
Abstract
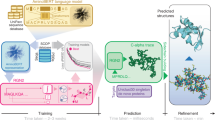
Predicting the tertiary structure of a protein from its primary sequence has been greatly improved by integrating deep learning and co-evolutionary analysis, as shown in CASP13 and CASP14. We describe our latest study of this idea, analysing the efficacy of network size and co-evolution data and its performance on both natural and designed proteins. We show that a large ResNet (convolutional residual neural networks) can predict structures of correct folds for 26 out of 32 CASP13 free-modelling targets and L/5 long-range contacts with precision over 80%. When co-evolution is not used, ResNet can still predict structures of correct folds for 18 CASP13 free-modelling targets, greatly exceeding previous methods that do not use co-evolution either. Even with only the primary sequence, ResNet can predict the structures of correct folds for all tested human-designed proteins. In addition, ResNet may fare better for the designed proteins when trained without co-evolution than with co-evolution. These results suggest that ResNet does not simply de-noise co-evolution signals, but instead may learn important protein sequence–structure relationships. This has important implications for protein design and engineering, especially when co-evolutionary data are unavailable.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Data availability
The PDB IDs of the human-designed proteins are available in Supplementary Data 4. The domain sequences determined by our own CASP13 server for the CASP13 targets are available in Supplementary Data 5. The official domain sequences of the CASP13 targets and their corresponding PDB IDs are available at the CASP13 web site, https://predictioncenter.org/casp13/index.cgi. The training data, including the multiple sequence alignment and ground truth files, are available at https://zenodo.org/record/4679643.
Code availability
The source code is available at https://github.com/j3xugit/RaptorX-3DModeling/ or https://doi.org/10.5281/zenodo.4642250 and the server is available at http://raptorx.uchicago.edu/. In addition to template-free protein structure prediction, this package also supports comparative protein structure modelling, that is, building protein 3D models from templates by deep learning.
References
De Juan, D., Pazos, F. & Valencia, A. Emerging methods in protein co-evolution. Nat. Rev. Genet. 14, 249–261 (2013).
Shrestha, R. et al. Assessing the accuracy of contact predictions in CASP13. Proteins 87, 1058–1068 (2019).
Abriata, L. A., Tamo, G. E. & Dal Peraro, M.A further leap of improvement in tertiary structure prediction in CASP13 prompts new routes for future assessments. Proteins 87, 1100–1112 (2019).
Wang, S. et al. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput. Biol. 13, e1005324 (2017).
Wang, S., Sun, S. Q. & Xu, J. B. Analysis of deep learning methods for blind protein contact prediction in CASP12. Proteins 86, 67–77 (2018).
Xu, J. Distance-based protein folding powered by deep learning. Proc. Natl Acad. Sci. USA 116, 16856–16865 (2019).
Xu, J. B. & Wang, S. Analysis of distance-based protein structure prediction by deep learning in CASP13. Proteins 87, 1069–1081 (2019).
Wang, S. et al. Folding membrane proteins by deep transfer learning. Cell Syst. 5, 202–211 (2017).
Zhu, J. W. et al. Protein threading using residue co-variation and deep learning. Bioinformatics 34, 263–273 (2018).
Senior, A. W. et al. Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (CASP13). Proteins 87, 1141–1148 (2019).
Ding, W. Z. & Gong, H. P. Predicting the real-valued inter-residue distances for proteins. Adv. Sci 7, 2001314 (2020).
Yang, J. Y. et al. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl Acad. Sci. USA 117, 1496–1503 (2020).
Greener, J. G., Kandathil, S. M. & Jones, D. T. Deep learning extends de novo protein modelling coverage of genomes using iteratively predicted structural constraints. Nat. Commun. 10, 3977 (2019).
Ovchinnikov, S. et al. Protein structure determination using metagenome sequence data. Science 355, 294–297 (2017).
Li, Y. et al. Ensembling multiple raw coevolutionary features with deep residual neural networks for contact-map prediction in CASP13. Proteins 87, 1082–1091 (2019).
Kandathil, S. M., Greener, J. G. & Jones, D. T. Prediction of interresidue contacts with DeepMetaPSICOV in CASP13. Proteins 87, 1092–1099 (2019).
Marks, D. S., Hopf, T. A. & Sander, C. Protein structure prediction from sequence variation. Nat. Biotechnol. 30, 1072 (2012).
Kamisetty, H., Ovchinnikov, S. & Baker, D. Assessing the utility of coevolution-based residue–residue contact predictions in a sequence- and structure-rich era. Proc. Natl Acad. Sci. USA 110, 15674–15679 (2013).
Seemayer, S., Gruber, M. & Söding, J. CCMpred—fast and precise prediction of protein residue–residue contacts from correlated mutations. Bioinformatics 30, 3128–3130 (2014).
Liu, Y. et al. Enhancing evolutionary couplings with deep convolutional neural networks. Cell Syst. 6, 65–74 (2018).
AlQuraishi, M. End-to-end differentiable learning of protein structure. Cell Syst. 8, 292–301 (2019).
Chaudhury, S., Lyskov, S. & Gray, J. J. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 26, 689–691 (2010).
Jones, D. T. et al. MetaPSICOV: combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics 31, 999–1006 (2015).
Eickholt, J. & Cheng, J. Predicting protein residue–residue contacts using deep networks and boosting. Bioinformatics 28, 3066–3072 (2012).
Steinegger, M. & Soding, J. Clustering huge protein sequence sets in linear time. Nat. Commun. 9, 2542 (2018).
Kim, D. E., Chivian, D. & Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531 (2004).
Xu, C. F. et al. Computational design of transmembrane pores. Nature 585, 129–134 (2020).
Lu, P. L. et al. Accurate computational design of multipass transmembrane proteins. Science 359, 1042–1046 (2018).
Pan, X. J. et al. Expanding the space of protein geometries by computational design of de novo fold families. Science 369, 1132–1136 (2020).
Chen, I. M. A. et al. The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic Acids Res. 49, D751–D763 (2021).
Steinegger, M., Mirdita, M. & Soding, J. Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat. Methods 16, 603–606 (2019).
Mitchell, A. L. et al. MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res. 48, D570–D578 (2020).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
Wang, G. L. & Dunbrack, R. L. PISCES: a protein sequence culling server. Bioinformatics 19, 1589–1591 (2003).
Remmert, M. et al. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9, 173–175 (2012).
Zhang, Y. & Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005).
Mirdita, M. et al. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 45, D170–D176 (2017).
Johnson, L. S., Eddy, S. R. & Portugaly, E. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11, 431 (2010).
Loshchilov, I. & Hutter, F. Decoupled weight decay regularization. In 7th International Conference on Learning Representations (ICLR, 2019).
Zhao, F. & Xu, J. A position-specific distance-dependent statistical potential for protein structure and functional study. Structure 20, 1118–1126 (2012).
Zhou, H. Y. & Zhou, Y. Q. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 11, 2714–2726 (2002); erratum 12, 2121 (2003).
Shen, M. Y. & Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 (2006).
Zhang, Y. & Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865–871 (2004).
Xu, J. R. & Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 26, 889–895 (2010).
Acknowledgements
We thank J. Yang and I. Anishchanka for their very helpful discussions, providing trRosetta results and helping with PyRosetta. We thank I. Anishchanka for providing the MSAs built by the Baker human group for the CASP14 targets. This work is supported by National Institutes of Health grant no. R01GM089753 (J.X.) and National Science Foundation grant no. DBI1564955 (J.X.). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
J.X. conceived the whole project, implemented and tested the code, and wrote the manuscript. M.M. studied the gradient-based energy minimization algorithm and revised the manuscript. J.L. studied the deep learning algorithms, trained some ResNet models and generated the RGN results.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Machine Intelligence thanks Jeffrey Gray, Sai Pooja Mahajan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Distance matrices predicted for T0969-D1 by deep ResNet.
Distance matrices predicted for T0969-D1 by deep ResNet when co-evolution is not used (left) and used (right). Only distance predictions less than 15Å are displayed in color. In each picture, native distance and predicted distance are shown below and above the diagonal, respectively.
Supplementary information
Supplementary Information
Supplementary Fig. 1 and Table 1.
Supplementary Data 1
Data for the ablation study of contact prediction accuracy on CASP13 FM targets.
Supplementary Data 2
Detailed 3D modelling accuracy on CASP13 FM targets.
Supplementary Data 3
Data for the ablation study of 3D modelling accuracy on CASP13 FM targets.
Supplementary Data 4
Detailed 3D modelling accuracy on human-designed proteins.
Supplementary Data 5
CASP13 FM domain sequences defined by the RaptorX server in the CASP13 session.
Rights and permissions
About this article

Cite this article
Xu, J., McPartlon, M. & Li, J. Improved protein structure prediction by deep learning irrespective of co-evolution information. Nat Mach Intell 3, 601–609 (2021). https://doi.org/10.1038/s42256-021-00348-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-021-00348-5
This article is cited by
-
Computational drug development for membrane protein targets
Nature Biotechnology (2024)
-
Generating mutants of monotone affinity towards stronger protein complexes through adversarial learning
Nature Machine Intelligence (2024)
-
Deep transfer learning for inter-chain contact predictions of transmembrane protein complexes
Nature Communications (2023)
-
Multi-domain and complex protein structure prediction using inter-domain interactions from deep learning
Communications Biology (2023)
-
AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms
Communications Biology (2023)
