
Abstract
Effective molecular representation learning is of great importance to facilitate molecular property prediction. Recent advances for molecular representation learning have shown great promise in applying graph neural networks to model molecules. Moreover, a few recent studies design self-supervised learning methods for molecular representation to address insufficient labelled molecules; however, these self-supervised frameworks treat the molecules as topological graphs without fully utilizing the molecular geometry information. The molecular geometry, also known as the three-dimensional spatial structure of a molecule, is critical for determining molecular properties. To this end, we propose a novel geometry-enhanced molecular representation learning method (GEM). The proposed GEM has a specially designed geometry-based graph neural network architecture as well as several dedicated geometry-level self-supervised learning strategies to learn the molecular geometry knowledge. We compare GEM with various state-of-the-art baselines on different benchmarks and show that it can considerably outperform them all, demonstrating the superiority of the proposed method.
Similar content being viewed by others

Main
Molecular property prediction has been widely considered as one of the most critical tasks in computational drug and materials discovery, as many methods rely on predicted molecular properties to evaluate, select and generate molecules1,2. With the development of deep neural networks (DNNs), molecular representation learning exhibits a great advantage over feature engineering-based methods, which has attracted increasing research attention to tackle the molecular property prediction problem.
Graph neural networks (GNNs) for molecular representation learning have recently become an emerging research area, which regard the topology of atoms and bonds as a graph, and propagate messages of each element to its neighbours3,4,5,6. However, one major obstacle to hinder the successful application of GNNs (and DNNs) in molecule property prediction is the scarity of labelled data, which is also a common research challenge in natural language processing7,8 and computer vision9,10 communities. Inspired by the success of self-supervised learning, recent studies4,11 start to use large-scale unlabelled molecules in a self-supervised methodology to pre-train the molecular representation and then use a small number of labelled molecules to fine tune the models, achieving substantial improvements.
Existing self-supervised learning techniques for GNNs4,11 only consider the topology information of the molecules, neglecting the molecular geometry, that is, the three-dimensional spatial structure of a molecule. These works conduct self-supervised learning by masking and predicting in nodes, edges or contexts in the topology4,11. Yet these tasks only enable the model to learn the laws of molecular graph such as which atom/group could be connected to a double bond, and lack the ability to learn the molecular geometry knowledge, which plays an important role in determining molecules’ physical, chemical and biological activities. For example, the water solubility (a critical metric of drug-likeness) of the two molecules illustrated in Fig. 1 is different due to their differing geometries, even though they have the same topology. Cis-platin and trans-platin are another example of molecules with the same topology but different geometries: cis-platin is a popular chemotherapy drug used to treat a number of cancers, whereas trans-platin has no cytotoxic activity12.
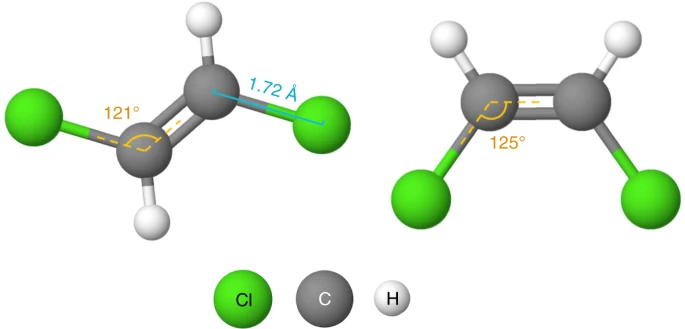
The two chlorine atoms are on different sides in trans-1,2-dichloroethene (left) but the same side in cis-1,2-dichloroethene (right).
Although incorporating geometric information into graph architectures to benefit some molecular property estimation tasks has attracted research attention in recent years13,14,15,16,17, there is still a demand to utilize the molecular geometry information to develop a self-supervised learning paradigm for property prediction. We argue that adopting the self-supervised learning to estimate the geometry can contribute to facilitating the model’s capacity in predicting various properties. Self-supervised learning can take advantage of the large-scale unlabelled molecules with coarse three-dimensional spatial structures to better learn the molecular representation, where the coarse three-dimensional spatial structures can be efficiently calculated by cheminformatics tools such as RDKit (https://www.rdkit.org/). By geometry-level self-supervised learning, the pre-trained model is capable of inferring the molecular geometry by itself.
To this end, we propose a novel geometry-enhanced molecular representation learning method (GEM). First, to make the message passing sensitive to geometries, we model the effects of atoms, bonds and bond angles simultaneously by designing a geometry-based GNN architecture (GeoGNN). The architecture consists of two graphs: the first graph regards the atoms as nodes and the bonds as edges, whereas the second graph regards the bonds as nodes and the bond angles as edges. Second, we pre-train the GeoGNN to learn the chemical laws and the geometries from large-scale molecules with coarse three-dimensional spatial structures, designing various geometry-level self-supervised learning tasks. To verify the effectiveness of the proposed GEM, we compared it with several state-of-the-art (SOTA) baselines on 15 molecular property prediction benchmarks, among which GEM achieves 14 SOTA results.
Our contributions can be summarized as follows:
-
We propose a novel geometry-based GNN to encode both the topology and geometry information of molecules.
-
We design multiple geometry-level self-supervised learning tasks to learn the molecular spatial knowledge from large-scale molecules with coarse spatial structures.
-
We evaluated GEM thoroughly on various molecular property prediction datasets. Experimental results demonstrate that GEM considerably outperforms competitive baselines on multiple benchmarks.
Preliminaries
Graph-based molecular representation
A molecule consists of atoms and the neighbouring atoms are connected by chemical bonds, which can be represented by a graph \(G=({{{\mathcal{V}}}},{{{\mathcal{E}}}})\), where \({{{\mathcal{V}}}}\) is a node set and \({{{\mathcal{E}}}}\) is an edge set. An atom in the molecule is regarded as a node \(v\in {{{\mathcal{V}}}}\) and a chemical bond in the molecule is regarded as an edge \((u,v)\in {{{\mathcal{E}}}}\) connecting atoms u and v.
Graph neural networks are message-passing neural networks18, making them useful for predicting molecular properties. Following the definitions of the previous GNNs19, the features of a node v are represented by xv and the features of an edge (u, v) are represented by xuv. Taking node features, edge features and the graph structure as inputs, a GNN learns the representation vectors of the nodes, where the representation vector of a node v is denoted by hv. A GNN iteratively updates a node’s representation vector by aggregating the messages from the node’s neighbours. Finally, the representation vector hG of the entire graph can be obtained by pooling over the representation vectors {hv} of all the nodes at the last iteration. The representation vector of the graph hG is utilized to estimate the molecular properties.
Pre-training methods for GNNs
In the molecular representation learning community, recently several works4,11,20 have explored the power of self-supervised learning to improve the generalization ability of GNN models on downstream tasks. They mainly focus on two kinds of self-supervised learning tasks: the node-level (edge-level) tasks and the graph-level tasks.
The node-level self-supervised learning tasks are devised to capture the local domain knowledge. For example, some studies randomly mask a portion of nodes or sub-graphs and then predict their properties by the node/edge representation. The graph-level self-supervised learning tasks are used to capture the global information, like predicting the graph properties by the graph representation. Usually, the graph properties are domain-specific knowledge, such as experimental results from biochemical assays or the existence of molecular functional groups.
The GEM framework
This section introduces the details of our proposed geometry-enhanced molecular representation learning method (GEM), which includes two parts: a novel geometry-based GNN and various geometry-level self-supervised learning tasks.
GeoGNN
We propose a GeoGNN that encodes molecular geometries by modelling the atom–bond–angle relations, distinguishing them from traditional GNNs, which only consider the relationship between atoms and bonds.
For a molecule, we denote the atom set as \({{{\mathcal{V}}}}\), the bond set as \({{{\mathcal{E}}}}\), and the bond angle set as \({{{\mathcal{A}}}}\). We introduce atom–bond graph G and bond–angle graph H for each molecule, as illustrated in Fig. 2a. The atom–bond graph is defined as \(G=({{{\mathcal{V}}}},{{{\mathcal{E}}}})\), where atom \(u\in {{{\mathcal{V}}}}\) is regarded as the node of G and bond \((u,v)\in {{{\mathcal{E}}}}\) as the edge of G, connecting atoms u and v. Similarly, the bond–angle graph is defined as \(H=({{{\mathcal{E}}}},{{{\mathcal{A}}}})\), where bond \((u,v)\in {{{\mathcal{E}}}}\) is regarded as the node of H and bond angle \((u,v,w)\in {{{\mathcal{A}}}}\) as the edge of H, connecting bonds (u, v) and (v, w). We use xu as the initial features of atom u, xuv as the initial features of bond (u, v), and xuvw as the initial features of bond angle (u, v, w). The atom–bond graph G and the bond–angle graph H—as well as atom features, bond features and bond angle features—are taken as the inputs of GeoGNN.
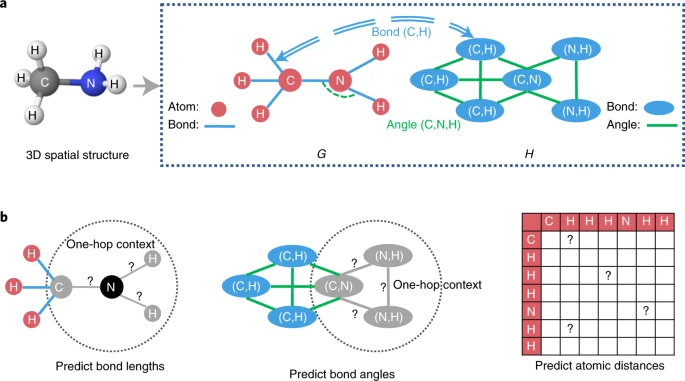
a, In atom–bond graph G, the chemical bonds are regarded as edges, connecting the atoms. In the bond–angle graph H, the bond angles are regarded as edges, and a bond angle connects two chemical bonds and three atoms. The double-dash arcs indicate the correspondence between the elements in the two graphs. b, Demonstration of geometry-level self-supervised learning tasks. The black circle represents the selected atom, whereas the grey circles in graph G represent the neighbouring masked atoms, the grey lines in graph G and the grey ovals in graph H represent the neighbouring masked bonds, and the grey lines in graph H represent the neighbouring masked bond angles.
GeoGNN learns the representation vectors of atoms and bonds iteratively. For the kth iteration, the representation vectors of atom u and bond (u, v) are denoted by hu and huv, respectively. To connect the atom–bond graph G and bond–angle graph H, the representation vectors of the bonds are taken as the communication links between G and H. In the first step, the bonds’ representation vectors are learned by aggregating messages from the neighbouring bonds and corresponding bond angles in the bond–angle graph H. In the second step, the atoms’ representation vectors are learned by aggregating messages from the neighbouring atoms and the corresponding bonds in the atom–bond graph G. Finally, the molecular representation hG is obtained by pooling over the atoms’ representations. See the Methods for details on the GeoGNN architecture.
Geometry-level self-supervised learning tasks
To further boost the generalization ability of GeoGNN, we propose three geometry-level self-supervised learning tasks to pre-train GeoGNN: (1) the bond lengths prediction; (2) the bond angles prediction; (3) the atomic distance matrices prediction. The bond lengths and bond angles describe the local spatial structures, whereas the atomic distance matrices describe the global spatial structures.
Local spatial structures
Bond lengths and angles are the most important molecular geometrical parameters: the former is the distance between two joint atoms in a molecule, reflecting the bond strength between the atoms, whereas the latter is the angle connecting two consecutive bonds, including three atoms, describing the local spatial structure of a molecule.
To learn the local spatial structures, we construct self-supervised learning tasks that predict bond lengths and angles. First, for a molecule, we randomly select 15% of atoms. For each selected atom, we extract the one-hop neighbourhood of this atom, including the adjacent atoms and bonds, as well as the bond angles formed by that selected atom. Second, we mask the features of these atoms, bonds and bond angles in the one-hop neighbourhood. The representation vectors of the extracted atoms and bonds at the final iteration of GeoGNN are used to predict the extracted bond lengths and bond angles. Self-supervised learning tasks based on bond lengths and bond angles are shown on left and middle of Fig. 2b. We design a regression loss function that penalizes the error between the predicted bond lengths/angles and the labels, whose details can be referred to in the Methods. The task of predicting the local spatial structures can be seen as a node-level self-supervised learning task.
Global spatial structures
Except for the tasks for learning local spatial structures, we also design the atomic distance matrices prediction task for learning the global molecular geometry. We construct the atomic distance matrix for each molecule based on the three-dimensional coordinates of the atoms. We then predict the elements in the distance matrix, shown on the right of Fig. 2b.
Note that for two molecules with the same topological structures, the spatial distances between the corresponding atoms could vary greatly; thus, for a molecule, rather than take predicting atomic distance matrix as a regression problem, we take it as a multi-class classification problem by projecting the atomic distances into 30 bins with equal stride. Details on the designed loss function can be found in the Methods. The task predicting the bond lengths can be seen as a special case of the task predicting the atomic distances. The former focuses more on the local spatial structures, whereas the latter focuses more on the distribution of the global spatial structures.
To pre-train GeoGNN, we consider both the local spatial structures and global spatial structures for each molecule by summing up the corresponding loss functions.
Experiments
To thoroughly evaluate the performance of GEM, we compare it with multiple SOTA methods on multiple benchmark datasets from MoleculeNet21 with various molecular property prediction tasks, such as physical, chemical and biophysics.
Pre-training settings
Datasets
We use 20 million unlabelled molecules sampled from Zinc1522, a public access database that contains purchasable drug-like compounds, to pre-train GeoGNN. We randomly sample 90% of the molecules for training and the remaining for evaluation.
Self-supervised learning task settings
We utilize geometry- and graph-level tasks to pre-train GeoGNN. For the former, we utilize the Merck molecular force field (MMFF94)23 function in RDKit to obtain the simulated three-dimensional coordinates of the atoms in the molecules. The geometric features of the molecule—including bond lengths, bond angles and atomic distance matrices—are calculated by the simulated three-dimensional coordinates. We predict the molecular fingerprints for the graph-level tasks. The graph-level tasks can be formulated as a set of binary classification problems, where each bit of the fingerprints corresponds to one binary classification problem. Two kinds of fingerprints are used: (1) the molecular access system (MACCS) key24 and (2) the extended-connectivity fingerprint (ECFP)25.
Molecular property prediction settings
Datasets and splitting method
We conduct experiments on multiple molecular benchmarks from the MoleculeNet21, including both classification and regression tasks26,27,28,29,30,31. Following the previous work11, we split all the datasets with scaffold split32, which splits molecules according to the their scaffold (molecular substructure). Scaffold split is a more challenging splitting method and can better evaluate the generalization ability of the models on out-of-distribution data samples.
GNN architecture
We use the AGGREGATE and COMBINE functions defined in the graph isomorphism network (GIN)19. Residual connections33, layer normalization34 and graph normalization35 are incorporated into GIN to further improve the performance. We also use the average pooling as the READOUT function to obtain the graph representation.
Evaluation metrics
As suggested by the MoleculeNet21, we use the average ROC-AUC36 as the evaluation metric for the classification datasets. ROC-AUC (area under the receiver operating characteristic curve) is used to evaluate the performance of binary classification tasks, for which higher is better. With respect to the regression datasets, we use root mean square error (RMSE) for FreeSolv37, ESOL38 and Lipo39, whereas we use mean average error (MAE) for QM740, QM841 and QM942. We execute four independent runs for each method and report the mean and the standard deviation of the metrics.
Baselines
We compare the proposed method with various competitive baselines. D-MPNN43, AttentiveFP44, SGCN16, DimeNet17 and HMGNN6 are the GNNs without pre-training, among which, SGCN, DimeNet and HMGNN incorporate three-dimensional geometry information; N-Gram45, PretrainGNN11 and GROVER4 are the methods with pre-training. N-Gram assembles the node embeddings in short walks in the graph and then leverages Random Forest or XGBoost to predict the molecular properties. PretrainGNN implements several types of self-supervised learning tasks, among which we report the best result. GROVER integrates GNN into Transformer with two self-supervised tasks, and we report the results of GROVERbase and GROVERlarge with different network capacity.
Experimental results
Overall performance
The overall performance of GEM along with other methods is summarized in Table 1. We have the following observations: (1) GEM achieves SOTA results on 14 out of 15 datasets. On the regression tasks, GEM achieves an overall relative improvement of 8.8% on average compared with the previous SOTA results in each dataset. On the classification tasks, GEM achieves an overall relative improvement of 4.7% on the average ROC-AUC compared with the previous SOTA result from D-MPNN. (2) GEM achieves more substantial improvements on the regression datasets than the classification datasets. We guess that the regression datasets focus on predicting the quantum chemical properties, which are highly correlated to molecular geometries.
Contribution of GeoGNN
We investigate the effect of GeoGNN without pre-training on the regression datasets, including the properties of quantum mechanics and physical chemistry, which are highly correlated to molecular geometries. GeoGNN is compared with multiple GNN architectures, including: (1) the commonly used GNN architectures, GIN19, GAT46 and GCN47; (2) recent works incorporating three-dimensional molecular geometry, SGCN16, DimeNet17 and HMGNN6; (3) the architectures specially designed for molecular representation, D-MPNN43, AttentiveFP44 and GTransformer4. From Table 2, we can conclude that GeoGNN considerably outperforms other GNN architectures on all the regression datasets since GeoGNN incorporates geometrical parameters even though the three-dimensional coordinates of the atoms are simulated. The overall relative improvement is 7.9% compared with the best results of previous methods.
Contribution of geometry-level tasks
To study the effect of the proposed geometry-level self-supervised learning tasks, we apply different types of self-supervised learning tasks to pre-train GeoGNN on the regression datasets. In Table 3, ‘Without pre-train’ denotes the GeoGNN network without pre-training, ‘Geometry’ denotes our proposed geometry-level tasks, ‘Graph’ denotes the graph-level task that predicts the molecular fingerprints and ‘Context’4 denotes a node-level task that predicts the atomic context. In general, the methods with geometry-level tasks are better than that without it. Furthermore, ‘Geometry’ performs better than ‘Geometry + Graph’ in the regression tasks, which may due to the weak connection between molecular fingerprints and the regression tasks.
Pre-trained representations visualization
To intuitively observe the representations that the self-supervised tasks (without downstream fine-tuning) have learned, we visualize the representations by mapping them to the two-dimensional space by t-SNE algorithm48, whose details can be found in the Supplementary Information. The Davies Bouldin index49 is calculated to measure the separation of clusters. The lower the Davies Bouldin index, the better the separation of the clusters. Here we test whether the pre-training methods are able to distinguish molecules with valid geometry (generated from RDKit) from molecules with invalid geometry (random generated). We randomly select 1,000 molecules from ZINC. For each molecule, we generate the valid and invalid geometry. As shown in Fig. 3a, both the graph-level and geometry-level pre-training methods can better distinguish the valid geometry from invalid geometry compared to not pre-trained. Besides, the geometry-level pre-training can further decrease the Davies Bouldin Index to 2.63, compared with 7.88 of the graph-level.

a, A pre-trained representation visualization comparing different self-supervised methods. The valid geometry cluster contains molecules with geometry generated by RDKit, whereas the invalid geometry cluster contains those with randomly generated geometry. b, A representation visualization comparing different qualities of geometries. The high cluster contains molecules with high HOMO–LUMO gaps, whereas the low cluster contains those with low HOMO–LUMO gaps. c, MAE difference (the lower the better) on QM9 between baseline GeoGNN with other GeoGNN variants.
Impact of the quality of geometry
To investigate the impact of the quality of geometry, we first compare GeoGNN, which adopts the default force field MMFF, with GeoGNN (UFF), which adopts the universal force field (UFF)50, on dataset QM9. GeoGNN and GeoGNN (UFF) achieve similar performance, as shown in Fig. 3c. The impact of more precise three-dimensional coordinates provided by dataset QM9 (calculated by DFT51) is also investigated. GeoGNN (precise 3D) achieves a great improvement of about 12% compared with the baseline GeoGNN.
Furthermore, Fig. 3b shows the representation visuals for different qualities of molecular geometry. GeoGNN (without 3D) is a variant of GeoGNN that masks all the geometry features with zeros, GeoGNN is the baseline that utilizes coarse three-dimensional coordinates, and GeoGNN (precise 3D) utilizes precise 3D coordinates generated by DFT. We equally divide 2,000 molecules from QM9 into two clusters, one with high HOMO–LUMO gaps and the other with low HOMO–LUMO gaps. We test the ability of different models to distinguish these two group of molecules. Visually, we observe that GeoGNN can better separate the clusters than GeoGNN (without 3D), whereas GeoGNN (precise 3D) works better than GeoGNN. The differences in Davies Bouldin index support the observations.
Contributions of atom–bond and bond–angle graphs
We evaluate the contributions of the atom–bond and bond–angle graphs in GeoGNN on dataset QM9, as shown in Fig. 3c. Atom–bond graph utilizes the atom–bond graph only and pool over the representations of the atoms to estimate the properties, whereas bond–angle graph utilizes the bond–angle graph only and pools over the representations of bonds. GeoGNN, which consists of both the atom–bond and bond–angle graphs, performs better than the above two variants, indicating that both the atom–bond and bond–angle graphs contribute to the performance.
Related work
Molecular representation
Current molecular representations can be categorized into three types: molecular fingerprints, sequence-based representations and graph-based representations.
Molecular fingerprints
Molecular fingerprints such as ECFP25 and MACCS24 are molecular descriptors. Fingerprints are handcrafted representations—widely used by traditional machine learning methods3,52,53,54—that encode a molecule into a sequence of bits according to the molecules’ topological substructures. Although fingerprints can represent the presence of the substructures in the molecules, they suffer from bit collisions and vector sparsity, limiting their representation power.
Sequence-based representations
Some studies3,55 take SMILES strings56 that describe the molecules by strings as inputs, and leverage sequence-based models such as Recurrent Neural Networks and Transformer57,58 to learn the molecular representations; however, it is difficult for sequence-based methods to comprehend the syntax of SMILES. For example, two adjacent atoms may be far apart in the text sequence. Besides, a small change in a SMILES string can lead to a large change in the molecular structure.
Graph-based representations
Many works3,4,5,6,18 have showcased the great potential of graph neural networks on modelling molecules by taking each atom as a node and each chemical bond as an edge. For example, AttentiveFP44 proposes to extend graph attention mechanism to learn aggregation weights. Meanwhile, a group of studies have tried to incorporate three-dimensional geometry information: (1)13,14,15 take partial geometry information as features, such as atomic distances; (2)16 proposed a spatial graph convolution that uses relative position vectors between atoms as input features; (3)17 proposed a message passing scheme based on bonds and transform messages from angles.
Pre-training for GNNs
Self-supervised learning7,8,9,10,59 has achieved great success in natural language processing, computer vision and other domains; it trains unlabelled samples in a supervised manner to alleviate the overfitting issue and improve data utilization efficiency. Some studies4,11 recently applied self-supervised learning methods to GNNs for molecular property prediction to overcome the insufficiency of the labelled samples. These works learn the molecular representation vectors by exploiting the node- and graph-level tasks, where the node-level tasks learn the local domain knowledge by predicting the node properties and the graph-level tasks learn the global domain knowledge by predicting biological activities. Although existing self-supervised learning methods can boost the generalization ability, they neglect the spatial knowledge that is strongly related to the molecular properties.
Conclusion
Efficient molecular representation learning is crucial for molecular property prediction. Existing works that apply pre-training methods for molecular property prediction fail to utilize the molecular geometries described by bonds, bond angles and other geometrical parameters. To this end, we design a geometry-based GNN and multiple geometry-level self-supervised learning methods capture the molecular spatial knowledge. Extensive experiments were conducted to verify the effectiveness of GEM, comparing it with multiple competitive baselines. GEM considerably outperforms other methods on multiple benchmarks. In the future we will try to adopt the proposed framework to more molecular tasks, especially the protein–ligand affinity prediction task that requires lots of three-dimensional samplings.
Methods
Preliminary for GNNs
Graph neural networks is a message passing neural networks. More concretely, given a node v, its representation vector \({\mathbf{h}}_{v}^{(k)}\) at the kth iteration is formalized by
where \({{{\mathcal{N}}}}(v)\) is the set of neighbours of node v, AGGREGATE(k) is the aggregation function for aggregating messages from a node’s neighbourhood, and COMBINE(k) is the update function for updating the node representation. We initialize \({\mathbf{h}}_{v}^{(0)}\) by the feature vector of node v, that is, \({\mathbf{h}}_{v}^{(0)}={\mathbf{x}}_{v}\).
READOUT function is introduced to integrate the nodes’ representation vectors at the final iteration so as to gain the graph’s representation vector hG, which is formalized as
where K is the number of iterations. In most cases, READOUT is a permutation invariant pooling function, such as summation and maximization. The graph’s representation vector hG can then be used for downstream task predictions.
GeoGNN
The GeoGNN architecture encodes the molecular geometries by modelling two graphs: the atom–bond and bond–angle graphs, under which the representation vectors of atoms and bonds are learned iteratively. More concretely, the representation vectors of atom u and bond (u, v) for the kth iteration are denoted by hu and huv, respectively. We initialize \({\mathbf{h}}_{u}^{(0)}={\mathbf{x}}_{u}\) and \({\mathbf{h}}_{uv}^{(0)}={\mathbf{x}}_{uv}\).
Given bond (u, v), its representation vector \({\mathbf{h}}_{uv}^{(k)}\) at the kth iteration is formalized by
Here, \({{{\mathcal{N}}}}(u)\) and \({{{\mathcal{N}}}}(v)\) denote the neighbouring atoms of u and v, respectively; \(\{(u,w):w\in {{{\mathcal{N}}}}(u)\}\cup \{(v,w):w\in {{{\mathcal{N}}}}(v)\}\) are the neighbouring bonds of (u, v). AGGREGATEbond−angle is the message aggregation function and COMBINEbond−angle is the update function for bond–angle graph H. In this way, the information from the neighbouring bonds and the corresponding bond angles is aggregated into \({\mathbf{a}}_{uv}^{(k)}\). The representation vector of bond (u, v) is then updated according to the aggregated information. With the learned representation vectors of the bonds from bond–angle graph \({{{\mathcal{H}}}}\), given an atom u, its representation vector \({\mathbf{h}}_{u}^{(k)}\) at the kth iteration can be formalized as
Similarly, \({{{\mathcal{N}}}}(u)\) denotes the neighbouring atoms of atom u, AGGREGATEatom−bond is the message aggregation function for atom–bond graph G, and COMBINEatom−bond is the update function. For atom u, messages are aggregated from the neighbouring atoms and the corresponding bonds. Note that, the messages of the bonds are learned from the bond–angle graph H. The aggregated messages then update the representation vector of atom u.
The representation vectors of the atoms at the final iteration are integrated to gain the molecular representation vector hG by the READOUT function, which is formalized as
where K is the number of iterations. The molecule’s representation vector hG is used to predict the molecular properties.
Geometry-level self-supervised learning tasks
Local spatial structures
The self-supervised tasks for local spatial information are designed to learn two important molecular geometrical parameters, the bond lengths and the bond angles. The loss functions of the self-supervised tasks are defined as follows:
Here, \({L}_{\mathrm{length}}({{{\mathcal{E}}}})\) is the loss function for bond lengths, with \({{{\mathcal{E}}}}\) as the set of bonds; \({L}_{\mathrm{angle}}({{{\mathcal{A}}}})\) is the loss function of bond angles, with \({{{\mathcal{A}}}}\) as set of angles; K is the number of iterations for GeoGNN; flength(⋅) is the network predicting the bond lengths; and fangle(⋅) is the network predicting the bond angles; luv denotes the length of the bond connecting atoms u and v; ϕuvw denotes the degree of the bond angle connecting bonds (u, v) and (v, w).
Global spatial structures
The self-supervised tasks for global spatial information are designed to learn the atomic distance matrices between all atom pairs. Each element of the distance matrices is the three-dimensional distance between two atoms. We use duv to denote the distance between two atoms u and v in the molecule. For the atomic distance prediction task, we clip the distance with the range from 0 Å to 20 Å and project it into 30 bins with equal stride. The loss function of the self-supervised tasks is defined as follows:
where \({{{\mathcal{V}}}}\) is the set of atoms, fdistance(⋅) is the network predicting the distribution of atomic distances, the bin(⋅) function is used to discretize the atomic distance duv into a one-hot vector and \({\mathrm{log}}(\cdot )\) is the logarithmic function.
Data availability
The self-supervised data used in our study are publicly available in ZINC (https://zinc.docking.org/tranches/home/), whereas the downstream benchmarks can be downloaded from MoleculeNet (https://moleculenet.org/datasets-1).
Code availability
The source code of this study providing the geometry-based GNN and several geometry-level self-supervised learning methods is freely available at GitHub (https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/pretrained_compound/ChemRL/GEM) to allow replication of the results. The version used for this publication is available at https://doi.org/10.5281/zenodo.5781821.
References
Shen, J. & Nicolaou, C. A. Molecular property prediction: recent trends in the era of artificial intelligence. Drug Discov. Today Technol. 32–33, 29–36 (2020).
Wieder, O. et al. A compact review of molecular property prediction with graph neural networks. Drug Discov. Today Technol. 37, 1–12 (2020).
Huang, K. et al. DeepPurpose: a deep learning library for drug-target interaction prediction. Bioinformatics 36, 5545–5547 (2020).
Rong, Y. et al. Self-supervised graph transformer on large-scale molecular data. In Advances in Neural Information Processing Systems 33: Annual Conference on Neural Information Processing Systems 2020 (eds Larochelle, H. et al.) 12559–12571 (NeurIPS 2020).
Shindo, H. & Matsumoto, Y. Gated graph recursive neural networks for molecular property prediction. Preprint at https://arxiv.org/abs/1909.00259 (2019).
Shui, Z. & Karypis, G. Heterogeneous molecular graph neural networks for predicting molecule properties. In 20th IEEE International Conference on Data Mining (eds Plant, C. et al.) 492–500 (IEEE, 2020).
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies (eds Burstein, J. et al.) 4171–4186 (Association for Computational Linguistics, 2019).
He, P., Liu, X., Gao, J. & Chen, W. DeBERTa: decoding-enhanced BERT with disentangled attention. In 9th International Conference on Learning Representations (ICLR, 2021).
Doersch, C., Gupta, A. & Efros, A. A. Unsupervised visual representation learning by context prediction. In International Conference on Computer Vision (IEEE Computer Society, 2015).
Gidaris, S., Singh, P. & Komodakis, N. Unsupervised representation learning by predicting image rotations. In 6th International Conference on Learning Representations (ICLR, 2018).
Hu, W. et al. Strategies for pre-training graph neural networks. In 8th International Conference on Learning Representations (ICLR, 2020).
Peleg-Shulman, T., Najajreh, Y. & Gibson, D. Interactions of cisplatin and transplatin with proteins: comparison of binding kinetics, binding sites and reactivity of the pt-protein adducts of cisplatin and transplatin towards biological nucleophiles. J. Inorg. Biochem. 91, 306–311 (2002).
Schütt, K. et al. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions. In Advances in Neural Information Processing Systems 30: Annual Conference on Neural Information Processing Systems 2017 (eds Guyon, I. et al.) 991–1001 (NeurIPS, 2017).
Li, J., Xu, K., Chen, L., Zheng, Z. & Liu, X. GraphGallery: a platform for fast benchmarking and easy development of graph neural networks based intelligent software. In 43rd IEEE/ACM International Conference on Software Engineering: Companion Proceedings 13–16 (IEEE, 2021).
Maziarka, L. et al. Molecule attention transformer. Preprint at https://arxiv.org/abs/2002.08264 (2020).
Danel, Tomasz et al. Spatial graph convolutional networks. In Neural Information Processing—27th International Conference, ICONIP 2020 Vol. 1333 (eds Yang, H. et al.) 668–675 (Springer, 2020).
Klicpera, J., Groß, J. & Günnemann, S. Directional message passing for molecular graphs. In 8th International Conference on Learning Representations (ICLR, 2020).
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E. Neural message passing for quantum chemistry. In Proc. 34th International Conference on Machine Learning Vol. 70 (eds Precup, D. & Teh, Y. W.) 1263–1272 (PMLR, 2017).
Xu, K., Hu, W., Leskovec, J. & Jegelka, S. How powerful are graph neural networks? In 7th International Conference on Learning Representations (ICLR, 2019).
Sun, F.-Y., Hoffmann, J., Verma, V. & Tang, J. Infograph: unsupervised and semi-supervised graph-level representation learning via mutual information maximization. In 8th International Conference on Learning Representations (ICLR, 2020).
Wu, Z. et al. Moleculenet: a benchmark for molecular machine learning. Chem. Sci. 9, 513–530 (2018).
Sterling, T. & Irwin, J. J. ZINC 15—ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015).
Halgren, T. A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 17, 490–519 (1996).
Durant, J. L., Leland, B. A., Henry, D. R. & Nourse, J. G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 42, 1273–1280 (2002).
Rogers, D. & Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754 (2010).
Subramanian, G., Ramsundar, B., Pande, V. & Denny, R. A. Computational modeling of β-secretase 1 (bace-1) inhibitors using ligand based approaches. J. Chem. Inf. Model. 56, 1936–1949 (2016).
Martins, I. F., Teixeira, A. L., Pinheiro, L. & Falcão, A. O. A Bayesian approach to in silico blood–brain barrier penetration modeling. J. Chem. Inf. Model. 52, 1686–1697 (2012).
Richard, A. M. et al. Toxcast chemical landscape: paving the road to 21st century toxicology. Chem. Res. Toxicol. 29, 1225–1251 (2016).
Gayvert, K. M., Madhukar, N. S. & Elemento, O. A data-driven approach to predicting successes and failures of clinical trials. Cell Chem. Biol. 23, 1294–1301 (2016).
Huang, R. et al. Editorial: Tox21 challenge to build predictive models of nuclear receptor and stress response pathways as mediated by exposure to environmental toxicants and drugs. Front. Environ. Sci. 3, 85 (2017).
Kuhn, M., Letunic, I., Jensen, L. J. & Bork, P. The SIDER database of drugs and side effects. Nucl. Acids Res. 44, 1075–1079 (2016).
Ramsundar, B., Eastman, P., Walters, P. & Pande, V. Deep Learning for the Life Sciences: Applying Deep Learning to Genomics, Microscopy, Drug Discovery, and More (O’Reilly Media, 2019).
He, K., Zhang, X., Ren, S. & Sun, J. Deep residual learning for image recognition. In 2016 IEEE Conference on Computer Vision and Pattern Recognition 770–778 (IEEE, 2016).
Ba, L. J., Kiros, J. R. & Hinton, G. E. Layer normalization. In NIPS 2016 Deep Learning Symposium recommendation (NIPS, 2016).
Chen, Y., Tang, X., Qi, X., Li, C.-G. & Xiao, R. Learning graph normalization for graph neural networks. Preprint at https://arxiv.org/abs/2009.11746 (2020).
Bradley, A. P. The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recognit. 30, 1145–1159 (1997).
Mobley, D. L. & Guthrie, J. P. FreeSolv: a database of experimental and calculated hydration free energies, with input files. J. Comput. Aided Mol. Des. 28, 711–720 (2014).
Delaney, J. S. ESOL: estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Model. 44, 1000–1005 (2004).
Gaulton, A. et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucl. Acids Res. 40, 1100–1107 (2012).
Blum, L. C. & Reymond, J.-L. 970 Million druglike small molecules for virtual screening in the chemical universe database GDB-13. J. Am. Chem. Soc. 131, 8732–8733 (2009).
Ramakrishnan, R., Hartmann, M., Tapavicza, E. & AnatoleVonLilienfeld, O. Electronic spectra from TDDFT and machine learning in chemical space. J. Chem. Phys. 143, 084111 (2015).
Ruddigkeit, L., van Deursen, R., Blum, L. C. & Reymond, J.-L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model. 52, 2864–2875 (2012).
Yang, K. et al. Analyzing learned molecular representations for property prediction. J. Chem. Inf. Model. 59, 3370–3388 (2019).
Xiong, Z. et al. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 63, 8749–8760 (2020).
Liu, S., Demirel, M. F. & Liang, Y. N-gram graph: simple unsupervised representation for graphs, with applications to molecules. In Advances in Neural Information Processing Systems 32: Annual Conference on Neural Information Processing Systems 2019 (eds Wallach, H. M. et al.) 8464–8476 (NeurIPS, 2019).
Velickovic, P. et al. Graph attention networks. In 5th International Conference on Learning Representations (ICLR, 2017).
Kipf, T. N. & Welling, M. Semi-supervised classification with graph convolutional networks. In 5th International Conference on Learning Representations (ICLR, 2017).
van der Maaten, L. Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res. 15, 3221–3245 (2014).
Davies, D. L. & Bouldin, D. W. A cluster separation measure. IEEE Trans. Pattern Anal. Mach. Intell 1, 224–227 (1979).
Rappé, A. K., Casewit, C. J., Colwell, K. S., Goddard, W. A. III & Skiff, W. M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 114, 10024–10035 (1992).
Gross, E.K.U. & Dreizler, R. M. Density Functional Theory Vol. 337 (Springer, 2013).
Cereto-Massagué, A. et al. Molecular fingerprint similarity search in virtual screening. Methods 71, 58–63 (2015).
Coley, C. W., Barzilay, R., Green, W. H., Jaakkola, T. S. & Jensen, K. F. Convolutional embedding of attributed molecular graphs for physical property prediction. J. Chem. Inf. Model. 57, 1757–1772 (2017).
Duvenaud, D. et al. Convolutional networks on graphs for learning molecular fingerprints. In Advances in Neural Information Processing Systems 28: Annual Conference on Neural Information Processing Systems (eds Cortes, C. et al.) 2224–2232 (NeurIPS, 2015).
Goh, G. B., Hodas, N. O., Siegel, C. & Vishnu, A. SMILES2Vec: an interpretable general-purpose deep neural network for predicting chemical properties. Preprint at https://arxiv.org/abs/1712.02034 (2018).
Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988).
Zaremba, W., Sutskever, I. & Vinyals, O. Recurrent neural network regularization. Preprint at https://arxiv.org/abs/1409.2329 (2014).
Vaswani, A. et al. Attention is all you need. In Advances in Neural Information Processing Systems 30: Annual Conferenceon Neural Information Processing Systems 2017 5998–6008 (NeurIPS, 2017).
Li, P. et al. Learn molecular representations from large-scale unlabeled molecules for drug discovery. Preprint at https://arxiv.org/abs/2012.11175 (2020).
Acknowledgements
This work is supported by National Engineering Research Center of Deep Learning Technology and Applications.
Author information
Authors and Affiliations
Contributions
X.F., F.W., H. Wu and H. Wang led the research. L.L., X.F. and F.W. contributed technical ideas. L.L., J.L., D.H., S.Z. and X.F. developed the proposed method. X.F., L.L., S.Z. and J.Z. developed analytics. X.F., L.L, F.W., J.L., D.H., S.Z. and J.Z. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Details of the proposed networks and experiments.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fang, X., Liu, L., Lei, J. et al. Geometry-enhanced molecular representation learning for property prediction. Nat Mach Intell 4, 127–134 (2022). https://doi.org/10.1038/s42256-021-00438-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-021-00438-4
This article is cited by
-
Improving chemical reaction yield prediction using pre-trained graph neural networks
Journal of Cheminformatics (2024)
-
Prediction of chemical reaction yields with large-scale multi-view pre-training
Journal of Cheminformatics (2024)
-
QuanDB: a quantum chemical property database towards enhancing 3D molecular representation learning
Journal of Cheminformatics (2024)
-
Unsupervised manifold embedding to encode molecular quantum information for supervised learning of chemical data
Communications Chemistry (2024)
-
Bidirectional generation of structure and properties through a single molecular foundation model
Nature Communications (2024)
