data analysis is available in a public repository at the following https://doi.org/10.5281/zenodo.5771725.
References
Li, X., Song, Y., Wong, G. & Cui, J. Bat origin of a new human coronavirus: there and back again. Sci. China Life Sci. 63, 461–462 (2020).
Meyer, B., Drosten, C. & Müller, M. A. Serological assays for emerging coronaviruses: challenges and pitfalls. Virus Res. 194, 175–183 (2014).
Lau, E. H. Y. et al. Neutralizing antibody titres in SARS-CoV-2 infections. Nat. Commun. 12, 1–7 (2021).
Hotez, P. J., Corry, D. B., Strych, U. & Bottazzi, M. E. COVID-19 vaccines: neutralizing antibodies and the alum advantage. Nat. Rev. Immunol. 20, 399–400 (2020).
Garcia-Beltran, W. F. et al. COVID-19-neutralizing antibodies predict disease severity and survival. Cell 184, 476–488.e11 (2021).
Khoury, D. S. et al. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 27, 1205–1211 (2021).
Bewley, K. R. et al. Quantification of SARS-CoV-2 neutralizing antibody by wild-type plaque reduction neutralization, microneutralization and pseudotyped virus neutralization assays. Nat. Protoc. 16, 3114–3140 (2021).
Dale, J. L. & Peters, D. Protein composition of the virions of five plant rhabdoviruses. Intervirology 16, 86–94 (1981).
Schmidt, N. J., Dennis, J. & Lennette, E. H. Plaque reduction neutralization test for human cytomegalovirus based upon enhanced uptake of neutral red by virus infected cells. J. Clin. Microbiol. 4, 61–66 (1976).
Abbad, A. et al. Middle East respiratory syndrome coronavirus (MERS-CoV) neutralising antibodies in a high-risk human population, Morocco, November 2017 to January 2018. Eurosurveillance 24, 1–8 (2019).
R, Y. et al. Development and effectiveness of pseudotyped SARS-CoV-2 system as determined by neutralizing efficiency and entry inhibition test in vitro. Biosaf. Health 2, 226–231 (2020).
Lisdat, F. & Schäfer, D. The use of electrochemical impedance spectroscopy for biosensing. Anal. Bioanal. Chem. 391, 1555–1567 (2008).
van der Helm, M. W. et al. Non-invasive sensing of transepithelial barrier function and tissue differentiation in organs-on-chips using impedance spectroscopy. Lab Chip 19, 452–463 (2019).
Gerasimenko, T. et al. Impedance spectroscopy as a tool for monitoring performance in 3D models of epithelial tissues. Front. Bioeng. Biotechnol. 7, 474 (2020).
Faria, G. C., Duong, D. T. & Salleo, A. On the transient response of organic electrochemical transistors. Org. Electron. 45, 215–221 (2017).
Faria, G. C. et al. Organic electrochemical transistors as impedance biosensors. MRS Commun. 4, 189–194 (2014).
Spanu, A., Martines, L. & Bonfiglio, A. Interfacing cells with organic transistors: a review of in vitro and in vivo applications. Lab on a Chip 21, 795–820 (2021).
Yeung, S. Y., Gu, X., Tsang, C. M., Tsao, S. W. & Hsing, I. ming. Engineering organic electrochemical transistor (OECT) to be sensitive cell-based biosensor through tuning of channel area. Sens. Actuators A Phys. 287, 185–193 (2019).
Rivnay, J. et al. Organic electrochemical transistors for cell-based impedance sensing. Appl. Phys. Lett. 106, 043301 (2015).
Ferro, M. P. et al. Effect of E cigarette emissions on tracheal cells monitored at the air–liquid interface using an organic electrochemical transistor. Adv. Biosyst. 3, 1800249 (2019).
Lieberth, K. et al. Monitoring reversible tight junction modulation with a current-driven organic electrochemical transistor. Adv. Mater. Technol. 6, 2000940 (2021).
Lingstedt, L. V. et al. Monitoring of cell layer integrity with a current-driven organic electrochemical transistor. Adv. Healthc. Mater. 8, 1900128 (2019).
Ramuz, M., Hama, A., Rivnay, J., Leleux, P. & Owens, R. M. Monitoring of cell layer coverage and differentiation with the organic electrochemical transistor. J. Mater. Chem. B 3, 5971–5977 (2015).
Curto, V. F. et al. Organic transistor platform with integrated microfluidics for in-line multi-parametric in vitro cell monitoring. Microsyst. Nanoeng. 3, 17028 (2017).
Ramuz, M. et al. Combined optical and electronic sensing of epithelial cells using planar organic transistors. Adv. Mater. 26, 7083–7090 (2014).
Decataldo, F. et al. Organic electrochemical transistors: smart devices for real‐time monitoring of cellular vitality. Adv. Mater. Technol. 4, 1900207 (2019).
Decataldo, F. et al. Organic electrochemical transistors for real‐time monitoring of in vitro silver nanoparticle toxicity. Adv. Biosyst. 4, 1900204 (2020).
Pizzorno, A. et al. Characterization and treatment of SARS-CoV-2 in nasal and bronchial human airway epithelia. Cell Rep. Med. 1, 100059 (2020).
Emeny, J. M. & Morgan, M. J. Regulation of the interferon system: evidence that vero cells have a genetic defect in interferon production. J. Gen. Virol. 43, 247–252 (1979).
Kwon, D. et al. The effect of fetal bovine serum (FBS) on efficacy of cellular reprogramming for induced pluripotent stem cell (iPSC) generation. Cell Transplant. https://doi.org/10.3727/096368915X689703 (2016).
El-Ensahsy, H. A. et al. Serum concentration effects on the kinetics and metabolism of HeLa-S3 cell growth and cell adaptability for successful proliferation in serum free medium. World Appl. Sci. J. 6, 608–615 (2009).
Ozturk, S. S. & Palsson, B. O. Examination of serum and bovine serum albumin as shear protective agents in agitated cultures of hybridoma cells. J. Biotechnol. 18, 13–28 (1991).
Guo, K. et al. Rapid single-molecule detection of COVID-19 and MERS antigens via nanobody-functionalized organic electrochemical transistors. Nat. Biomed. Eng. https://doi.org/10.1038/s41551-021-00734-9 (2021).
Liu, H. et al. Ultrafast, sensitive, and portable detection of COVID-19 IgG using flexible organic electrochemical transistors. Sci. Adv. 7, 8387–8402 (2021).
Paulose, A. K. et al. A rapid detection of COVID-19 viral RNA in human saliva using electrical double layer-gated field-effect transistor-based biosensors. Adv. Mater. Technol. https://doi.org/10.1002/ADMT.202100842 (2021).
Ditte, K. et al. Rapid detection of SARS-CoV-2 antigens and antibodies using OFET biosensors based on a soft and stretchable semiconducting polymer. ACS Biomater. Sci. Eng. https://doi.org/10.1021/ACSBIOMATERIALS.1C00727 (2021).
Charretier, C. et al. Robust real-time cell analysis method for determining viral infectious titers during development of a viral vaccine production process. J. Virol. Methods 252, 57–64 (2018).
Tian, D. et al. Novel, real-time cell analysis for measuring viral cytopathogenesis and the efficacy of neutralizing antibodies to the 2009 influenza A (H1N1) virus. PLoS ONE 7, e31965 (2012).
Fang, Y., Ye, P., Wang, X., Xu, X. & Reisen, W. Real-time monitoring of flavivirus induced cytopathogenesis using cell electric impedance technology. J. Virol. Methods 173, 251–258 (2011).
Maestro Pro | Axion Biosystems. https://www.axionbiosystems.com/products/systems/maestro-pro?camp=techEU&gclid=Cj0KCQjw5JSLBhCxARIsAHgO2ScGZCPAk270jRfNh8BfOZansrMKX0wk9i8P86-CkMwmVEhXoyZhXKAaAr9PEALw_wcB.
Shi, J. et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 368, 1016–1020 (2020).
de Morais, H. A. et al. Natural Infection by SARS-CoV-2 in companion animals: a review of case reports and current evidence of their role in the epidemiology of COVID-19. Front. Vet. Sci. 7, 591216 (2020).
Jara, L.M. et al. Evidence of neutralizing antibodies against SARS-CoV-2 in domestic cats living with owners with a history of COVID-19 in Lima - Peru. One Heal. (Amsterdam, Netherlands) 13, https://doi.org/10.1016/J.ONEHLT.2021.100318 (2021).
Ogando, N. S. et al. SARS-coronavirus-2 replication in Vero E6 cells: replication kinetics, rapid adaptation and cytopathology. J. Gen. Virol. 101, 925–940 (2020).
Takayama, K. In vitro and animal models for SARS-CoV-2 research. Trends Pharmacol. Sci. 41, 513–517 (2020).
Ammerman, N. C., Beier-Sexton, M. & Azad, A. F. Growth and maintenance of vero cell lines. Curr. Protoc. Microbiol. 11, A.4E.1–A.4E.7 (2008).
Pierro, A. et al. Seroprevalence of West Nile virus–specific antibodies in a cohort of blood donors in Northeastern Italy. Vector Borne Zoonotic Dis. https://home.liebertpub.com/vbz (2011).
Reed, L. J. & Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27, 493–497 (1938).
Smith, M. R. R., Schirtzinger, E. E., Wilson, W. C. & Davis, A. S. Rift valley fever virus: propagation, quantification, and storage. Curr. Protoc. Microbiol. 55, e92 (2019).
Ramakrishnan, M. A. Determination of 50% endpoint titer using a simple formula. World J. Virol. 5, 85 (2016).
Author information
Authors and Affiliations
Contributions
F.D. contributed to literature investigation, device fabrication, and testing, together with electrical measurements and data analysis; L.G. contributed in cell maintenance, seeding, and qRT-PCR measurements and analysis; M.M. contributed in q-RT-PCR analysis and cell maintenance; C.G. contributed with ideas, cytofluorimeter analysis, and supervising; M.B., G.D., and F.T. contributed with suggestions and ideas; F.F. and D.L. contributed in cell maintenance and supervising; M.T. and M.C. contributed with ideas and supervising; C.C. contributed supervising the work and results; A.S., B.F., A.L., and V.S. contributed supervising as project leaders. All authors have given approval to the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
There is a patent pending on the reported technology, number 102021000023354. The inventors are Dr. Decataldo Francesco, Dr. Marta Tessarolo, Dr. Catia Giovannini, Prof. Vittorio Sambri, Prof. Alessandra Scagliarini, and Prof. Beatrice Fraboni. The remaing authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Rona Chandrawati and John Plummer. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Decataldo, F., Grumiro, L., Marino, M.M. et al. Fast and real-time electrical transistor assay for quantifying SARS-CoV-2 neutralizing antibodies. Commun Mater 3, 5 (2022). https://doi.org/10.1038/s43246-022-00226-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-022-00226-6
