
Abstract
Mussel-inspired hydrogels have gained attention for underwater applications, including treatment of wastewater. However, they are typically limited by poor mechanical properties, short-term mechanical stability and by not being reusable. Here, we develop a mechanically stable and self-healing hydrogel with high mechanical strength for the degradation of dyes in wastewater, based on cellulose-derived co-polydopamine@Pd nanoparticles. A dynamic catechol redox system was achieved by reversible conversion between semiquinone and quinone/hydroquinone radicals, endowing the hydrogel with stable mechanical properties and self-healing behavior. Furthermore, a graphene oxide membrane is covalently grafted on to the hydrogel surface, which regulates its water permeability and intercepts some metal ions or large particles, protecting the hydrogel structure. High catalytic activity for anionic and cationic dyes is achieved, with the degradation rate reaching more than 95% after multiple cycles without significant deterioration in performance or hydrogel structure. Our work demonstrates a route to achieve mechanically stable hydrogels for degradation of organic dyes in wastewater.
Similar content being viewed by others

Introduction
Underwater application is essential for materials in many areas, and thus is a ubiquitous and indispensable requirement. Hydrogels have attracted a great deal of interest in a wide range of tissue engineering1,2, chemical engineering3,4, and biomedicine5,6. Scientists have been fascinated by the multifunctional hydrogels that can be used underwater, such as for water purification7,8,9, organics adsorption or degradation10,11,12,13,14, and biological and medical purposes15. However, it is difficult to keep the mechanical properties and stability-hold ability because water molecules can greatly weaken the contribution of van der Waals’ interaction when it was fully immersed in water16,17,18. More seriously, a complex water environment, especially one containing metal ions, may affect the structure of the hydrogels and cause salt deposition19,20,21,22. This is a common issue that would further reduce the underwater application efficiency and life. The existing technologies have difficulty meeting the requirements of underwater applications with high efficiency and effectiveness in in situ reaction separation and recycling.
To address these challenges, mussel-inspired hydrogels with polydopamine (PDA), a typical example of super-toughness hydrogels prepared by catechol chemistry, can be used in a water environment23,24,25,26,27. Such hydrogels can effectively enhance the underwater mechanical property due to the formation of covalent/noncovalent bonds28,29,30. Among them, the construction of dynamic bond is an effective means to realize strong mechanical and self-healing18,31. However, the hydrogels functionalized by pure PDA generally do not meet the actual demand for mechanical properties32,33. In addition, overoxidation of catechol groups causes a worsening of the mechanical properties of the hydrogel34, which is vital to controlling the oxidation degree of catechol groups35,36,37. On the other hand, it is necessary to assemble a chemical stability membrane for the selective passage of different ions and substances38,39,40,41. Previous reports suggested that an effective combination of the catalytic performance, long-term mechanical property, and selective permeation would benefit from an integrative strategy. However, are these achievements technically possible?
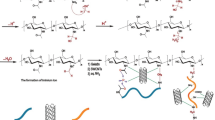
To verify the feasibility of the above-described strategy, we develop a cellulose-based hydrogel for dye degradation in water. This hydrogel is constructed by grafted polymers, that is, coagulating them from an aqueous precursor solution containing cellulose–graft–polydopamine loaded with Pd NPs (DACO-PDA@Pd NPs), AA, and SA. As shown in Fig. 1, the polymers form an interpenetrating network through the cross-linking of covalent and noncovalent bonds. The dynamic redox catechol system between semiquinone and quinone/hydroquinone radicals, triggered by the Pd NPs in the interhydrogel network, endows the hydrogel with an excellent ductility, reversibility, durability, and self-healing ability. Furthermore, the prepared hydrogel is completely coated with graphene oxide sheets to achieve its strong hydrophilicity, stability, and selective permeability using PNIPAM chains as a medium, which is noted as p(DACO-DA@Pd NPs-co-AA-SA)@P-GOM. This is a promising approach for degrading organic matter, such as anionic dyes (Congo red, CR) and cationic dyes (methylene blue, MB).

a Cross-linking point between DACO-PDA@Pd NPs with PAA and SA. b Immobilization form of Pd NPs on DACO-PDA and a brief diagram of the DACO-PDA@Pd NPs. c Cross-linking between the PNIPAm-co-GO membrane and the surface of the p(DACO-PDA@Pd NPs-co-AA-SA) hydrogel, that is, the PNIPAm-co-GO membrane (P-GOM) was coated outside of the hydrogel. d PNIPAm-co-GO membrane (P-GOM).
Results and discussion
Fabrication rationale of the hydrogel
To construct a reversible hydrogel system, the preparation method was crucial, and the mechanism diagram is shown in Fig. 2. This method can be achieved in three steps. First, Pd NPs are immobilized on the DACO-PDA fibers to form DACO-PDA@Pd NPs rich in hydrophilic groups based on previously reported work42,43,44. Second, the phenolic hydroxyls on the DACO-PDA@Pd NPs were converted into semiquinone radicals, and then were oxidized to the corresponding quinone/hydroquinone in the process of converting Pd2+ into Pd NPs (Fig. 2a). Finally, SA and AA are mixed with the undried DACO-PDA@Pd NPs to construct a catechol-conjugated alginate hydrogel with high toughness owing to the quinone–catechol-reversible reaction under an ambient environment (Fig. 2b, c).

a Immobilization of Pd NPs on the surface of the DACO-PDA, and the generation of free radicals by the redox reaction between the DACO-PDA@Pd NPs and APS. Schematic diagrams of the molecular structure model b before and c after polymerization. The following models show what each stage represents. d SEM and e TEM images of Pd NPs loaded on the surface of DACO-PDA fibrils. f, g SEM images of the pure PAA and DACO-PDA-PAA@Pd NPs hydrogels, respectively.
In this system, the introduction of multifunctional groups and the successful loading of Pd NPs on the surface of the DACO-PDA were essential to the construction of hydrogel network structure. The grafting method of DACO-PDA fibers and the immobilization mechanism of Pd NPs are shown in Supplementary Fig. 1. The DACO-PDA chains were rich in carboxyl (9.64 mmol g−1, Supplementary Figs. 2–4, Supplementary Tables 1 and 2), aldehyde (5.29 mmol g−1), and hydroxyl groups. Among them, the negatively charged carboxyl groups could attract the positively charged Pd(II) by electrostatic interaction; meanwhile, the aldehyde groups and some hydroxyls acted as reductants. The Pd NPs were distributed on the surface with a diameter range of 15–40 nm (Fig. 2d, e, Supplementary Figs. 5–7)45. These Pd NPs displayed clear codirectionally arranged lattice fringes, demonstrating the single-crystalline character of the Pd NP crystal (Supplementary Fig. 8)46. In contrast to the characteristic peaks of DACO, the characteristic peaks of DACO-PDA@Pd NPs at 40.1°, 46.6°, 68.9°, and 82.5° correspond to Pd (111), Pd (200), Pd (220), Pd (311), and Pd (222), respectively, proving the production of Pd NPs. The changes of characteristic peaks in XRD and XPS spectra confirmed the deposition of Pd NPs onto DACO-PDA (Supplementary Figs. 9 and 10). The short reaction time (less than 2 h) and mild–moderate reducing condition (approximately 80 °C) made this stratgy an ideal green-reaction system.
Based on the above results, preparing tough hydrogel using DACO-PDA-PAA@Pd NPs was another crucial step. In this system, a large number of carboxyl and hydroxyl groups on the DACO chains acted as active sites of hydrogen bonds or ionic bonds, thus achieving cross-linking. Compared with the samples without Pd NPs, the appearance of the strong peak of DACO-PDA@Pd NPs/APS indicated that many quinone/semiquinone-free radicals were generated in the solution, characterized by electron spin resonance (ESR) (Supplementary Fig. 11). These results directly showed that only the DACO-PDA@Pd NP solution could generate enough free radicals to trigger the polymerization of free radicals (Supplementary Fig. 12). The composite hydrogels showed an interwoven microfiber structure after freeze drying, owing to the uniform distribution of catechol groups and Pd NPs in the DACO-PDA-PAA@Pd NPs (Fig. 2f, g, Supplementary Figs. 13 and 14). This finding was attributed to the synergistic effect of the catechol groups in the DACO-PDA@Pd NPs (which can generate catechol and quinone groups during the redox reaction) and –COOH (in PAA and DACO, which can interact with various surfaces by electrostatic interaction) (Supplementary Fig. 15). Furthermore, the appearance of C–O (C–OH) and C = O indicated the presence of catechol groups (Supplementary Fig. 16), and the changes in the N1s and Pd regions revealed the successful construction (Supplementary Figs. 17 and 18). Therefore, as a result of the synergistic effects of the carboxyl groups in the PAA and catechol groups in the DACO-PDA@Pd NP chains, the hydrogels were formed and the Pd NPs worked as nanoreinforcement to improve the mechanical properties of the hydrogels. In addition, hydrogels could form covalent bonds with some surfaces containing amines or thiols or noncovalent bonds (e.g., hydrogen bonds, π–π stacking, or metal coordination) with special solid surfaces (Fig. 2b, c).
Mechanical and interfacial assembly properties of hydrogel
To characterize the mechanical strength of these hydrogels, compressive and tensile tests were carried out. Compared with the pure PAA and SA hydrogel without resistance, the addition of DACO-PDA@Pd NPs significantly enhanced the strength of the hydrogels (Fig. 3a, Supplementary Fig. 19). It can withstand a high pressure and quickly recover its original shape without damage after removing the compressive load (Supplementary Fig. 20). The almost-unchanged load–unloaded compression curves after five cycles showed that the DPAS hydrogel had good recoverability (Fig. 3b). Similarly, the hydrogel can also withstand a high tensile stress (Fig. 3d, e). The tensile stress–strain curve increased with increasing DACO-PDA@Pd NP content, and reached a maximum value at the DP0.05A2S0.4 (Supplementary Fig. 21); 0.05, 2, and 0.4 represent the mass ratio of each factor. The mechanical properties of DPAS hydrogels were in stark contrast to those of pure hydrogels. On the other hand, under the action of the outer load, the freeze-dried hydrogels exhibited a regular layered structure under pressure (Fig. 3c) and an interwoven microfibril structure under tension by cross-linking (Fig. 3f). The improved mechanical properties attributed to the cross-linking of covalent bonds initiated by hydrophilic functional groups on DACO-PDA@Pd NPs. They also endowed the catechol-conjugated mixed hydrogel with a highly flexibility and toughness. Similarly, the adhesiveness properties of the hydrogels to pigskin were also measured (Supplementary Fig. 22a). The adhesion strength of the hydrogels increased with the DACO-DA@Pd NPs contents and had a maximum value of 34.7 kPa ± 2.6 kPa at 0.05 wt% DACO-PDA@Pd NPs. Furthermore, the hydrogel still kept good adhesion properties even after 15 cycles (Supplementary Fig. 22b). The results were attributed to the construction of various covalent and noncovalent bonds between catechol groups of DACO-PDA@Pd NPs and carboxyl groups of PAA/SA. The mechanical and adhesiveness properties ensured the high-level demand for strength and bending degree of hydrogels during underwater application.

a The compressive loading–unloading curves of various hydrogels. b The compressive loading–unloading curves of 5 cycles of DPAS hydrogel. c SEM image showing the layered stacking structure of the DPAS hydrogel in compression. d The typical tensile stress–strain curves of various hydrogels. e The tensile loading–unloading curves of five cycles of DPAS hydrogel. f SEM image showing the microfibril structure of DPAS hydrogel when stretching. g Fracture-recovery test of the hydrogel. h Tensile images and i the corresponding stretch magnification of the hydrogel after cutting and followed by self-healing for 2 h. j Tensile stress–strain curves of the original and healed hydrogel after healing for 2 h. (The hydrogels are expressed as DPA, DPAS, and DPAS@P-GOM gels, where DP, A, S, and GO indicate the hydrogels containing DACO-PDA@Pd NPs, acrylic acid, sodium alginate, and graphene oxide, respectively. For example, DPAS corresponds to the p(DACO-DA@Pd NPs-co-AA-SA) hydrogel.).
In addition to the excellent mechanical strength and adhesiveness properties, the dynamic catechol chemistry endowed the hydrogel with a good self-healing ability. Hydrogels placed in a petri dish were fractured manually with a sharp knife and kept at room temperature without any externally applied influences. All the visible fractures disappeared completely within 2 h (Fig. 3g). When the healed hydrogel was stretched, an obvious tensile pattern could be observed in the fractured part, but no breaking occurred (Fig. 3h, i). Regrading the tensile properties, the cut hydrogel after self-healing for 2 h nearly recovered to ~90% (Fig. 3j). The self-healing ability was higher than that of other hydrogels in the literature47,48,49. These results demonstrated that the dynamic bond could effectively facilitate the reversible recombination at the fracture interface and resulted in good self-healing properties.
To investigate the kinetics behavior of the hydrogel in water, we further explored the wettability and selective ion permeability properties of the sample surface. For the outside coating layer, in contrast to the DPAS hydrogel with rough micropore structure, a dense hydrophilic layer was formed when the hydrogel was grafted with GO by PNIPAm (DPAS@P-GOM). The appearance of new peaks at 1100–1500 cm−1 by Fourier transform-infrared spectroscopy (FT-IR) (Supplementary Fig. 23) indicated the fracture of C = C bonds on the surface of the GO, further showing the successful modification of the surface50,51. Plainly, the water contact angle (WCA) decreased from 74.9° to 33.5° with the addition of SA and further decreased to 0° with the modification of the surface by GO (Fig. 4a). The introduction of hydrophilic groups and the construction of a superabsorbent interface allow a reversible transition between superhydrophilicity and hydrophobicity, as shown in Fig. 4b. In addition, in this structure, the P-GOM acted as a functional coating that changed the materials in contact with the feed liquid, acting as a protective layer and antifouling coating (Fig. 4c–f)38,52,53. Consequently, the rejection of metal ions could avoid the changes in the hydrogel structure because hydrogels could form covalent bonds with some metal ions containing amines or thiols or noncovalent bonds (e.g., hydrogen bonds, π–π stacking, or metal coordination) (Fig. 4g).

a Photographs showing the wettability of DPA, DPAS, and DPAS@P-GOM (left) and the corresponding water contact angle (right). b Water contact angle (WCA) cyclicity of various hydrogels. SEM images of DPAS hydrogel, c, e unmodified and d, f modified by GO immersing in water, and the corresponding enlarged images. g Transport pathways of the water molecules, organics, and metal ions in the DPAS hydrogel coated by P-GO functional membranes (DPAS@P-GOM).
Degradation of organic dyes and the reusability of hydrogel
To explore the underwater application performance of DPAS@P-GOM hydrogel, the discoloration of dyes (CR and MB dissolved in tap water) was monitored in the presence of NaBH4. As the reaction proceeded, a continuous discoloration of CR solution by NaBH4 was observed, reaching up to approximately 98% after ~20 min, as shown in Fig. 5a. In contrast, it occurred at an extremely slow rate in the absence of DPAS@P-GOM hydrogel, such that only a slight color change was observed after 60 min (Supplementary Fig. 24). In this system, CR molecules diffused into the hydrogel and came into contract with the Pd NPs, creating an electron transport “relay station”, where the Pd NPs accepted electrons from the BH4− and passed them to the CR. The BH4− solution liberated hydrogen in the existence of Pd NPs and reduced the azo bond to imine, which eventually led to the degradation of the CR into small molecules that were colorless and exhibited a low toxicity (Supplementary Fig. 25a, b). The final molecules in the CR degradation in our case were confirmed by liquid chromatography–mass spectrometry (LC–MS) and gas chromatography–mass spectrometry (Supplementary Figs. 26 and 27)54. Similarly, as shown in Fig. 5b, the MB solution was also discolored in the presence of DPAS@P-GOM hydrogel and NaBH4 within 3.5 min. Again, the Pd NPs acted as an electron transport “relay station” in this system, and more surface sites of the Pd NPs, BH4−, and water molecules were involved in the fast discoloration (Supplementary Fig. 25c, Supplementary Fig. 28). It has been proven that the DPAS@P-GOM hydrogel has effective degradation performance for dyes.

The reduction of a CR and b MB degraded by DPAS@P-GOM hydrogel. Reduction of c CR and d MB for five successive cycles with the same batch of DPAS@P-GOM hydrogel. SEM images of the freeze-dried e DPAS@P-GOM and f DPAS hydrogel in CR solution for five cycles. g Catalytic activity of the DPAS@P-GOM hydrogel in the redox reaction of CR and NaBH4.
To further understand the reusability of the DPAS@P-GOM hydrogel, recycling experiments were carried out subsequently. For the CR solution, the extension of the reduction time from 20 min for the first cycle to 23 min for the fifth cycle showed good reusability of the DPAS@P-GOM hydrogel (Fig. 5c). On the other hand, for MB solution, the reduction time increased from 3.5 min for the first cycle to 4 min for the fifth cycle, and the degradation efficiency remained over 99% in each case (Fig. 5d). However, the sites occupied by dye molecules increased gradually over time, which made the reduction reaction of each molecule “queue up” and led to a certain delay. After five cycles, no obvious changes in morphology of DPAS@P-GOM hydrogel have been observed (Fig. 5e) with chemical structure monitoring by FT-IR (Supplementary Fig. 29), confirming its stability. In contrast, the DPAS hydrogel was seriously damaged after five cycles (Fig. 5f). This was mainly because salt deposition destroyed the structure of DPAS hydrogel, resulting in a prolonged delay during the cycle and the reduction in degradation efficiency39,51. These results confirmed that the DPAS@P-GOM hydrogel had good stability and exhibited excellent reusability for the degradation of dyes.
Since Pd NPs were the controlling factor in this system, the leaching of Pd from the hydrogel carrier was also considered and identified by inductively coupled plasma MS analysis (detection limit of 0.4 ppt). After five cycles, the content of leached Pd in the water cannot be determined (i.e., below the minimum value of the instrument). Furthermore, after 50 cycles, it is still difficult to detect the existence of Pd in the water. These results showed that Pd can be well immobilized on the composite cellulose (inside the hydrogel) and also verifies the stability and reusability of the composite hydrogel. To explore the role of Pd NPs in this system, the sample was separated from the reaction solution, and a slow change of degradation yield was observed (Fig. 5g). However, when Pd NPs were introduced into the above system again, the reaction rate increased, and the final degradation efficiency was equal to the control group. These results illustrated that Pd NPs immobilized on the surface of the DACO-PDA (or hydrogel interior) that act as the reaction “controllable” reaction sites.
In summary, we developed a tough and self-healing hydrogel with a high durability, reversibility, and stability, suitable for long-term underwater applications. The DACO-PDA@Pd NPs were developed to maintain a dynamic balance between the catechol and the quinone/semiquinone groups to polymerize the AA monomers. The hydrogel herein displayed high mechanical strength and self-healing owing to the reversible quinone–catechol reaction under an ambient environment. These properties maintained the stability of the 3D framework and avoided volume collapse by constructing a dense layer with an interior network structure. Moreover, the grafted P-GOM layer on the surface of the hydrogel could realize the high permeation of water and organics while rejecting the entry of metal ions to maintain the stability of the hydrogel structure. In short, the effective combination of reusable, stable, self-healing, and catalysis characteristics makes this hydrogel particularly suitable for underwater application, suggesting a great potential for use in water purification.
Methods
Materials and chemicals
Sodium periodate, sodium chlorite (NaClO2), hydrochloric acid (HCl), dopamine hydrochloride (DA), sodium alginate (SA), acrylic acid (AA), graphene oxide (GO, 1 wt%), poly(N-isopropylacrylamide) (PNIPAm), ammonium persulfate (APS, 98.0%), palladium chloride (PdCl2, 99.9%), sodium borohydride (NaBH4), disodium hydrogen phosphate (Na2HPO4), sodium dihydrogen phosphate (NaH2PO4), dialysis bag (MWCO = 8000), and ethanol were acquired from Aladdin (Shanghai, China). The water used throughout the hydrogel preparation process was deionized, and the dye degradation experiment process was tap water.
Preparation of cellulose nanofibrils
Bamboo pulp was obtained by a refined cooking process with active oxygen and solid alkali (CAOSA) developed by the authors’ group55. The bamboo pulp was reformed by mechanical treatment and used as the original cellulose fiber, which has been described in detail in our earlier work42,43. Then, adding NaClO2 to the solution, the mixture was adjusted to pH 5.8 with PBS and stirred for 24 h. Then NaBH4 was added and stirred for another 1 h to form a uniform emulsion, until no further decrease in pH, and dialyzed against for 2 days. The obtained supernatant was referred to as DACO. The concentration of DACO suspensions was 2.3 wt% by freeze drying. The details of preparation of DACO are listed in Supplementary Table 1.
Preparation of DACO-PDA@Pd NPs
The DACO suspension was mixed with dopamine (DA) for 12 h at room temperature. Then, NaBH4 was added, and the mixture was further stirred for 1 h to obtain DACO-PDA after dialyzing. The details of preparation of DACO-PDA are listed in Supplementary Table 2. The PdCl2 solution (20 mM, HCl) was slowly dripped into DACO-PDA under violent stirring at 80 °C for 2 h. The dialysis products were marked as DACO-PDA@Pd NPs. The details of the preparation of DACO-PDA@Pd NPs are listed in Supplementary Table 3.
Preparation of the hydrogels
AA, SA, APS, and DACO-PDA@Pd NPs solution were mixed to prepare a homogeneous solution under magnetic stirring at room temperature, and then injected into a reaction mold. The final products were placed in a beaker for 2 h to obtain p(DACO-DA@Pd NPs-co-AA-SA) hydrogel at 60 °C (Supplementary Tables 4 and 5).
Surface modification of the hydrogels
The above hydrogel (5 × 1 × 0.1 cm, ~0.5 g) was immersed into 10 mL of the aqueous solution containing GO. Poly(N-isopropylacrylamide) (PNIPAm) and ammonium persulfate (APS, 98.0%) were added into the above solution and stored at room temperature (~25 °C) for at least 12 h. The as-prepared product was further polymerized at 60 °C for 2 h and abbreviated as p(DACO-DA@Pd NPs-co-AA-SA)@P-GOM. Finally, the product was dialyzed in deionized water for 48 h to remove the unreacted molecules.
Characterization
The aldehyde content of the DAC or DACO was calculated using an elemental analyzer. The functional groups or chemical bonds of the samples were analyzed by Fourier transform-infrared spectroscopy (FT-IR, Nicolet iS5, Thermo Scientific, USA). The morphology of the Pd NPs loaded on DACO-PDA was scanned with a high-resolution transmission electron microscope (TEM, JEM-2100, JEOL Ltd., Japan) and a Field Emission Scanning Electron Microscope (FE-SEM, SUPRA 55VP, ZEISS, Germany). The elemental analysis was exhibited by energy-dispersive X-ray spectroscopy attached to the SEM. The crystal was determined by an X-ray powder diffractometer (XRD, Rigaku Ultima IV, Japan). UV–vis (UV-2600, Shimadzu, Japan) was used to monitor the reduction of Pd(II) by DACO-PDA and catalytic performance of the hydrogels. An ESR Spectrometer (EMX-10/12, Bruker, Germany) was used to monitor the free radicals in this system. The mechanical and adhesiveness properties of hydrogels were tested by an Instron 4465 instrument. The products of dye degradation were detected by LC–MS.
Removal of organic dyes
Methylene blue (MB) and Congo red (CR) were selected to evaluate the catalytic activity of the p(DACO-DA@Pd NPs-co-AA-SA)@P-GOM hydrogels. The water used throughout the dye degradation experiment process was tap water. The hydrogel was added into 15 mL of a mixed solution containing 10 mL of 30 mg L−1 dyes and 5 mL of 40 mg L−1 NaBH4 in a flask and stirred throughout the determination. During the determination, a solution of 3 ml was transferred to a colorimetric dish of the UV–vis spectrophotometer that can monitor the amount of adsorption of p(DACO-DA@Pd NPs-co-AA-SA)@P-GOM hydrogels by recording the time-dependent absorbance spectra of MB (525–725 nm) and CR (250–600 nm), and calculated the number of reduced dye moles (mol) per mole of Pd (mol−1) per minute (min−1). After the test, the liquid was repoured into the mixed liquid and stirred, and repeated determination was carried out. As a contrast, experiments were also carried out without Pd NPs or NaBH4.
To study the recycling performance of the p(DACO-DA@Pd NPs-co-AA-SA)@P-GOM hydrogels, the dye-adsorbed hydrogel was extruded with tweezers and placed into a solution containing the same concentration of dye and NaBH4. The as-prepared hydrogels (2 cm × π 0.52) were immobilized in a funnel. The mixed solution contained 10 mL of 30 mg L−1 dyes, and 5 mL of 40 mg/L NaBH4 was permeated by the porous hydrogel driven by gravity. The hydrogel properties of five repeated compression/discoloration were monitored by UV–vis absorption spectroscopy. The reaction rate and discoloration efficiency of each cycle was calculated by taking MB at 665 nm and CR at 495 nm as the maximum absorption peaks.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Yuk, H. et al. Dry double-sided tape for adhesion of wet tissues and devices. Nature 575, 169 (2019).
Guo, J. L. et al. Modular, tissue-specific, and biodegradable hydrogel cross-linkers for tissue engineering. Sci. Adv. 5, eaaw7396 (2019).
Chen, C. et al. Highly flexible and efficient solar steam generation device. Adv. Mater. 29, 1701756 (2017).
Sutar, P., Suresh, V. M., Jayaramulu, K., Hazra, A. & Maji, T. K. Binder driven self-assembly of metal-organic cubes towards functional hydrogels. Nat. Commun. 9, 3587 (2018).
Chen, W. et al. Stimuli-responsive nucleic acid-based polyacrylamide hydrogel-coated metal-organic framework nanoparticles for controlled drug release. Adv. Funct. Mater. 28, 1705137 (2018).
Zhu, Y., Zhang, Q., Shi, X. & Han, D. Hierarchical hydrogel composite interfaces with robust mechanical properties for biomedical applications. Adv. Mater. 31, 1804950 (2019).
Bethke, K. et al. Functionalized cellulose for water purification, antimicrobial applications, and sensors. Adv. Funct. Mater. 28, 1800409 (2018).
Geng, H. et al. Plant leaves inspired sunlight-driven purifier for high-efficiency clean water production. Nat. Commun. 10, 1512 (2019).
Hou, N. et al. Facile preparation of self-assembled hydrogels constructed from poly-cyclodextrin and poly-adamantane as highly selective adsorbents for wastewater treatment. Soft Matter. 15, 6097–6106 (2019).
Song, X. et al. A bioinspired strategy towards super-adsorbent hydrogel spheres via self-sacrificing micro-reactors for robust wastewater remediation. J. Mater. Chem. A 7, 21386–21403 (2019).
Wang, J. et al. A targeted “Capture” and “Removal” scavenger toward multiple pollutants for water remediation based on molecular recognition. Adv. Sci. 3, 1500289 (2016).
Zhu, J. et al. A facile preparation method for new two-component supramolecular hydrogels and their performances in adsorption, catalysis, and stimuli-response. RSC Adv. 9, 22551–22558 (2019).
Feng, Y. et al. Facile synthesis of Ag/Pd nanoparticle-loaded poly(ethylene imine) composite hydrogels with highly efficient catalytic reduction of 4-nitrophenol. ACS Omega 5, 3725–3733 (2020).
Meng, Y. et al. Self-assembled copper/cobalt-containing polypyrrole hydrogels for highly efficient ORR electrocatalysts. J. Mol. Liq. 298, 112010 (2019).
Han, L., Wang, M., Prieto-Lopez, L. O., Deng, X. & Cui, J. Self-hydrophobization in a dynamic hydrogel for creating nonspecific repeatable underwater adhesion. Adv. Funct. Mater. 30, 1907064 (2020).
Ma, Y. et al. Remote control over underwater dynamic attachment/detachment and locomotion. Adv. Mater. 30, 1801595 (2018).
Ostrowska-Czubenko, J. & Gierszewska-Drużyńska, M. Effect of ionic crosslinking on the water state in hydrogel chitosan membranes. Carbohyd. Polym. 77, 590–598 (2009).
Chakma, P. & Konkolewicz, D. Dynamic covalent bonds in polymeric materials. Angew. Chem. Int. Ed. 58, 9682–9695 (2019).
Guo, H., Sanson, N., Hourdet, D. & Marcellan, A. Thermoresponsive toughening with crack bifurcation in phase-separated hydrogels under isochoric conditions. Adv. Mater. 28, 5857–5864 (2016).
Sato, K. et al. Phase-separation-induced anomalous stiffening, toughening, and self-healing of polyacrylamide gels. Adv. Mater. 27, 6990–6998 (2015).
Liu, J. et al. Soy protein-based polyethylenimine hydrogel and its high selectivity for copper ion removal in wastewater treatment. J. Mater. Chem. A. 5, 4163–4171 (2017).
Xing, Q. et al. Increasing mechanical strength of gelatin hydrogels by divalent metal ion removal. Sci. Rep. 4, 04706 (2014).
Yang, B. et al. In vivo residue-specific dopa-incorporated engineered mussel bioglue with enhanced adhesion and water resistance. Angew. Chem. Int. Ed. 53, 13360–13364 (2014).
Cholewinski, A., Yang, F. K. & Zhao, B. Algae-mussel-inspired hydrogel composite glue for underwater bonding. Mater. Horiz. 6, 285–293 (2019).
North, M. A., Del Grosso, C. A. & Wilker, J. J. High strength underwater bonding with polymer mimics of mussel adhesive proteins. ACS Appl. Mater. Interfaces 9, 7866–7872 (2017).
Hofman, A. H., van Hees, I. A., Yang, J. & Kamperman, M. Bioinspired underwater adhesives by using the supramolecular toolbox. Adv. Mater. 30, 1704640 (2018).
Han, L. et al. Tough, self-healable and tissue-adhesive hydrogel with tunable multifunctionality. NPG Asia Mater. 9, e372 (2017).
Liu, Y., Ai, K. & Lu, L. Polydopamine and its derivative materials: synthesis and promising applications in energy, environmental, and biomedical fields. Chem. Rev. 114, 5057–5115 (2014).
Yang, X., Yan, L., Ma, J., Bai, Y. & Shao, L. Bioadhesion-inspired surface engineering constructing robust, hydrophilic membranes for highly-efficient wastewater remediation. J. Membr. Sci. 591, 117353 (2019).
Sun, H. et al. Novel mussel-inspired zwitterionic hydrophilic polymer to boost membrane water-treatment performance. J. Membr. Sci. 582, 1–8 (2019).
Jeon, I., Cui, J., Illeperuma, W. R. K., Aizenberg, J. & Vlassak, J. J. Extremely stretchable and fast self-healing hydrogels. Adv. Mater. 28, 4678–4683 (2016).
GhavamiNejad, A., Park, C. H. & Kim, C. S. In situ synthesis of antimicrobial silver nanoparticles within antifouling zwitterionic hydrogels by catecholic redox chemistry for wound healing application. Biomacromolecules 17, 1213–1223 (2016).
Shin, J. et al. Tissue adhesive catechol-modified hyaluronic acid hydrogel for effective, minimally invasive cell therapy. Adv. Funct. Mater. 25, 3814–3824 (2015).
Park, J. P. et al. Vanadyl-catecholamine hydrogels inspired by ascidians and mussels. Chem. Mater. 27, 105–111 (2015).
Gan, D. et al. Plant-inspired adhesive and tough hydrogel based on Ag-Lignin nanoparticles-triggered dynamic redox catechol chemistry. Nat. Commun. 10, 1487 (2019).
Zhao, Y. et al. Bio-inspired reversible underwater adhesive. Nat. Commun. 8, 2218 (2017).
Ponzio, F. et al. Oxidant control of polydopamine surface chemistry in acids: a mechanism-based entry to superhydrophilic-superoleophobic coatings. Chem. Mater. 28, 4697–4705 (2016).
Huang, L. et al. Reduced graphene oxide membranes for ultrafast organic solvent nanofi ltration. Adv. Mater. 28, 8669–8674 (2016).
Yang, Q. et al. Ultrathin graphene-based membrane with precise molecular sieving and ultrafast solvent permeation. Nat. Mater. 16, 1198 (2017).
Lee, H. A., Ma, Y., Zhou, F., Hong, S. & Lee, H. Material-independent surface chemistry beyond polydopamine coating. Acc. Chem. Res. 52, 704–713 (2019).
Jiao, T. et al. Self-assembly reduced graphene oxide nanosheet hydrogel fabrication by anchorage of chitosan/silver and its potential efficient application toward dye degradation for wastewater treatments. ACS Sustain. Chem. Eng. 3, 3130–3139 (2015).
Yan, G. et al. Stability of soluble dialdehyde cellulose and the formation of hollow microspheres: optimization and characterization. ACS Sustain. Chem. Eng. 7, 2151–2159 (2019).
Yan, G. et al. Stable and biocompatible cellulose-based CaCO3 microspheres for tunable pH-responsive drug delivery. ACS Sustain. Chem. Eng. 24, 19824–19831 (2019).
Yan, G. et al. Recent advances on sustainable cellulosic materials for pharmaceutical carrier applications. Carbohyd. Polym. https://doi.org/10.1016/j.carbpol.2020.11649 (2020).
Wu, X. et al. A novel reagentless approach for synthesizing cellulose nanocrystal-supported palladium nanoparticles with enhanced catalytic performance. J. Mater. Chem. A 1, 8645–8652 (2013).
Gu, J., Hu, C., Zhang, W. & Dichiara, A. B. Reagentless preparation of shape memory cellulose nanofibril aerogels decorated with Pd nanoparticles and their application in dye discoloration. Appl. Catal. B 237, 482–490 (2018).
Wei, Z. et al. Novel biocompatible polysaccharide-based self-healing hydrogel. Adv. Funct. Mater. 25, 1352–1359 (2015).
Miao, T., Fenn, S. L., Charron, P. N. & Oldinski, R. A. Self-healing and thermoresponsive dual-cross-linked alginate hydrogels based on supramolecular inclusion complexes. Biomacromolecules. 16, 3740–3750 (2015).
Hong, S. H. et al. Dynamic bonds between boronic acid and alginate: hydrogels with stretchable, self-healing, stimuli-responsive, remoldable, and adhesive properties. Biomacromolecules. 19, 2053–2061 (2018).
Ghawanmeh, A. A., Ali, G. A. M., Algarni, H., Sarkar, S. M. & Chong, K. F. Graphene oxide-based hydrogels as a nanocarrier for anticancer drug delivery. Nano Res. 12, 973–990 (2019).
Fathizadeh, M., Xu, W. L., Zhou, F., Yoon, Y. & Yu, M. Graphene oxide: a novel 2-dimensional material in membrane separation for water purification. Adv. Mater. Interfaces. 4, 1600918 (2017).
Lin, L. & Grossman, J. C. Atomistic understandings of reduced graphene oxide as an ultrathin-film nanoporous membrane for separations. Nat. Commun. 6, 8335 (2015).
Fan, J., Luo, J. & Wan, Y. Aquatic micro-pollutants removal with a biocatalytic membrane prepared by metal chelating affinity membrane chromatography. Chem. Eng. J. 327, 1011–1020 (2017).
Mondal, A., Adhikary, B. & Mukherjee, D. Room-temperature synthesis of air stable cobalt nanoparticles and their use as catalyst for methyl orange dye degradation. Colloid. Surface A 482, 248–257 (2015).
Jiang, Y. et al. Cooking with active oxygen and solid alkali: a promising alternative approach for lignocellulosic biorefineries. ChemSusChem 10, 3982–3993 (2017).
Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (Nos. 21978248 and 21676223), the special fund for Fujian Ocean High-Tech Industry Development (No. FJHJF-L-2018-1), China, and the Natural Science Foundation of Fujian Province of China (No. 2019J06005). The authors also acknowledge the useful discussion with Dr. Youhui Lin in the College of Phyical Science and Technology, Xiamen University.
Author information
Authors and Affiliations
Contributions
The authors acknowledge the support of College of Energy, Xiamen University, China. G.Y., W.H., X.Z., and L.L. designed the experiment. G.Y., Y.F., and H.W. performed the experiment and data analysis. G.Y., Y.F., X.T., and Y.S. contributed to the tests and characterizations. All authors commented on the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yan, G., Feng, Y., Wang, H. et al. Interfacial assembly of self-healing and mechanically stable hydrogels for degradation of organic dyes in water. Commun Mater 1, 41 (2020). https://doi.org/10.1038/s43246-020-0043-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-020-0043-0
This article is cited by
-
Terahertz Detection of Acid Blue 113 Dye Using Hybrid Hydrogels
Journal of Infrared, Millimeter, and Terahertz Waves (2024)
-
Highly Flexible and Broad-Range Mechanically Tunable All-Wood Hydrogels with Nanoscale Channels via the Hofmeister Effect for Human Motion Monitoring
Nano-Micro Letters (2022)
-
Influence of Genipin and Multi-walled Carbon Nanotubes on the Dye Capture Response of CS/PVA Hybrid Hydrogels
Journal of Polymers and the Environment (2022)
