
Abstract
Patients with cancer have higher COVID-19 morbidity and mortality. Here we present the prospective CAPTURE study, integrating longitudinal immune profiling with clinical annotation. Of 357 patients with cancer, 118 were SARS-CoV-2 positive, 94 were symptomatic and 2 died of COVID-19. In this cohort, 83% patients had S1-reactive antibodies and 82% had neutralizing antibodies against wild type SARS-CoV-2, whereas neutralizing antibody titers against the Alpha, Beta and Delta variants were substantially reduced. S1-reactive antibody levels decreased in 13% of patients, whereas neutralizing antibody titers remained stable for up to 329 days. Patients also had detectable SARS-CoV-2-specific T cells and CD4+ responses correlating with S1-reactive antibody levels, although patients with hematological malignancies had impaired immune responses that were disease and treatment specific, but presented compensatory cellular responses, further supported by clinical recovery in all but one patient. Overall, these findings advance the understanding of the nature and duration of the immune response to SARS-CoV-2 in patients with cancer.
Similar content being viewed by others


Main
Patients with cancer are at increased risk of severe outcomes from coronavirus disease 2019 (COVID-19)1,2, with risk factors including general features (such as increased age, male sex, obesity and comorbidities) as well as cancer-specific features (such as hematological and thoracic malignancies, progressive cancer and poor performance status)3,4,5,6,7,8. The precise effects of anticancer treatments on the course and outcome of SARS-CoV-2 infection are yet to be fully understood, with different reports yielding conflicting results5,7,9,10. Understanding the immune response to SARS-CoV-2 in this heterogeneous population, spanning multiple malignancy types and numerous treatment regimens, is crucial for optimal clinical management of those patients during the ongoing pandemic.
Previous studies established the features of the acute immune response to SARS-CoV-2 in patients with cancer: (1) patients with solid tumors show high seroconversion rates; (2) patients with hematological cancer show impaired humoral immunity, especially those on anti-CD20 therapy; and (3) higher CD8+ T cell counts in patients with hematological malignancies are associated with improved survival11,12,13. In contrast to the studies above, our cohort consisted mainly of convalescent patients with a range of COVID-19 presentations, from asymptomatic to severe disease. Furthermore, we present an integrated analysis of functional immune response, including SARS-CoV-2-specific T cells and neutralizing antibodies, and cross-protection against emerging variants of concern (VOCs).
CAPTURE (COVID-19 antiviral response in a pan-tumor immune monitoring study) is a prospective, longitudinal cohort study initiated in response to the global SARS-CoV-2 pandemic and its impact on patients with cancer14. The study aims were to evaluate the impact of cancer and cancer therapies on the immune response to SARS-CoV-2 infection and COVID-19 vaccination. Here, we report findings from the SARS-CoV-2 infection cohort of the CAPTURE study.
Results
Patient demographics and baseline characteristics
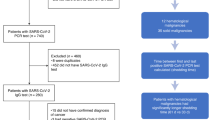
Between 4 May 2020 and 31 March 2021 (database lock), 357 unvaccinated patients with cancer were evaluable with a median followup of 154 d (interquartile range (IQR), 63–273 d). Their median age was 59 years, 54% were male, 89% had a diagnosis of solid malignancy, and the majority (64%) had advanced disease (Table 1). Overall, 118 patients (33%; 97 with solid cancers and 21 with hematological malignancies) were classified as SARS-CoV-2 positive according to our case definition (positive SARS-CoV-2 RT–PCR (PCR with reverse transcription) and/or ELISA (enzyme-linked immunosorbent assay) for S1-reactive antibodies at or before study enrollment) and were included in the analysis (Fig. 1a,b; Methods). Distinct from a population screening program, the intentional recruitment of patients with suspected or confirmed SARS-CoV-2 infection within the study framework (Methods) led to a higher proportion of infected patients than the community prevalence in the United Kingdom within the same time frame. The most common comorbidities were hypertension (27%), obesity (21%) and diabetes mellitus (11%); no significant baseline differences were observed between patients with solid malignancies and those with hematological malignancies (Table 2 and Supplementary Table 1). Overall, 88% of patients received systemic anti-cancer therapy (SACT) in the 12 weeks before infection (51% chemotherapy, 21% targeted therapy, 12% immune-checkpoint inhibitors (CPIs) and 5% anti-CD20), and 10% had radiotherapy and 13% underwent surgery in the 12 weeks before infection. Response to the most recent anti-cancer intervention is shown in Table 2.

a, Patients with cancer irrespective of cancer type, stage or treatment were recruited. Follow-up schedules for patients with cancer were bespoke to their COVID-19 status and account for their clinical schedules (inpatients, every 2–14 d; outpatients, every clinical visit maximum every 3–6 weeks in year one and every 6 months in year two and at the start of every or every second cycle of treatment). Clinical data, ONP swabs and blood were collected at each study visit. Viral antigen testing (RT–PCR on swabs), antibody (ELISA and flow cytometric assay), T cell response and IFN-γ activation assays were performed. b, Distribution of SARS-CoV-2 infection and S1-reactive antibody status and COVID-19 severity in patients with cancer. In total, 357 patients with cancer were recruited between 4 May 2020 and 31 March 2021. SARS-CoV-2 infection status by RT–PCR and S1-reactive antibodies were analyzed at recruitment and in serial samples. RT–PCR results before recruitment were extracted from electronic patient records. The COVID-19 case definition included all patients with either RT–PCR-confirmed SARS-CoV-2 infection or S1-reactive antibodies. c, Viral shedding in 43 patients with serial positive swabs. Solid bars indicate time to the last positive test and dotted lines denote the time from the last positive test to the first negative test. d, Distribution of symptoms in 118 patients with COVID-19. Bar graph denotes the number of patients. Each row in the lower graph denotes one patient. ONP, oronasopharyngeal; WGS, whole-genome sequencing; RTx, radiotherapy; HSCT, human stem cell transplant; GI, gastrointestinal.
Viral shedding and lineage
SARS-CoV-2 infection was confirmed by SARS-CoV-2 RT–PCR in 95 of 118 patients (81%). Repeat testing was not mandated by study protocol, but 40% of the patients (47 of 118) had longitudinal swabs during the course of routine clinical care. Within this group, the estimated median duration of viral shedding (Methods) was 12 d (range, 6–80 d) (Fig. 1c and Table 3), with evidence of prolonged shedding in patients with hematological malignancies (median 21 d, versus 12 d in patients with solid cancers) (Extended Data Fig. 1a). Duration of viral shedding did not correlate with COVID-19 severity (r = 0.04, P = 0.7). We performed viral sequencing in 52 RT–PCR-positive samples with Ct < 32 (Methods), of which 44 of 52 passed sequencing quality control. The Alpha VOC accounted for the majority of infections in our cohort between December 2020 and March 2021, consistent with community prevalence in the United Kingdom at that time (Extended Data Fig. 1b).
Clinical correlates of COVID-19 severity in patients with cancer
Overall, 94 patients (80%) were symptomatic, of whom 52 (44%) had mild illness, 36 (31%) had moderate illness and 6 (5%) had severe illness (as per the World Health Organization (WHO) severity scale15; Table 3); 24 patients (20%) were asymptomatic (WHO score 1). Among all patients (n = 118), fever (47%), cough (42%), dyspnea (31%) and gastrointestinal symptoms (12%) were the most common presenting symptoms (Fig. 1d), with a median of 2 symptoms reported (range, 0–7). In patients with a clear date of symptom resolution (n = 77), median duration of symptoms was 18 d (IQR, 11–30 d). Three patients met the criteria of long COVID (symptomatic >90 d since presentation of disease (POD)), all following severe COVID-19 requiring care in an intensive therapy unit.
Thirty-three patients (28%) were hospitalized due to COVID-19, with a median duration of inpatient stay of 9 d (range, 1–120 d); twenty-seven (23%) required supplemental oxygen, and seven (6%) were admitted to an intensive care unit, with one (1%) requiring mechanical ventilation and inotropic support (Table 3). Thirteen patients (11%) were treated with corticosteroids (>10 mg prednisolone equivalent), and three patients (3%) received treatment with a monoclonal antibody to IL-6. Nine patients (8%) had a thromboembolic complication. At database lock, eleven SARS-CoV-2-positive patients (9%) died of progressive cancer, and two patients (2%) died due to recognized complications of COVID-19 (Table 3).
The risk of moderate and severe COVID-19 was associated with hematological malignancies, whereas the risk of severe COVID-19 in solid malignancies was associated with progressive disease under SACT (Supplementary Table 2), in line with previous reports7,8,12. We found no association between COVID-19 severity, cancer stage, performance status, sex, age, obesity, smoking status or comorbidities across the whole cohort, in contrast to reports from cancer registries, which largely reflected patients who were hospitalized with COVID-19 (refs. 4,6,7,8,16) and the general population17. Furthermore, our relatively small cohort size probably also contributed to the lack of association with these factors.
Cytokine profiles and disease severity during infection
Owing to our study design (Fig. 1a), recruitment was biased toward patients within the convalescent stage of SARS-CoV-2 infection. Only 27 patients (23%) were recruited while still SARS-CoV-2 RT–PCR positive, and 3 (3%) became SARS-CoV-2 RT–PCR positive after recruitment to CAPTURE. Cytokine/chemokine profiling indicated only a non-significant increase in cytokine concentrations in SARS-CoV-2-infected patients (eight with solid tumors and six with hematological malignancies) relative to that in uninfected patients with cancer (n = 5) (Extended Data Figure 1c,d; Methods). Notably, the concentrations of interferon (IFN)-γ, interleukin (IL)-18, IL-6, IL-8, IL-9, IP-10 and macrophage inflammatory protein (MIP)1-β correlated with severe disease (Extended Data Fig. 1e,f). The concentration of IFN-γ and IL-18 in serum was significantly higher in patients with hematological malignancies than in those with solid cancer during acute infection (Extended Data Fig. 1g).
S1-reactive SARS-CoV-2 antibody response in patients with cancer
We evaluated total S1-reactive antibody titers by ELISA at multiple time points during follow-up (with two median samples per patient (range, 1–11)) in 112 patients; 6 patients (5%) were excluded, as blood samples were unavailable or were obtained after COVID-19 vaccination. In total, 93 of 112 patients (83%) had detectable antibodies. S1 antibodies were detectable in 74 of 89 symptomatic patients (83%) and in 19 of 23 asymptomatic patients (83%). S1-reactive antibody titers were associated with COVID-19 severity (P = 0.074) (Fig. 2a).

a, S1-reactive antibody titers (AbT) by COVID-19 severity (n = 112 patients). Significance was tested by Kruskal–Wallis test, P = 0.074. b, S1-reactive antibody titers by cancer type (solid, n = 92; hematological, n = 20). Significance was tested by two-sided Wilcoxon–Mann–Whitney U-test, P = 0.011. c, NAb titers (NAbT) by COVID-19 severity (n = 112 patients). Significance was tested by Kruskal–Wallis test, P = 0.0027. d, NAb titers by cancer type (solid, n = 92; hematological, n = 20). Significance was tested by two-sided Wilcoxon–Mann–Whitney U-test, P = 0.052. Boxes indicate 25th and 75th percentiles, line indicates median and whiskers indicate 1.5 × IQR. Dots represent individual samples. Dotted lines and gray boxes denote the limit of detection. e, Multivariate binary logistic regression evaluating association with lack of NAb in patients with cancer (n = 112). Wald z-statistic was used two calculate two-sided P values. *P = 0.038. f, Multivariate binary logistic regression evaluating the association of lack of NAb in patients with solid cancer (n = 92). g, Multivariate binary logistic regression evaluating the association of lack of NAb in patients with solid cancer (n = 92). Dot denotes odds ratio (blue, positive odds ratio; red, negative odds ratio); whiskers indicate 1.5 × IQR. h, NAb titers against WT, Alpha, Beta and Delta VOCs in patients (n = 112) infected with WT SARS-CoV-2 or Alpha VOC. Violin plots denote density of data points. Point range denotes median and 25th and 75th percentiles. Dots represent individual samples. Significance was tested by Kruskal–Wallis test, P = 3.5 × 10−7, two-sided Wilcoxon–Mann–Whitney U-test with Bonferroni correction (post hoc test) was used for pairwise comparisons. P values are denoted in the graph. i,j, S1-reactive antibody titers (i) and NAb titers (j) after onset of disease (n = 97 patients). Blue line denotes loess regression line with 95% confidence bands in gray. Black dots denote patients with one sample and colored dots denote patients with serial samples (n = 51 patients). Samples from individual patients are connected. Dotted lines and gray areas at the bottom indicate limit of detection.
Nineteen patients (17%), with median follow up of 22 d (range, 0–301 d), had no evidence of S1-reactive antibodies following a positive SARS-CoV-2 RT–PCR. Lack of seroconversion was significantly associated with hematological malignancies: 9 of 20 patients (45%) with hematological malignancies versus 10 of 92 patients (11%) with solid malignancies did not seroconvert (chi-squared test, P = 0.0002). In addition, S1-reactive antibody titers were significantly lower in patients with hematological malignancies than in those with solid malignancies (Fig. 2b). Two patients with long COVID had no evidence of seroconversion at any point during follow-up (followed for 222 d and 235 d after disease onset, respectively).
We conducted a sensitive flow cytometric assay on serum from a subset of patients with S1-reactive antibodies(n = 40; Extended Data Figs. 2a and 3) and detected S-specific IgG in 38 of 40 patients (95%) (Extended Data Fig. 2b) and IgM in 23 of 40 patients (58%) (Extended Data Fig. 2c). IgG and IgM levels significantly correlated with S1-reactive antibody titers (P < 0.0001) (Extended Data Fig. 2e,f). S-reactive IgA was detected in only four convalescent patients (10%) (Extended Data Fig. 2d), consistent with its role in the early response to SARS-CoV-2 infection17.
Finally, we evaluated matched pre-pandemic serum samples from 47 patients: 10 with and 37 without evidence of S1-reactive antibodies in post-pandemic serum. We found no evidence of S1-reactive antibodies in the pre-pandemic serum of any patient (Extended Data Fig. 2g). However, S-reactive IgG or IgM was detected in serum in 18 patients without confirmed SARS-CoV-2 infection, indicating cross-reactivity to seasonal human coronaviruses, with more frequent cross-recognition of the S domain than of the more conserved S1 domain, as reported in individuals without cancer18.
NAbs against SARS-CoV-2 VOCs in patients with cancer
We assessed neutralizing antibodies (NAbs) in all patients using a high-throughput a live-virus neutralization assay (Methods), against wild-type (WT) SARS-CoV-2 and the Alpha (B.1.1.7), Beta (B.1.351) and Delta (B.1.617.2) VOCs, and results are presented as titers (the reciprocal of serum required to inhibit 50% of viral replication (IC50)). NAb titers below 40 were considered undetectable (Methods).
We detected NAbs against WT SARS-CoV-2 in 88 of 93 patients (95%) with S1-reactive antibodies (in 77 of 82 (94%) with solid tumors and in 11 of 11 (100%) with hematological malignancy). NAbs were detected in 4 of 19 RT-PCR-positive patients without S1-reactive antibodies (21%) (in 2 of 10 (20%) with solid cancer and in 2 of 9 (22%) with hematological malignancy). NAb titers were significantly associated with COVID-19 severity (Fig. 2c).
In a binary logistic regression model including all patients with cancer (n = 112), presence of hematological malignancy, but not comorbidities, age, sex or COVID-19 severity, was associated with undetectable NAbs (Fig. 2e). Accordingly, median NAb titers against WT SARS-CoV-2 were lower in patients with hematological malignancies than in those with solid cancer (Fig. 2d).
In patients with solid cancer (n = 92), cancer type, stage, progressive disease and cancer therapy (Fig. 2f,g) were not associated with undetectable NAbs. Due to limited sample size, patients with hematological malignancies (n = 20) could not be evaluated by a multivariate model.
In patients infected with WT SARS-CoV-2 (n = 85) or the Alpha VOC (n = 27), the proportion of patients with detectable NAbs against VOCs was significantly lower than those with detectable NAbs against WT SARS-CoV-2 (WT, 92 of 112 (82%); Alpha, 89 of 112 (79%); Beta, 77 of 112 (69%); Delta, 73 of 112 (65%); chi-squared test, P = 0.009), and the median NAb titers against Beta and Delta were significantly lower than those against WT and Alpha (Fig. 2h). The proportion of patients with detectable NAbs against all variants was significantly lower in patients with hematological malignancies than in those with solid cancer (WT, 86% versus 65%, chi-squared test, P = 0.03; Alpha, 84% versus 60%, chi-squared test, P = 0.02; Beta, 74% versus 45%, chi-squared test, P = 0.01; Delta, 71% versus 40%, chi-squared test, P = 0.009).
There was a significant correlation between the levels of S1-reactive antibody and NAb titers against WT SARS-CoV-2 and all VOCs (P < 0.0001) (Extended Data Fig. 2h). However, the proportion of patients with detectable S1-reactive antibodies without detectable NAbs was greater for the VOCs than for WT SARS-CoV-2 (WT, 5 of 93 (5%); Alpha, 7 of 93 (8%); Beta, 17 of 93 (18%); Delta, 20 of 93 (22%); chi-squared test, P = 0.002).
SARS-CoV-2 antibody response lasts up to 11 months
Next, we assessed antibody kinetics in 59 of 97 patients (61%) (n = 45 with solid cancer and n = 14 with hematological malignancy) with detectable S1-reactive antibodies in whom the time of disease onset was known (median of two time points per patient (range, 2–10); median length of follow-up, 181 d (range, 8–336 d)). Five patients were followed for more than 300 d. Follow-up samples collected after COVID-19 vaccination were excluded.
Thirty-three (56%) had S1-reactive antibodies at the time of enrollment (median of 69 d post onset of disease (POD) (range, 3–217 d); Fig. 2i), and a further five (8%) seroconverted within 13–117 d POD. S1-reactive antibody titers showed a weak declining trend, and 14 of 59 patients (24%) became seronegative 24–313 d POD (including 2 of 5 patients with delayed seroconversion). Most of those (12 of 14) had solid cancer and no clinical features that could conceivably account for the short-lived antibody response. Two patients with hematological malignancy who did not seroconvert included one with a diagnosis of T cell acute lymphoblastic leukemia who had a stem cell transplant complicated by chronic graft-versus-host disease after recovering from COVID-19, and and one patient with plasmablastic lymphoma treated with anti-CD20 prior to SARS-CoV-2 infection.
NAbs against all variants were detected as early as day 1 in the 59 patients (Fig. 2j and Extended Data Fig. 4a–c) and as late as day 217 POD, and NAb titers remained stable overall up to 336 d. In the group of patients who sero-reverted (S1 antibodies became undetectable during follow-up), NAbs against WT SARS-CoV-2 remained detectable in 10 of 14 (71%) (Alpha, 9 of 14 (64%); Beta, 2 of 14 (14%); Delta, 2 of 14 (14%)).
SARS-CoV-2-specific T cells are detected in patients with cancer
Peripheral blood mononuclear cell (PBMC)-stimulation assays (Methods) were performed in 110 of 112 patients who were SARS-CoV positive (81 with solid cancer and 19 with hematological malignancy; Extended Data Fig. 3b); 12 of 112 samples were excluded (for lack of PBMC collection, low viability, or no detection of CD3+ cells in activation-induced marker). SARS-CoV-2-specific CD4+ and CD8+ T cells (SsT cells; identified by activation-induced markers OX40, CD137 and CD69)19 were quantified at the first time point after seroconversion, at a median of 59 d POD (range, 1–292 d) (Fig. 3a,b). We detected SARS-CoV-2-specific CD4+ T cells in 77 of 100 patients (77%) and CD8+ T cells in 49 of 100 patients (49%) (Fig. 3c–f). CD8+ T cells levels were consistently lower than CD4+ T cell levels (Extended Data Fig. 5a), a result also noted in participants without cancer20,21,22, possibly reflecting our use of 15-mer peptide pools for stimulation, which could favor detection of CD4 responses over that of CD8 responses.

a,b, Representative plots of CD4+CD137+OX40+ (CD4+) and CD8+CD137+CD69+ (CD8+) T cells in a patient with confirmed COVID-19 and a cancer patient without COVID-19 after in vitro stimulation with S, M and N peptide pools, positive control Staphylococcal enterotoxin B (SEB) or negative control (NC). c,d, Frequency of SARS-CoV-2-specific CD4+ (c) and CD8+ (d) T cells in patients with solid malignancies (n = 83). e,f, Frequency of SARS-CoV-2-specific CD4+ (e) and CD8+ (f) T cells in patients with hematological malignancies (n = 21). The stimulation index was calculated by dividing the percentage of positive cells in the stimulated sample by the percentage of positive cells in NC. To obtain the total number of SsT cells, the sum of cells activated by S, M and N was calculated (SMN). Boxes indicate the 25th and 75th percentiles, the line indicates the median and whiskers indicate 1.5 × IQR. Dots represent individual samples. Dotted lines and gray boxes denote the limit of detection.
CD4+ T cells were detected in 81% of patients with solid malignancies and in 58% of patients with hematological malignancies (Fig. 3c,e). CD8+ T cells were detected in similar proportions of patients with solid malignancies and those with hematological malignancies (51% and 42%, respectively) (Fig. 3d,f).
Consistent with functional activation of SsT cells, after in vitro stimulation of PBMCs, we detected increased levels of IFN-γ in culture supernatants, which correlated with the number of SsT cells (Extended Data Fig. 5b). IFN-γ levels did not differ between patients with solid cancers versus those with hematological malignancies (Extended Data Fig. 5c).
Finally, to account for the lack of matched pre-infection samples in our cohort, and given reports of cross-reactive T cell responses to other human coronaviruses in healthy individuals19,23, we extended the T cell assay to 12 patients with cancer without confirmed SARS-CoV-2 infection. Cross-reactive CD4+ T cells were detected in 7 of 12 participants and CD8+ T cells were detected in 3 of 12 participants, but the overall level SsT cells was significantly lower in uninfected patients than in patients with confirmed SARS-CoV-2 infection (P < 0.05) (Extended Data Fig. 5d,e).
SsT cell compensation in patients without humoral response
Patients with hematological malignancies had a wide range of S1, NAb and SsT cell responses (Fig. 4a,b,c). Among patients with leukemia, NAbs were detected in 9 of 11 and SsT cells were detected in 5 of 10 evaluable patients (2 had both CD4+ and CD8+ responses; 2 had CD4+ only responses; and 1 had a CD8+ only response). Among patients with myeloma, two of three had NAbs, and two of three had detectable SsT cells (CD4+ and CD8+). Finally, two of six patients with lymphoma had detectable NAbs, whereas SsT cells were detected in five of six (three had both CD4+ and CD8+ responses; one had a CD4+ only response; and one had a CD8+ only response). Overall, SsT cell levels were higher in patients with lymphoma than in those with leukemia (Fig. 4c). Four of five patients with lymphoma given anti-CD20 treatment had no NAb response (Fig. 4d,e). The fifth patient with plasmablastic lymphoma had detectable NAb titers against WT SARS-CoV-2 and the Alpha VOC. In contrast, NAb titers against Beta and Delta and S1-reactive antibody titers were detected at only one time point before the patient sero-reverted at 37 d POD. One additional patient with a diagnosis of acute myeloid leukemia, with a history of allogeneic stem-cell transplant and treatment with anti-CD20, had no NAbs, and SsT cells could not be evaluated. Four of five patients with lymphoma given anti-CD20 treatment had detectable SsT cells, and their levels of SsT cells were not lower than those in patients not treated with anti-CD20 (Fig. 4f). In patients with solid malignancies, the levels of NAbs and SsT cells did not differ significantly by tumor type (Figs. 2f and 4g, respectively).

a, S1-reactive antibody titers in patients with leukemia (n = 11), myeloma (n = 4) and lymphoma (n = 6). b, NAb titers in patients with leukemia (n = 10), myeloma (n = 4) and lymphoma (n = 6). c, CD4+ and CD8+ cells T cells in patients with leukemia (n = 10), myeloma (n = 4) and lymphoma (n = 6). The stimulation index was calculated by dividing the percentage of CD4+CD137+OX40+ (CD4+) and CD8+CD137+CD69+ (CD8+) T cells in the stimulated sample by the percentage of positive cells in the NC. Significance was tested by Kruskal–Wallis test, where P < 0.05 was considered significant. d, S1-reactive antibody titers in patients with hematological malignancy receiving anti-CD20 treatment (n = 6) versus other SACT (n = 15). e, NAb titers in patients with hematological malignancy receiving anti-CD20 treatment (n = 6) versus other SACT (n = 15). Significance was tested by two-sided Wilcoxon–Mann–Whitney U-test, where P < 0.05 was considered significant. f, Comparison of CD4+/CD8+ T cells between patients with hematological malignancies on anti-CD20 therapy (n = 5, administered within 6 months) and not on anti-CD20 therapy (n = 15). Significance was tested by two-sided Wilcoxon–Mann–Whitney U-test, where P < 0.05 was considered significant. g, CD4+ and CD8+ cells T cells in patients with solid malignancies (n = 81) by cancer subtype. Boxes indicate 25th and 75th percentiles, the line indicates the median and whiskers indicate 1.5 × IQR. Dots represent individual patient samples. Dotted lines and gray boxes denote the limit of detection. Significance was tested by Kruskal–Wallis test, where P < 0.05 was considered significant.
Overall, we observed a discordance between antibody responses and T cell responses among patients with hematological malignancy. First, a greater proportion of patients with detectable NAb titers had detectable CD4+ T cells than that of patients without detectable NAb titers (among NAb-positive patients, 9 of 13 (69%); among NAb-negative patients, 2 of 6 (33%)), and we observed no correlation of S-reactive CD4+ T cell levels and NAb titers (Extended Data Fig. 5g). Second, 2 of 6 patients with undetectable NAb titers (33%) still had detectable CD8+ T cells, compared with 7 of 13 patients with detectable NAb titers (54%) (Supplementary Table 3).
Among patients with solid cancer, the proportion of patients with detectable CD4+ T cells or detectable CD8+ T cells was lower for those with undetectable NAb titers (CD4+, 8 of 12 (67%); CD8+, 2 of 12 (17%)) than for those with detectable NAb titers (CD4+, 58 of 69 (84%); CD8+, 39 of 69 (57%)) (Supplementary Table 3). We further observed a significant correlation of S-reactive CD4+ T cell levels and NAb titers against WT SARS-CoV-2 (Extended Data Fig. 5f).
Finally, following stimulation with S and N pools, we observed that patients with hematological malignancy exhibited higher levels of N-reactive CD8+ T cells than S-reactive CD8+ T cells (Fig. 3e,f), whereas similar levels were observed in patients with solid cancer (Fig. 3c,d), indicating that antigens other than spike (S) may contribute to T cell responses in patients with hematological malignancies.
T cell responses are impacted in CPI-treated patients
Next, we evaluated features associated with impaired T cell responses to SARS-CoV-2 in patients with cancer. We found no association between lack of SsT cells and the presence of solid or hematological malignancies, or the number of comorbidities, age, sex or COVID-19 severity (Fig. 5a,b). In patients with solid malignancies, those on CPIs (n = 13) had significantly reduced levels of SARS-CoV-2-reactive CD4+ T cells (Fig. 5c), and in a binary logistic regression model, lack of SARS-CoV-2-reactive CD4+ (but not CD8+) T cells was associated with CPI therapy within 3 months of SARS-CoV-2 infection (Fig. 5d,e).

a,b, Multivariate binary logistic regression analysis evaluating associations between SARS-CoV-2-specific CD4+ (a) and CD8+ (b) T cells with cancer diagnosis (solid versus hematological malignancies), comorbidities, age, sex and COVID-19 disease severity in 100 patients. Wald z-statistic was used to calculate two-sided P values. *P = 0.038. c,d, Multivariate binary logistic regression analysis evaluating associations between SARS-CoV-2-specific CD4+ (c) and CD8+ (d) T cells with anticancer intervention, age, sex and COVID-19 disease severity in patients with solid cancer (n = 81). Wald z-statistic was used to calculate two-sided P values. *P = 0.045. Dot denotes odds ratio (blue and red dots indicate positive or negative odds ratios, respectively); whiskers indicate 1.5 × IQR. e, Comparison of SARS-CoV-2-specific CD4+/CD8+ T cells between patients with solid malignancies taking CPIs (n = 13, administered within 3 months) and not on CPIs (n = 68). Boxes indicate the 25th and 75th percentiles, line indicates the median and whiskers indicate 1.5 × IQR. Dots represent individual samples. Significance was tested by two-sided Wilcoxon–Mann–Whitney U-test (P = 0.038 and P = 0.53).
Immune responses are lower than in those without cancer
We compared S1-reactive and NAb responses in patients with cancer with those of a control cohort of 21 healthcare workers (HCWs) with SARS-CoV-2 infection who were recruited to CAPTURE. We applied the same case definition as that for patients with cancer (positive SARS-CoV-2 RT–PCR and/or ELISA for S1-reactive antibodies at or prior to study enrolment). Of note, HCWs were not matched to patients with cancer by age, and they represented an overall younger cohort, with a median age of 43 years (IQR, 40–52 years). Seven HCWs (33%) were asymptomatic, and fourteen HCWs (67%) reported mild symptoms, among whom eight (38%) had SARS-CoV-2 infection confirmed by RT-PCR; none were hospitalized. Twenty-one HCWs (100%) had detectable S1-reactive antibodies, and twelve (57%) had detectable NAbs against WT SARS-CoV-2, a lower proportion than that of patients with cancer. S1-reactive antibody titers and NAb titers against WT SARS-CoV-2 were numerically lower in HCWs than in patients with solid malignancies but higher than in patients with hematological malignancies (Extended Data Fig. 6a,b).
We also evaluated SARS-CoV-2-reactive CD4+ and CD8+ T cell levels in the same cohort of HCWs (CD4+ T cells, 16 of 19 (84%); CD8+ T cells, 10 of 19 (53%)). We observed a similar proportion of HCWs with detectable SsT cells as noted for patients with solid cancer. The median SsT cell levels were numerically higher in HCWs than in all patients with cancer (Extended Data Fig. 6c,d).
Discussion
Results from this prospective, longitudinal study of 118 patients with cancer and SARS-CoV-2 infection indicated that most patients with solid tumors developed a functional and probably durable (up to 11 months) humoral immune response to SARS-CoV-2 infection, as well as an anti-SARS-CoV-2-specific T cell response. Patients with hematological malignancies had significantly lower seroconversion rates, and impaired immune responses that were related to both disease and treatment (anti-CD20), although with evidence of compensation, consistent with prior reports11.
Our findings largely relate to patients with a diagnosis of solid cancer (82% of the cohort), the majority of whom had evidence of seroconversion (89%). Absence of or a delay in seroconversion was observed in 10% of patients with solid tumors, in line with data reported from smaller prospective studies from the United Kingdom (95%, n = 22)12 and Italy (88%, n = 28)24 and comparable to results in individuals without cancer. We did not observe an obvious impact of solid cancer characteristics on the likelihood of seroconversion. Recent studies of people without cancer demonstrated a clear relationship between neutralizing responses and recovery from infection25, as well as vaccine efficacy26,27. In our cohort, 94% of seroconverted patients with solid tumors also had detectable NAbs (to WT SARS-CoV-2 or the Alpha VOC, consistent with the causative variant). Notably, although we observed a weak decline in S1-reactive antibody titers, NAb titers were stable for at least 7 months and in some cases up to 11 months of follow-up. Discordance was specifically observed in 14 patients with declining S1-reactive antibodies, indicating that these patients had persistent NAbs that were not detected by the S1 ELISA28.
Longer follow-up was limited by COVID-19 vaccination, which commenced in the United Kingdom in December 2020 (ref. 29). In individuals without cancer, the reported durability of both SARS-CoV-2-specific IgG and NAbs varies substantially20,30,31,32,33, and direct comparison of our data to those reports is challenging.
We compared antibody and cellular responses in patients with cancer with those of 21 HCWs with SARS-CoV-2 infection. Antibody and cellular responses in patients with hematological malignancies were all lower than those of HCWs. We note that the HCWs all had asymptomatic or mild COVID-19. The association of antibody responses with COVID-19 severity in this cohort and in healthy individuals34 may explain why we observed numerically higher S1-reactive antibody and NAb titers in patients with solid cancer than in HCWs. We found that SsT cell levels were numerically lower in patients with cancer; cellular responses in healthy individuals show an inverse association with age and disease severity21. In summary, the comparison with HCWs confirmed that patients with hematological malignancies had impaired immune responses, while the majority of patients with solid cancer had responses comparable to those of healthy individuals.
In line with data for SARS-CoV-2 convalescent patients without cancer35, we found that neutralizing activity against the Alpha, Beta, and Delta VOCs was decreased. Of note, we showed that 35% of patients with cancer previously infected with WT or Alpha SARS-CoV-2 lacked NAbs against Delta. In patients who sero-reverted, NAbs against WT SARS-CoV-2 were still detected, while NAb titers against Delta, if present, declined over time. This raises concerns about considering a history of prior infection with one variant as evidence of functional immunity against VOCs. Finally, given that the majority of patients with cancer have generally been prioritized for COVID-19 vaccines, protection against evolving variants is critically relevant in the context of COVID-19-vaccine-induced immunity29.
During acute SARS-CoV-2 infection, patients with cancer were previously shown to have depleted T cells that showed markers of activation and exhaustion and correlated with COVID-19 severity, but SsT cells were not evaluated12. In our cohort, at a median of 54 d POD, SsT cells (including functional IFN-γ-secreting SsT cells) were present in the majority of evaluated patients with solid malignancies (76%) or hematological malignancies (52%). In both the acute phase and the convalescent phase of SARS-CoV-2 infection, a substantial proportion of SARS-CoV-2-specific CD4+ T cells are follicular helper T cells21,36, which are required for IgG and neutralizing responses by B cells37. Accordingly, in our study, the level of CD4+ T cells was significantly correlated with S1-reactive antibody titers in patients with solid tumors, which probably reflected activation of B cells following a follicular helper T cell response. Lack of detectable T cell responses to SARS-CoV-2 was not associated with any cancer-specific factors other than treatment withCPI within 3 months of SARS-CoV-2 infection (in solid tumors). We found that CPI treatment was associated with a lower level of SARS-CoV-2-specific CD4+ T cells but not that of CD8+ T cells. It was previously shown that PD-1 blockade during acute viral infection can increase viral clearance by promoting CD8+ T cell proliferation but can also impair CD8+ T cell memory differentiation, thereby impairing long-term immunity38. Although the impact of PD-1 blockade on the CD4+ T cell response to acute infection is less well understood, PD-1 signaling regulates the expansion of CD4+ T cells after an immunogenic stimulus39, which offers a potential explanation for our finding of lower CD4+ T cell levels. Overall, this finding warrants validation in larger datasets, but this is unlikely to be clinically important, given the lack of consistent association between CPIs and COVID-19 outcomes5,6,7,8.
We found an inverse relationship between antibody responses and SsT cell responses in patients with hematological malignancies, whereby patients with leukemia had more pronounced antibody responses but impaired SsT cell responses, and the opposite was true for patients with lymphoma. Furthermore, we found SsT cells in four of five evaluable patients on anti-CD20 treatment, none of whom had detectable humoral responses. In total, all but one patient with hematological malignancy had mild or moderate COVID-19 disease from which they recovered. This suggests that SsT cell responses, specifically CD8+ T cells and non–spike-specific SsT cells, can at least partially compensate for defective humoral responses. In a recent study, 10 of 13 patients with hematological malignancy and COVID-19 had detectable SsT cells, the levels of which were associated with improved survival (including in those on anti-CD20 therapy)11. T cells have been shown to play a crucial role in the clearance of acute SARS-CoV infection in preclinical animal models40,41, and early induction of functional SsT cells are associated with rapid viral clearance and mild COVID-19 disease in patients without cancer42. VOCs are not suspected to escape SsT cell responses, due to their highly multi-antigenic properties43. The emerging data from our study and that of others44 suggest that T cell responses are probably important in those with hematological malignancies and may offer protection from severe COVID-19 in the absence of humoral responses. Notably, SsT cells were shown to be induced by COVID-19 vaccines in both populations without cancer45,46 and those with cancer29 and to have activity against VOCs43. Overall, our data on natural immunity also bear relevance to vaccination approaches in this population, especially the context of heterologous vaccination that aims to optimally boost humoral and cellular responses47,48,49.
This report has several limitations. First, the lack of a broader matched control group without cancer prevents direct comparisons between populations with cancer and those without cancer. Second, as recruitment to CAPTURE commenced in May 2020, which marked the end of the first wave of SARS-CoV-2 infections in the United Kingdom, most of the participants initially recruited were infected prior to study enrollment and were evaluated in the convalescent phase. The fact that we recorded only two COVID-19-related deaths may reflect this, as well as the relatively low proportion of lung and hematological malignancies, which are the two cancer groups with increased COVID-19-related mortality)3,4,5,6. Furthermore, all but 1 patient with hematological malignancy in our cohort recovered, whereas 11 of 18 patients with blood cancer died due to COVID-19 at our institution50 before enrollment into CAPTURE commenced. Thus, it is possible that the patients with hematological malignancy in our analysis are not entirely representative of this population. Nevertheless, the cohort as a whole provides a broader representation of all patients with cancer than do those of other studies. Another limitation pertains to our SsT cell evaluation, which was performed at a single time point, and therefore the proportion of patients with a T cell response may be underestimated. Also, although we did not assay against viral peptide pools for other human viruses (such as EBV or CMV), reports suggest that specific patterns of activation markers can be detected on SARS-CoV-2-specific T cells51. Finally, our analyses are probably underpowered for robust detection of differences in immune responses between cancer subtypes.
In summary, our data suggest that patients with solid malignancies develop humoral and cellular immunity to SARS-CoV-2, with NAbs detectable for up to 11 months. In line with other reports11,12, we found that patients with hematological malignancies had impaired humoral responses, and this was associated with disease subtype and anti-CD20 treatment. However, such patients frequently had detectable SsT cell responses. Finally, we found that neutralizing activity against VOCs was reduced in patients infected with WT SARS-CoV-2, which raises concerns about the protection afforded by natural immunity to new SARS-CoV-2 VOCs. Whether such responses can be boosted by COVID-19 vaccines remains under investigation in the vaccine cohort of our study, which includes the currently predominant Delta VOC29.
Methods
Study design
CAPTURE (NCT03226886) is a prospective, longitudinal cohort study that commenced recruitment in May 2020 at the Royal Marsden NHS Foundation Trust. The study design has been previously published14. In brief, adult patients with current or history of invasive cancer are eligible for enrollment (Fig. 1a). Inclusion criteria are intentionally broad and patients are approached irrespective of cancer type, stage or treatment. Patients with confirmed or suspected SARS-CoV-2 infection are targeted with broader recruitment in the course of routine clinical care (asymptomatic cases). Patients are screened at each study visit and classified as SARS-CoV-2-negative or SARS-CoV-2-positive on the basis of a laboratory case definition of RT–PCR-positive result and/or S1-reactive antibodies (details below). The primary end point is to describe the population characteristics of patients with cancer who are positive and negative for SARS-CoV-2. The secondary end points include the impact of COVID-19 on long-term survival and intensive care unit admission rates. Exploratory end points pertain to characterizing clinical and immunological determinants of COVID-19 in patients with cancer.
CAPTURE was approved as a sub-study of TRACERx Renal (NCT03226886). TRACERx Renal was initially approved by the NRES Committee London on 17 January 2012. The TRACERx Renal sub-study CAPTURE was submitted as part of Substantial Amendment 9 and approved by the Health Research Authority on 30 April 2020 and the NRES Committee London on 1 May 2020. CAPTURE is being conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice and applicable regulatory requirements. All patients provided written informed consent to participate.
Study schedule and follow-up
Clinical data and sample collection for participating patients with cancer is performed at baseline and at clinical visits per standard-of-care management during the first year of follow-up; frequency varies depending on in- or outpatient status and systemic anticancer treatment regimens. For inpatients, study assessments are repeated every 2–14 d. For outpatients, the follow-up study assessments are aligned with clinically indicated hospital attendances. The frequency of study assessments in the first year for patients on anticancer therapies are as follows: every cycle for CPIs or targeted therapies; every second cycle for chemotherapy; every outpatient appointment (maximum 6 weekly) for patients on endocrine therapy or in surveillance or routine cancer care follow-up. Patient-reported data are collected 3-monthly via an online questionnaire. In year two to five of follow-up, the frequency of study assessments is reduced (Supplementary Information).
Data and sample sources
Patient-reported outcome data are collected using PROFILES (patient-reported outcomes following initial treatment and long-term evaluation of survivorship; https://profiles-study.rmh.nhs.uk/). PROFILES is a web-based questionnaire administration and management system designed for the study of physical and psychosocial impacts of cancer and its treatment. Online questionnaires for baseline and follow-up assessments were designed to record data for patients with cancer participating in CAPTURE. Collected self-reported data include ethnicity, smoking status, alcohol consumption, recent travel history, occupation, exercise habits, dietary habits, previous medical history, autoimmune disease (self or next of kin), vaccination history, concomitant medication, self-shielding status, previous SARS-CoV-2 tests, SARS-CoV-2 tests in household members and current and recent symptoms. Further demographic, epidemiological and clinical data (such as cancer type, cancer stage and treatment history) are collected from the internal electronic patient record system and entered into detailed case report forms in a secure electronic database. For information on anticancer intervention and response to most recent anticancer intervention, data are collected reflective of the time of SARS-CoV-2 infection as per the definition above, where available or at the time of enrollment if data of disease onset is unknown (for example asymptomatic infections defined by positive serological positivity but negative/no RT–PCR results).
The study samples collected consisted of blood samples, oropharyngeal swabs and archival and excess material from routine clinical investigations. A detailed sampling schedule and methodology has been previously described14. Surplus serum from patient biochemistry samples taken as part of routine care were also retrieved and linked to the study IDs before being anonymization and study analysis. Collected data and study samples were de-identified and stored with only the study-specific study identification number. For self-reported data, a PROFILES member number was used, which is generated automatically.
WHO classification of severity of COVID-19
We classified severity of COVID-19 according to the WHO clinical progression scale (0–10)15: uninfected, uninfected with no viral RNA detected (0); asymptomatic, viral RNA and/or S1-reactive IgG detected (1); mild (ambulatory), symptomatic and independent (2); symptomatic and assistance needed (3); moderate (hospitalized), no oxygen therapy (if hospitalized for isolation only, record status as for ambulatory patient) (4); oxygen by mask or nasal prongs (5); severe (hospitalized), oxygen by noninvasive ventilation or high flow (6); intubation and mechanical ventilation, pO2/FIO2 ≥ 150 or SpO2/FIO2 ≥ 200 (7); mechanical ventilation, pO2/FIO2 < 150 (SpO2/FIO2 < 200) or vasopressors (8); mechanical ventilation, pO2/FIO2 < 150 and vasopressors, dialysis or extracorporeal membrane oxygenation (9); and dead (10).
Cell lines and viruses
SUP-T1 cells stably transfected with spike or control vectors were obtained from M.P. and L.M. Vero E6 cells were obtained from the National Institute for Biological Standards and Control, United Kingdom. The SARS-CoV-2 isolate hCoV-19/England/02/2020 was obtained from the Respiratory Virus Unit, Public Health England, United Kingdom and propagated in Vero E6 cells.
Handling of oronasopharyngeal swabs, RNA isolation and RT–PCR
SARS-CoV-2 RT–PCR was performed from ONP swabs using a diagnostics assay established at the Francis Crick Institute52. The complete standard operating procedure is available at https://www.crick.ac.uk/research/covid-19/covid19-consortium. ONP swabs were collected in VTM medium, frozen within 24 h after collection and stored at −80 °C until processing. ONP swabs were handled in a CL3 laboratory inside a biosafety cabinet using appropriate personal protective equipment and safety measures, which were in accordance with a risk assessment and standard operating procedure approved by the safety, health and sustainability committee at the Francis Crick Institute. In brief, 100 µl of swab vial content was inactivated in 5 M guanidinium thiocyanate and RNA isolated using a completely automated kit-free, silica bead-based method.
PCR detection of SARS-CoV-2 was performed from 10 µl extracted RNA using two kits depending on the date of test. Up to 6 December 2020, samples were tested in duplicate using a real-time fluorescent RT–PCR kit for detecting 2019-nCoV (BGI). Positive, negative and extraction controls were included on each plate. Runs were regarded as valid when negative control Ct values were >37 and positive control Ct values were <37. Samples were only considered positive if Ct values in both replicates were <37. From 7 December 2020, tests were performed using a TaqPath COVID-19 CE-IVD RT–PCR kit (Thermo Fisher), this time without replication. Positive and negative controls were included on each plate and samples were reported as positive if 2 or 3 SARS-CoV-2 targets had a Ct value <37 and the internal control Ct < 32. With both kits, samples with non-exponential amplification were excluded from analysis.
Viral sequencing
All PCR-positive samples with ORF1ab Ct value <32 were selected for viral sequencing, representing 52 samples from 32 patients. Sequencing was performed either on Illumina or on Oxford Nanopore Technologies instruments. Oxford Nanopore libraries were prepared following the ARTIC nCoV-2019 sequencing protocol v3 (LoCost) (https://protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye) and then sequenced for 20 h on a MinION flowcell on a GridION instrument. The ncov2019-artic-nf pipeline (v.1.1.1; https://github.com/connor-lab/ncov2019-artic-nf) written in the Nextflow domain specific language (v.20.10.0)53 was used to perform quality control, variant calling and consensus sequence generation for the samples. The full command used was ‘nextflow run ncov2019-artic-nf–nanopolish–prefix $PREFIX–basecalled_fastq fastq_pass/–fast5_pass fast5_pass/–sequencing_summary sequencing_summary.txt–schemeVersion V3–minReadsPerBarcode 1–minReadsArticGuppyPlex 1 -with-singularity artic-ncov2019-nanopore.img -profile singularity,slurm -r v1.1.1’. Illumina libraries were prepared following the CoronaHiT protocol with minor modifications54, were pooled, and then were sequenced at 100-bp paired ends on HiSeq 4000. The nf-core/viralrecon pipeline (v.1.1.0)55 was used to perform quality control, variant calling and consensus sequence generation for the samples. The full command used was ‘nextflow run nf-core/viralrecon–input samplesheet.csv–genome ‘MN908947.3’–amplicon_bed nCoV-2019.artic.V3.bed–protocol ‘amplicon’–callers ivar–skip_assembly–skip_markduplicates–skip_fastqc–skip_picard_metrics–save_align_intermeds -profile crick -r 1.1.0’. Overall, 44 of 52 passed quality control (>50% consensus sequence) and the lineage was obtained using PANGOLIN (https://github.com/cov-lineages/pangolin). In the absence of sequencing data to confirm the causative SARS-CoV-2 variant, all patients tested with Thermo Fisher TaqPath RT–PCR kit that reported S-dropout were considered to be infected with the Alpha VOC.
Viral shedding
Duration of viral shedding was estimated from research and opportunistic swabs and was defined as the time from first positive swab to the last positive swab (preceded by at least one negative swab).
Handling of whole blood samples
All blood samples and isolated products were handled in a CL2 laboratory inside a biosafety cabinet using appropriate personal protective equipment and safety measures in accordance with a risk assessment and standard operating procedure approved by the safety, health and sustainability committee of the Francis Crick Institute. For indicated experiments, serum or plasma samples were heat-inactivated at 56 °C for 30 min before use, after which they were used in a CL1 laboratory.
Plasma and PBMC isolation
Whole blood was collected in EDTA tubes (VWR) and stored at 4 °C until processing. All samples were processed within 24 h. Time of blood draw, processing and freezing was recorded for each sample. Before processing, tubes were brought to room temperature. PBMCs and plasma were isolated by density-gradient centrifugation using pre-filled centrifugation tubes (pluriSelect). Up to 30 ml of undiluted blood was added on top of the sponge and centrifuged for 30 min at 1,000g at room temperature. Plasma was carefully removed then centrifuged for 10 min at 4,000g to remove debris, then aliquoted and stored at −80 °C. The cell layer was then collected and washed twice in PBS by centrifugation for 10 min at 300g at room temperature. PBMCs were resuspended in Recovery cell culture freezing medium (Thermo Fisher Scientific) containing 10% dimethylsulfoxide, placed overnight in CoolCell freezing containers (Corning) at −80 °C and then stored at −80 °C.
Serum isolation
Whole blood was collected in serum coagulation tubes (Vacuette CAT tubes, Greiner) for serum isolation and stored at 4 °C until processing. All samples were processed within 24 h. Time of blood draw, processing and freezing was recorded for each sample. Tubes were centrifuged for 10 min at 2,000g at 4 °C. Serum was separated from the clotted portion, aliquoted and stored at −80 °C.
S1-reactive IgG ELISA
Ninety-six-well MaxiSorp plates (Thermo Fisher Scientific) were coated overnight at 4 °C with purified S1 protein in PBS (3 μg ml−1 per well in 50 μl) and blocked for 1 h in blocking buffer (PBS, 5% milk, 0.05% Tween 20 and 0.01% sodium azide). Sera were diluted in blocking buffer (1:50 dilution). Fifty microliters of serum were added to the wells and incubated for 2 h at room temperature. After washing four times with PBS-T (PBS and 0.05% Tween 20), plates were incubated with alkaline phosphatase-conjugated goat anti-human IgG (1:1,000 dilution, Jackson ImmunoResearch) for 1 h. Plates were developed by adding 50 μl alkaline phosphatase substrate (Sigma Aldrich) for 15–30 min after six washes with PBS-T. Optical densities were measured at 405 nm on a microplate reader (Tecan). CR3022 (Absolute Antibodies) was used as a positive control. The cutoff for a positive response was defined as the mean negative value × (0.35 × mean positive value).
Flow cytometry for spike-reactive IgG, IgM and IgA
SUP-T1 cells were collected and counted and spike-expressing and control SUP-T1 cells were mixed in a 1:1 ratio. The cell mix was transferred into V-bottom 96-well plates at 20,000 cells per well. Cells were incubated with heat-inactivated serum diluted 1:50 in PBS for 30 min, washed with FACS buffer (PBS, 5% BSA and 0.05% sodium azide) and stained with FITC anti-IgG (clone HP6017, BioLegend), APC anti-IgM (clone MHM-88, BioLegend) and PE anti-IgA (clone IS11-8E10, Miltenyi Biotech) for 30 min (all antibodies diluted 1:200 in FACS buffer). Cells were washed with FACS buffer and fixed for 20 min in 1% PFA in FACS buffer. Samples were run on a Bio-Rad Ze5 analyzer running Bio-Rad Everest software v.2.4 and analyzed using FlowJo v.10.7.1 (Tree Star) analysis software. Spike-expressing and control SUP-T1 cells were gated and mean fluorescence intensity (MFI) of both populations was measured. MFI in control SUP-T1 cells was subtracted from MFI in spike-expressing SUP-T1 cells and resulting values were divided by MFI in control SUP-T1 cells to calculate the specific increase in MFI. Values >2 were considered positive.
Neutralizing antibody assay against SARS-CoV-2
Confluent monolayers of Vero E6 cells were incubated with SARS-CoV-2 WT or Alpha virus and twofold serial dilutions of heat-treated serum or plasma samples starting at 1:40 for 4 h at 37 °C, 5% CO2, in duplicate. The inoculum was then removed and cells were overlaid with viral growth medium. Cells were incubated at 37 °C, 5% CO2. At 24 h after infection, cells were fixed in 4% paraformaldehyde and permeabilized with 0.2% Triton-X-100/PBS. Virus plaques were visualized by immunostaining, as described previously for neutralization of influenza viruses using a rabbit polyclonal anti-NSP8 antibody used at 1:1,000 dilution and anti-rabbit-HRP conjugated antibody at 1:1,000 dilution and detected by action of horseradish peroxidase (HRP) on a tetramethyl benzidine-based substrate. Virus plaques were quantified and half-maximum infective dose was calculated.
High-throughput live virus microneutralization assay
High-throughput live virus microneutralization assays were performed for a subset of 37 patients for WT SARS-CoV-2, Alpha, Beta or Delta. High-throughput live virus microneutralization assays were performed as described previously56. Briefly, Vero E6 cells (Institute Pasteur) or Vero E6 cells expressing ACE2 and TMPRSS2 (VAT-1) (Centre for Virus Research)57 at 90–100% confluency in a 384-well format were first titrated with varying multiplicities of infection of each SARS-CoV-2 variant and varying concentrations of a control monoclonal nanobody to normalize for possible replicative differences between variants and select conditions equivalent to WT virus. Following this calibration, cells were infected in the presence of serial dilutions of patient serum samples. After infection (24 h Vero E6 Pasteur, 16 h VAT-1), cells were fixed with 4% final formaldehyde, permeabilized with 0.2% Triton-X-100 and 3% BSA in PBS (v/v) and stained for SARS-CoV-2 N protein using Alexa488-labeled-CR3009 antibody produced in-house and cellular DNA using DAPI. Whole-well imaging at 5× was carried out using an Opera Phenix (PerkinElmer) and fluorescent areas and intensity calculated using the Phenix-associated software Harmony 9 (PerkinElmer). Inhibition was estimated from the measured area of infected cells / total area occupied by all cells. The inhibitory profile of each serum sample was estimated by fitting a four-parameter dose–response curve executed in SciPy. Neutralizing antibody titers are reported as the fold-dilution of serum required to inhibit 50% of viral replication (IC50) and are further annotated if they lie above the quantitative (complete inhibition) range, below the quantitative range but still within the qualitative range (partial inhibition is observed but a dose–response curve cannot be fitted because it does not sufficiently span the IC50) or if they show no inhibition at all. IC50 values above the quantitative limit of detection of the assay (>25,600) were recoded as 3,000; IC50 values below the quantitative limit of the assay (<40) but within the qualitative range were recoded as 39 and data below the qualitative range (no response observed) were recoded as 35.
PBMC stimulation assay
PBMCs for in vitro stimulation were thawed at 37 °C and resuspended in 10 ml of warm complete medium (RPMI and 5% human AB serum) containing 0.02% benzonase. Viable cells were counted and 1 × 106 to 2 × 106 cells were seeded in 200 µl complete medium per well of a 96-well plate. Cells were stimulated with 4 µl per well PepTivator SARS-CoV-2 spike (S), membrane (M) or nucleocapsid (N) pools (synthetic SARS-CoV-2 peptide pools, consisting of 15-mer sequences with 11 amino acid overlap covering the immunodominant parts of the S protein and the complete sequence of the N and membrane M proteins), representing 1 µg ml−1 final concentration per peptide (Miltenyi Biotec). SEB (Merck, UK) was used as a positive control at 0.5 µg ml−1 final concentration, negative control was PBS containing dimethylsulfoxide at 0.002% final concentration. PBMCs were cultured for 24 h at 37 °C, 5% CO2.
Activation-induced marker assay
PBMC supernatants were collected for cytokine analysis after stimulation for 24 h. Cells were washed twice in warm PBMCs. Dead cells were stained with 0.5 µl per well Zombie dye V500 for 15 min at room temperature in the dark, then washed once with PBS containing 2% FCS (FACS buffer). A surface staining mix was prepared per well, containing 2 µl per well of each antibody for surface staining in 50:50 brilliant stain buffer (BD) and FACS buffer. PBMCs were stained with 50 µl surface staining mix per well for 30 min at room temperature in the dark. Cells were washed once in FACS buffer and fixed in 1% PFA in FACS buffer for 20 min, then washed once and resuspended in 200 µl PBS. All samples were acquired on a Bio-Rad Ze5 flow cytometer running Bio-Rad Everest software v.2.4 and analyzed using FlowJo v.10.7.1 (Tree Star). Compensation was performed with 20 µl antibody-stained anti-mouse Ig, κ/negative control compensation particle set (BD Biosciences). A total of 1 × 106 live CD19−CD14− cells were acquired per sample. Gates were drawn relative to the unstimulated control for each donor. T cell response is displayed as a stimulation index by dividing the percentage of activation-induced marker (AIM)-positive cells by the percentage of cells in the negative control. If negative control was 0, then the minimum value across the cohort was used. When S, M and N stimulation were combined, the sum of AIM-positive cells was divided by three times the percentage of positive cells in the negative control. A 1.5-fold increase in stimulation index was considered positive.
IFN-γ ELISA
IFN-γ ELISA was performed using the human IFN-γ DuoSet ELISA (R&D Systems) according to the manufacturer’s instructions. Briefly, 96-well plates were coated overnight with capture antibody, washed twice in wash buffer then blocked with reagent diluent for 2 h at room temperature. Then, 100 µl of PBMC culture supernatants were added and incubated for 1 h at room temperature and washed twice in wash buffer. Following that, 100 µl detection antibody diluted in reagent diluent was added per well and incubated for 2 h at room temperature. Plates were washed twice in wash buffer and 100 µl streptavidin-HRP dilution was added to the plates. Plates were incubated for 20 min in the dark at room temperature and then washed twice in wash buffer. The reaction was developed using 200 µl substrate solution for 20 min in the dark at room temperature then stopped with 50 µl stop solution. Optical density was measured at 450 nm on a multimode microplate reader (Berthold). Serial dilutions of standard were run on each plate. Concentrations were calculated by linear regression of standard concentrations ranging 0–600 pg ml−1 and normalized to the number of stimulated PBMC. The assay sensitivity was 5 pg ml−1.
Multiplex immune assay for cytokines and chemokines
The preconfigured multiplex Human Immune Monitoring 65-plex ProcartaPlex immunoassay kit (Invitrogen, Thermo Fisher Scientific) was used to measure 65 protein targets in plasma on the Bio-Plex platform (Bio-Rad Laboratories), using Luminex xMAP technology. Analytes measured included APRIL; BAFF; BLC; CD30; CD40L; ENA-78; eotaxin; eotaxin-2; eotaxin-3; FGF-2; fractalkine; G-CSF; GM-CSF; Gro-α; HGF; IFN-α; IFN-γ; IL-10; IL-12p70; IL-13; IL-15; IL-16; IL-17A; IL-18; IL-1α; IL-1β; IL-2; IL-20; IL-21; IL-22; IL-23; IL-27; IL-2R; IL-3; IL-31; IL-4; IL-5; IL-6; IL-7; IL-8; IL-9; IP-10; I-TAC; LIF; MCP-1; MCP-2; MCP-3; M-CSF; MDC; MIF; MIG; MIP-1α; MIP-1β; MIP-3α; MMP-1; NGF-β; SCF; SDF-1α; TNF-β; TNF-α; TNF-R2; TRAIL; TSLP; TWEAK; and VEGF-A. All assays were conducted as per the manufacturer’s recommendation.
Statistics and reproducibility
No statistical method was used to predetermine sample size but as many patients with SARS-CoV-2 infection were recruited as possible, including patients with no history of infection to identify patients in routine care with asymptomatic infection. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Data and statistical analysis were performed using FlowJo 10 and R v.3.6.1 in R studio v.1.2.1335. Gaussian distribution of baseline characteristics was tested by Kolmogorov–Smirnov test, and differences in patient groups were compared using chi-squared test, Mann–Whitney test or Kruskal–Wallis test, as appropriate. Statistical methods for each experiment are provided in the figure legends. Gaussian distribution was tested by Kolmogorov–Smirnov test. Mann–Whitney, Wilcoxon, Kruskal–Wallis, chi-squared, Fisher’s exact test and Friedman tests were performed for statistical significance. A P value <0.05 was considered significant. The ggplot2 package in R was used for data visualization and illustrative figures were created with BioRender.com. Data were usually plotted as single data points and box plots on a logarithmic scale. For box plots, boxes represent upper and lower quartiles, the line represents the median and whiskers represent 1.5 × IQR. Notches represent confidence intervals of the median. For correlation matrix analysis, Spearman rank correlation coefficients were calculated between all parameter pairs using the corrplot package in R without clustering. For pairwise correlation, Spearman rank correlation coefficients were calculated. Multivariate binary logistic regression analysis was performed using the glm function with the stats package in R.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All requests for raw and analyzed data and CAPTURE study protocol will be reviewed by the CAPTURE Trial Management Team, Skin and Renal Clinical Trials Unit, the Royal Marsden NHS Foundation Trust (CAPTURE@rmh.nhs.uk) to determine if the request is subject to confidentiality and data protection obligations. Materials used in this study will be made available upon request. There are restrictions to the availability based on limited quantities. Response to any request for data and/or materials will be given within a 28-d period. Data and materials that can be shared would then be released upon completion of a material transfer agreement. Source data are provided with this paper.
Code availability
No unpublished code was used in this study.
References
Saini, K. S. et al. Mortality in patients with cancer and coronavirus disease 2019: a systematic review and pooled analysis of 52 studies. Eur. J. Cancer 139, 43–50 (2020).
Williamson, E. J. et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 584, 430–436 (2020).
Garcia-Suarez, J. et al. Impact of hematologic malignancy and type of cancer therapy on COVID-19 severity and mortality: lessons from a large population-based registry study. J. Hematol. Oncol. 13, 133 (2020).
Garassino, M. C. et al. COVID-19 in patients with thoracic malignancies (TERAVOLT): first results of an international, registry-based, cohort study. Lancet Oncol. 21, 914–922 (2020).
Lee, L. Y. et al. COVID-19 mortality in patients with cancer on chemotherapy or other anticancer treatments: a prospective cohort study. Lancet 395, 1919–1926 (2020).
Kuderer, N. M. et al. Clinical impact of COVID-19 on patients with cancer (CCC19): a cohort study. Lancet 395, 1907–1918 (2020).
Grivas, P. et al. Association of clinical factors and recent anticancer therapy with COVID-19 severity among patients with cancer: a report from the COVID-19 and Cancer Consortium. Ann. Oncol. 32, 787–800 (2021).
Lee, L. Y. W. et al. COVID-19 prevalence and mortality in patients with cancer and the effect of primary tumour subtype and patient demographics: a prospective cohort study. Lancet Oncol. 21, 1309–1316 (2020).
Crolley, V. E. et al. COVID-19 in cancer patients on systemic anti-cancer therapies: outcomes from the CAPITOL (COVID-19 Cancer PatIenT Outcomes in North London) cohort study. Ther. Adv. Med. Oncol. 12, 1758835920971147 (2020).
Robilotti, E. V. et al. Determinants of COVID-19 disease severity in patients with cancer. Nat. Med. 26, 1218–1223 (2020).
Bange, E. M. et al. CD8+ T cells contribute to survival in patients with COVID-19 and hematologic cancer. Nat. Med. 27, 1280–1289 (2021).
Abdul-Jawad, S. et al. Acute immune signatures and their legacies in severe acute respiratory syndrome coronavirus-2 infected cancer patients. Cancer Cell 39, 257–275.e256 (2021).
Thakkar, A. et al. Seroconversion rates following COVID-19 vaccination among patients with cancer. Cancer Cell 39, 1081–1090.e2 (2021).
Au, L. et al. Cancer, COVID-19, and antiviral immunity: the CAPTURE study. Cell 183, 4–10 (2020).
Marshall, J. C. et al. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect. Dis. 20, e192–e197 (2020).
Lowe, K. E., Zein, J., Hatipoglu, U. & Attaway, A. Association of smoking and cumulative pack-year exposure with COVID-19 outcomes in the Cleveland Clinic COVID-19 registry. JAMA Intern. Med. 181, 709–711 (2021).
Rosenthal, N., Cao, Z., Gundrum, J., Sianis, J. & Safo, S. Risk factors associated with in-hospital mortality in a US national sample of patients with COVID-19. JAMA Netw. Open 3, e2029058 (2020).
Sterlin, D. et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci. Transl. Med. 13, eabd2223 (2021).
Grifoni, A. et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell https://doi.org/10.1016/j.cell.2020.05.015 (2020).
Dan, J. M. et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 371, eabf4063 (2021).
Moderbacher, C. R. et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell https://doi.org/10.1016/j.cell.2020.09.038 (2020).
Weiskopf, D. et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol.5, eabd2071 (2020).
Mateus, J. et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science https://doi.org/10.1126/science.abd3871 (2020).
Marra, A. et al. Seroconversion in patients with cancer and oncology health care workers infected by SARS-CoV-2. Ann. Oncol. 32, 113–119 (2021).
Dispinseri, S. et al. Neutralizing antibody responses to SARS-CoV-2 in symptomatic COVID-19 is persistent and critical for survival. Nat. Commun. 12, 2670 (2021).
Earle, K. A. et al. Evidence for antibody as a protective correlate for COVID-19 vaccines. Vaccine 39, 4423–4428 (2021).
Khoury, D. S. et al. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 27, 1205–1211 (2021).
Ju, B. et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 584, 115–119 (2020).
Fendler, A. et al. Adaptive immunity and neutralizing antibodies against SARS-CoV-2 variants of concern following vaccination in patients with cancer: the CAPTURE study. Nat. Cancer https://doi.org/10.1038/s43018-021-00274-w (2021).
Seow, J. et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-CoV-2 infection in humans. Nat. Microbiol. 5, 1598–1607 (2020).
Gaebler, C. et al. Evolution of antibody immunity to SARS-CoV-2. Nature 591, 639–644 (2021).
Wang, Z. et al. Naturally enhanced neutralizing breadth against SARS-CoV-2 one year after infection. Nature 595, 426–431 (2021).
Achiron, A. et al. Humoral immune response to COVID-19 mRNA vaccine in patients with multiple sclerosis treated with high-efficacy disease-modifying therapies. Ther. Adv. Neurol. Disord. https://doi.org/10.1177/17562864211012835 (2021).
Garcia-Beltran, W. F. et al. COVID-19 neutralizing antibodies predict disease severity and survival. Cell 184, 476–488.e11 (2021).
Vacharathit, V. et al. CoronaVac induces lower neutralising activity against variants of concern than natural infection. Lancet Infect. Dis. 21, 1352–1354 (2021).
Juno, J. A. et al. Humoral and circulating follicular helper T cell responses in recovered patients with COVID-19. Nat. Med. 26, 1428–1434 (2020).
Murugesan, K. et al. Interferon-γ release assay for accurate detection of severe acute respiratory syndrome coronavirus 2 T-cell response. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa1537 (2020).
Pauken, K. E. et al. The PD-1 pathway regulates development and function of memory CD8+ T cells following respiratory viral infection. Cell Rep. 31, 107827 (2020).
Konkel, J. E. et al. PD-1 signalling in CD4+ T cells restrains their clonal expansion to an immunogenic stimulus, but is not critically required for peptide-induced tolerance. Immunology 130, 92–102 (2010).
Zhao, J., Zhao, J. & Perlman, S. T cell responses are required for protection from clinical disease and for virus clearance in severe acute respiratory syndrome coronavirus-infected mice. J. Virol. 84, 9318–9325 (2010).
Muñoz-Fontela, C. et al. Animal models for COVID-19. Nature 586, 509–515 (2020).
Tan, A. T. et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 34, 108728 (2021).
Tarke, A. et al. Impact of SARS-CoV-2 variants on the total CD4+ and CD8+ T cell reactivity in infected or vaccinated individuals. Cell Rep. Med. https://doi.org/10.1016/j.xcrm.2021.100355 (2021).
Apostolidis, S.A. et al. Cellular and humoral immune responses following SARS-CoV-2 mRNA vaccination in patients with multiple sclerosis on anti-CD20 therapy. Nat. Med. https://doi.org/10.1038/s41591-021-01507-2 (2021).
Sahin, U. et al. COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature 586, 594–599 (2020).
Ewer, K. J. et al. T cell and antibody responses induced by a single dose of ChAdOx1 nCoV-19 (AZD1222) vaccine in a phase 1/2 clinical trial. Nat. Med. 27, 270–278 (2021).
Normark, J. et al. Heterologous ChAdOx1 nCoV-19 and mRNA-1273 vaccination. N. Engl. J. Med. 385, 1049–1051 (2021).
Barros-Martins, J. et al. Immune responses against SARS-CoV-2 variants after heterologous and homologous ChAdOx1 nCoV-19/BNT162b2 vaccination. Nat. Med. 27, 1525–1529 (2021).
Spencer, A.J. et al. Heterologous vaccination regimens with self-amplifying RNA and adenoviral COVID vaccines induce robust immune responses in mice. Nat. Commun. 12, 2893 (2021).
Angelis, V. et al. Defining the true impact of coronavirus disease 2019 in the at-risk population of patients with cancer. Eur. J. Cancer 136, 99–106 (2020).
Sekine, T. et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell 183, 158–168 (2020).
Aitken, J. et al. Scalable and robust SARS-CoV-2 testing in an academic center. Nat. Biotechnol. 38, 927–931 (2020).
Di Tommaso, P. et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319 (2017).
Baker, D. J. et al. CoronaHiT: high-throughput sequencing of SARS-CoV-2 genomes. Genome Med. 13, 21 (2021).
Ewels, P. A. et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 38, 276–278 (2020).
Faulkner, N. et al. Reduced antibody cross-reactivity following infection with B.1.1.7 than with parental SARS-CoV-2 strains. eLife 10, e69317 (2021).
Rihn, S. J. et al. A plasmid DNA-launched SARS-CoV-2 reverse genetics system and coronavirus toolkit for COVID-19 research. PLoS Biol. 19, e3001091 (2021).
Acknowledgements
We thank the CAPTURE trial team, including E. Carlyle, K. Edmonds and L. Del Rosario, as well as S. Agarwal, H. Ahmod, N. Ash, R. Dhaliwal, L. Dowdie, T. Foley, L. Holt, D. Kabir, M. O’Flaherty, M. Ndlovu, S. Ali, J. Korteweg, C. Lewis, K. Lingard, M. Mangwende, A. Murra, K. Peat, S. Sarker, N. Shaikh, S. Vaughan and F. Williams. We acknowledge support from the clinical and research teams at participating units at the Royal Marsden Hospital, including E. Black, A. Dela Rosa, C. Pearce, J. Bazin, L. Conneely, C. Burrows, T. Brown, J. Tai, E. Lidington, H. Hogan, A. Upadhyay, D. Capdeferro, I. Potyka, A. Drescher, F. Baksh, M. Balcorta, C. Da Costa Mendes, J. Amorim, V. Orejudos and L. Davison. We also thank volunteer staff at the Francis Crick Institute and the Crick COVID-19 Consortium and A. Lilley for help with neutralizing assays, A. Toncheva and staff at the cloning unit at Autolus, including J. Sillibourne, K. Ward, K. Lamb and P. Wu. We thank B. Stockinger for thoughtful review and comments on the manuscript. Due to the pace at which the field is evolving, we acknowledge researchers in COVID-19, particularly in furthering our understanding of SARS-CoV-2 infection and we apologize for work that was not cited. This research was funded in part, by the Cancer Research UK (grant no. C50947/A18176). This work was supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (CRUK) (FC001988, FC001218, FC001099, FC001002, FC001078, FC001169 and FC001030), the UK Medical Research Council (FC001988, FC001218, FC001099, FC001002, FC001078, FC001169 and FC001030) and the Wellcome Trust (FC001988, FC001218, FC001099, FC001002, FC001078, FC001169 and FC001030). For the purpose of Open Access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. TRACERx Renal is partly funded by the National Institute for Health Research (NIHR) Biomedical Research Centre (BRC) at the Royal Marsden Hospital and Institute of Cancer Research (A109). The CAPTURE study is sponsored by the Royal Marsden NHS Foundation Trust and funded from a grant from the Royal Marsden Cancer Charity. A.F. has received funding from the European Union’s Horizon 2020 Research and Innovation program under Marie Skłodowska-Curie grant no. 892360. L.A. is funded by the Royal Marsden Cancer Charity. S.T. is funded by CRUK (grant no. A29911); the Francis Crick Institute, which receives its core funding from CRUK (FC10988), the UK Medical Research Council (FC10988) and the Wellcome Trust (FC10988); the NIHR BRC at the Royal Marsden Hospital and Institute of Cancer Research (grant no. A109), the Royal Marsden Cancer Charity, the Rosetrees Trust (grant no. A2204), Ventana Medical Systems (grant nos. 10467 and 10530), the National Institute of Health (U01 CA247439) and Melanoma Research Alliance (award no. 686061). C. Swanton is a Royal Society Napier Research Professor (RP150154). His work is supported by the Francis Crick Institute, which receives its core funding from CRUK (FC001169), the UK Medical Research Council (FC001169) and the Wellcome Trust (FC001169). C. Swanton is funded by the CRUK (TRACERx, PEACE and CRUK Cancer Immunotherapy Catalyst Network), CRUK Lung Cancer Centre of Excellence, the Rosetrees Trust, Butterfield and Stoneygate Trusts, NovoNordisk Foundation (ID16584), Royal Society Research Professorship Enhancement Award (RP/EA/180007), the NIHR BRC at University College London Hospitals, the CRUK-UCL Centre, Experimental Cancer Medicine Centre and the Breast Cancer Research Foundation, USA. His research is supported by a Stand Up To Cancer (SU2C)-LUNGevity-American Lung Association Lung Cancer Interception Dream Team Translational Research Grant (SU2C-AACR-DT23-17). SU2C is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. C. Swanton also receives funding from the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013) Consolidator Grant (FP7-THESEUS-617844), European Commission ITN (FP7-PloidyNet 607722), an ERC Advanced Grant (PROTEUS) from the European Research Council under the European Union’s Horizon 2020 Research and Innovation program (835297) and Chromavision from the European Union’s Horizon 2020 Research and Innovation program (665233). R.W. has received Francis Crick Institute support from the Wellcome Trust (FC0010218), UKRI (FC0010218), CRUK (FC0010218) and research funding from the Wellcome Trust (203135 and 222754), Rosetrees Trust (M926) and the South African Medical Research Council.
Author information
Authors and Affiliations
Consortia
Contributions
S.T., L.A. and L.A.B. were responsible for conceptualization of the study. Methodology was developed by S.T., A.F., F.B., K.A.W., G.K. and R.H. M.G. was responsible for software. Formal analysis was conducted by A.F., L.A., S.T.C.S., G.K., K.A.W., R.J.W., R.H. and M.C. Investigations were conducted by A.F., L.A., F.B., S.T.C.S, B.S., C.G., W.X., B.W., K.A.W., M.C., A.A-D. and R.H. Resources were provided by S.T., A.F., L.A., L.A.B., F.B., S.T.C.S., B.S., C.G., B.W., W.X., M.C., G.C., M.P., L.M., R.S., C.G., H.F., M.G., F.G., O.C., T.S., Y.K., Z.T. and I.L. Data were curated by L.A.B., S.T.C.S., B.S., C.G., A.F. and L.A. Writing of the original draft was the responsibility of S.T., K.R., A.F., L.A. and S.T.C.S.; A.F., L.A., S.T.C.S., F.B., M.C., L.A.B., K.R., W.G., B.S., C.L.G., B.W., W.X., A.M.S., N.J.-H., G.H.C., M.P., L.M., K.W.N, E.C., K.E., L.D.R., S.S., K.L., M.M., L.H., H.A., R.S., C.G., H.R.F., A.A.-D., P.H., S.C., M.H., M.W., R.G., M.C., L.C., H.P., M.G., E.N., A.-.S., J.I.M., J.N., F.G., R.L.S., C.M., D.C., I.C., N.S., N.T., L.W., N.v.A., R.L.J., J.D., S. Banerjee, K.C.T., S.J., M.O.’B., O.C., K.H., S.B., J.B., A. Robinson, C. Stephenson, T.S., Y.K., Z.T., I.L., S.G., A.O., A. Reid, K.Y., A.J.S.F., L.P., S. Gandhi, S. Gamblin, C. Swanton, E.N., S.K., N.Y., K.A.W., A.S., R.H., G.K., J.L., R.J.W. and S.T. were responsible for review and editing of the manuscript. Visualization was carried out by A.F., S.T.C.S., S.T. and L.A. Supervision was conducted by S.T. Trial conduct was overseen by S.T., L.A., S.T.C.S., E.C., L.R., K.E., L.A.B., J.L., N.Y., A.R., E.N. and S.K.
Corresponding author
Ethics declarations
Competing interests
S.T. has received speaking fees from Roche, AstraZeneca, Novartis and Ipsen. S.T. has the following patents filed: Indel mutations as a therapeutic target and predictive biomarker PCTGB2018/051892 and PCTGB2018/051893 and Clear Cell Renal Cell Carcinoma Biomarkers P113326GB. N.Y. has received conference support from Celegene. A.R. received speaker’s fees from Janssen and AstraZeneca; travel support from Janssen and Astellas, and consultancy fees from AstraZeneca. D.C. received funding from MedImmune, AstraZeneca, Clovis, Eli Lilly, 4SC, Bayer, Celgene and Roche, and is on the advisory board of OVIBIO. J.L. has received research funding from Bristol-Myers Squibb, Merck, Novartis, Pfizer, Achilles Therapeutics, Roche, Nektar Therapeutics, Covance, Immunocore, Pharmacyclics and Aveo, and served as a consultant to Achilles, Astra Zeneca, Boston Biomedical, Bristol-Myers Squibb, Eisai, EUSA Pharma, GlaxoSmithKline, Ipsen, Imugene, Incyte, iOnctura, Kymab, Merck Serono, Nektar, Novartis, Pierre Fabre, Pfizer, Roche Genentech, Secarna and Vitaccess. I.C. has served as a consultant to Eli-Lilly, Bristol Meyers Squibb, MSD, Bayer, Roche, Merck Serono, Five Prime Therapeutics, Astra Zeneca, OncXerna, Pierre Fabre, Boehringer-Ingelheim, Incyte, Astella, GSK, Sotio and, Eisai; has received research funding from Eli-Lilly & Janssen-Cilag; and has received honorarium from Eli-Lilly, Eisai, Servier. A.O. acknowledges receipt of research funding from Pfizer and Roche; speakers fees from Pfizer, Seagen, Lilly and Astra Zeneca; is an advisory board member of Roche, Seagen and Astra Zeneca; and has received conference support from Leo Pharmaceuticals, Astra Zeneca/Diachi-Sankyo and Lilly. C. Swanton acknowledges grant support from Pfizer, Astra Zeneca, Bristol-Myers Squibb, Roche-Ventana, Boehringer-Ingelheim, Archer Dx (collaboration in minimal residual disease sequencing technologies) and Ono Pharmaceutical; is an Astra Zeneca Advisory Board member and Chief Investigator for the MeRmaiD1 clinical trial; has consulted for Amgen, Pfizer, Novartis, GlaxoSmithKline, MSD, Bristol-Myers Squibb, Celgene, Astra Zeneca, Illumina, Genentech, Roche-Ventana, GRAIL, Medicxi, Metabomed, Bicycle Therapeutics and the Sarah Cannon Research Institute; has stock options in Apogen Biotechnologies, Epic Bioscience, GRAIL and has stock options; and is co-founder of Achilles Therapeutics. C. Swanton holds European patents relating to assay technology to detect tumor recurrence (PCT/GB2017/053289); to targeting neoantigens (PCT/EP2016/059401), identifying patent response to immune-checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (human leukocyte antigen loss of heterozygosity) (PCT/GB2018/052004), predicting survival rates of patients with cancer (PCT/GB2020/050221), identifying patients who respond to cancer treatment (PCT/GB2018/051912), a US patent relating to detecting tumor mutations (PCT/US2017/28013) and both a European and US patent related to identifying insertion/deletion mutation targets (PCT/GB2018/051892). L.P. has received research funding from Pierre Fabre and honoraria from Pfizer, Ipsen, Bristol-Myers Squibb and EUSA Pharma. S.B. has received institutional research funding from AstraZeneca, Tesaro, GSK; speakers fees from Amgen, Pfizer, Astra Zeneca, Tesaro, GSK, Clovis, Takeda, Immunogen and Mersana; and has an advisory role for Amgen, AstraZeneca, Epsilogen, Genmab, Immunogen, Mersana, Merck Sharp & Dohme, Merck Serono, Oncxerna, Pfizer and Roche. N.T. has received advisory board honoraria from AstraZeneca, Bristol-Myers Squibb, Lilly, Merck Sharpe and Dohme, Novartis, Pfizer, Roche/Genentech, GlaxoSmithKline, Zentalis Pharmaceuticals, Repare Therapeutics and Arvinas; and research funding from AstraZeneca, BioRad, Pfizer, Roche/Genentech, Merck Sharpe & Dohme, Guardant Health, Invitae, Inivata, Personalis and Natera. M.O.B. is on advisory boards for Amgen, Pierre Fabre, Pharmamar, Puma, MSD and Roche. A.M.S. received and educational grant from Janssen-Cilag. N.S. received travel grants from AstraZeneca, BMS, Eli Lilly, Merck, Roche and MSD Oncology; received honoraria from Eli Lilly, Merck Serono, MSD Oncology, Pierre Fabre, Servier, GSK and Amgen; and is on the advisory board for Pfizer, AstraZeneca, Servier and MSD. N.S. received funding from AstraZeneca, BMS, Pfizer, NIHR EME, RMCC and RM/ICR BCR. R.L.J. received research support from Merck Sharp & Dohme and GSK; and received consultation fees from Adaptimmune, Astex, Athenex, Bayer, Boehringer Ingelheim, Blueprint, Cinigen, Eisai, Epizyme, Daichii, Deciphera, Immundesign, Immunicum, Karma Oncology, Lilly, Merck, Mundipharma, Pharmamar, Springworks, SynOx, Tracon, Upto Date and Merck Sharp & Dohme. A.F., L.A., S.T.C.S., F.B., M.C., L.A.B., K.R., W.G., B.S., C.L.G., B.W., W.X., N.J.-H., G.H.C., M.P., L.M., K.W.N., E.C., K.E., L.D.R., S.S., K.L., M.M., L.H., H.A., R.S., C.G., H.R.F., A.A.-D., P.H., S.C., M.H., M.W., R.G., M.C., L.C., H.P., M.G., E.N., A.-.S., J.I.M., J.N., F.G., R.L.S., C.M., D.C. L.W., N.v.A., J.D., K.C.T., S.J., O.C., K.H., S.B., J.B., A. Robinson, C. Stephenson, T.S., Y.K., Z.T., I.L., S.G., A.O., A. Reid, K.Y., A.J.S.F., S. Gamblin, C. Swanton, E.N., S.K., K.A.W., A.S., R.H., G.K. and R.J.W. declare no competing interests.
Additional information
Peer review information Nature Cancer thanks Ailong Huang, Sean Lim and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Duration of viral shedding and SARS-CoV-2 viral strain incidence in the CAPTURE cohort, and cyto/chemokine correlations with COVID-19 severity.
a) Duration to final positive swab in patients with solid (n = 32) vs haematological malignancies (n = 11). Boxes indicate the 25 and 75 percentiles, line indicates the median, and whiskers indicate the 10 and 90 percentiles. Dots represent individual samples. Significance was tested by two-sided Wilcoxon Mann-Whitney U test, p < 0.05 was considered significant. b) Change over time in the dominant SARS-CoV-2 strain as assessed in 44 patients with cancer with viral sequencing data. Cytokine levels related to c) T-cell and d) macrophage responses were measured in patients with cancer with acute SARS-CoV infection (blue bar, n = 13) using the human immune monitoring 65-plex ProcartaPlex immunoassay in serum samples. Samples were measured in duplicates. Control samples are sera from non-matched patients with cancer (pink bar, n = 5) without SARS-CoV-2 infection. Data are presented as the log10 of the concentration in pg ml−1. Correlation of cyto/chemokines related to e) T cells and f) macrophages with COVID-19 WHO severity score. g) Comparisons of cytokine levels between patients with haematological (orange, n = 8) and solid malignancies (aqua, n = 5). Boxes indicate the 25th and 75th percentiles, line indicates the median, and whiskers indicate the 10 and 90 percentiles. Dots represent individual samples. Significance was tested by Wilcoxon-Mann-Whitney U test, p-values are denoted in the graph.
Extended Data Fig. 2 Spike-reactive and neutralising antibody titers.
a) Flow cytometric assays to quantify S-reactive IgG and IgM and IgA levels in sera of patients (n = 109 for IgG and IgM, n = 51 for IgA) with S-reactive antibodies. b) S-reactive IgG, c) IgM, and d) IgA in SARS-CoV-2 positive compared with infection-naive patients (n = 40). Boxes indicate the 25 and 75 percentiles, line indicates the median, and whiskers indicate the 10th and 90th percentiles. e) Correlation between S1-reactive IgG and S1-reactive AbT in 40 patients and f) Correlation between S1-reactive IgM and S1-reactive AbT in 40 patients. Blue line denotes linear regression line with grey areas marking the 95% confidence band. Regression coefficient and p-values were computed by two-sided spearman regression, p < 0.05 was considered significant. g) S1-reactive antibodies in pre-pandemic sera of patients (n = 77). Boxes indicate the 25 and 75 percentiles, line indicates the median, and whiskers indicate the 10th and 90th percentiles. h) Correlation between NAbT against WT, Alpha, Beta, Delta and S1-reactive AbT in 112 patients. Blue line denotes linear regression line with grey areas marking the 95% confidence band. Regression coefficient and p-values were computed by two-sided spearman regression, p < 0.05 was considered significant.
Extended Data Fig. 3 Gating strategy.
Gating strategy for flow analysis of a) S-reactive IgG, IgG, IgA and b) AIM assay.
Extended Data Fig. 4 Longitudinal assessment of NAb against VOCs.
NAbT against a) Alpha, b) Beta, c) Delta over time POD. Black dots denote patients with only one sample, coloured dots denote patients with serial samples. Samples from individual patients are connected. Loess regression was performed and is displayed as a blue line with 95% confidence bands as grey area. Dotted lines and grey areas at graph bottom indicate limit of detection. POD, post onset of disease.
Extended Data Fig. 5 T-cell response in patients with cancer.
a) Comparison between SARS-CoV-2 reactive CD4 + and CD8 + T cells evaluated in 100 patients with cancer. Boxes indicate the 25th and 75th percentiles, the median is shown, and whiskers indicate the 10th and 90th percentiles. Dots represent individual samples, connected dots are from the same sample. Significance as tested by two-sided Wilcoxon Mann Whitney U-test, p-values < 0.05 was considered significant. b) Pairwise spearman correlation of CD4 + and CD8 + T cells and IFN-𝛾 concentration (pg/ml) in supernatants after stimulation of PBMCs (n = 82). Correlation is shown by color gradient. p-values for each correlation were calculated by two-sided t-test and were adjusted for multiple testing by Benjamini-Hochberg correction. P-value is denoted by size, in addition p-values < 0.05 are denoted by asterisk. *, p < 0.05. c) Comparison of IFN-𝛾 concentration (pg/ml) in patients with solid (n = 41) and haematological malignancies (n = 8). Only patients in which SsT cells were detected were included. Significance as tested by two-sided Wilcoxon Mann Whitney U-test, p-values < 0.05 was considered significant. d) CD4 + T cells in 12 patients with cancer without confirmed SARS-CoV-2 infection. e) CD8 + T cells in 12 patients with cancer without confirmed SARS-CoV-2 infection. Boxes indicate the 25 and 75 percentiles, line indicates the median, and whiskers indicate the 10 and 90 percentiles. Dots represent individual samples. Correlation of S1-reactive AbT and SARS-CoV-2-specific CD4 + T cells in patients with f) solid (n = 81) and g) haematological malignancies (n = 19). Blue line denotes linear regression line with grey areas marking the 95% confidence band. Regression coefficient and p-values were computed by two-sided spearman regression, p < 0.05 was considered significant. NC, negative control, SEB, Staphylococcal enterotoxin B, IFN, IFN-𝛾.
Extended Data Fig. 6 Comparison of antibody and T-cell responses to individuals without cancer.
Comparison of a) S1-reactive AbT and b) NAb against WT in HCW (n = 21), patients with solid malignancies (n = 92), and haematological malignancies (n = 20). Comparison of SARS-CoV-specific c) CD4+ and d) CD8+ T cells in HCW (n = 19), patients with solid malignancies (n = 81), and haematological malignancies (n = 19). Boxes indicate the 25 and 75 percentiles, line indicates the median, and whiskers indicate the 10 and 90 percentiles. Dots represent individual samples. Significance was tested by Kruskal Wallis test, p < 0.05 was considered significant, post-hoc test with Bonferroni correction was used for pairwise comparisons.
Supplementary information
Supplementary Information
Contains full list of both consortia (Crick COVID-19 Consortium and CAPTURE Study Consortium)
Supplementary Tables
Supplementary Tables 1–3.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fendler, A., Au, L., Shepherd, S.T.C. et al. Functional antibody and T cell immunity following SARS-CoV-2 infection, including by variants of concern, in patients with cancer: the CAPTURE study. Nat Cancer 2, 1321–1337 (2021). https://doi.org/10.1038/s43018-021-00275-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-021-00275-9
This article is cited by
-
Vaccination against SARS-CoV-2 in adults with a diagnosis of cancer: a short review
memo - Magazine of European Medical Oncology (2023)
-
A population-scale temporal case–control evaluation of COVID-19 disease phenotype and related outcome rates in patients with cancer in England (UKCCP)
Scientific Reports (2023)
-
Dynamic changes in peripheral lymphocytes and antibody response following a third dose of SARS-CoV-2 mRNA-BNT162b2 vaccine in cancer patients
Scientific Reports (2022)
-
COVID-19 vaccines in patients with cancer: immunogenicity, efficacy and safety
Nature Reviews Clinical Oncology (2022)
-
Predictive factors for the presence and long-term persistence of SARS-CoV-2 antibodies in healthcare and university workers
Scientific Reports (2022)
