
Abstract
The coronavirus disease 2019 (COVID-19) pandemic continues to represent a global public health issue. The viral main protease (Mpro) represents one of the most attractive targets for the development of antiviral drugs. Herein we report peptidyl nitroalkenes exhibiting enzyme inhibitory activity against Mpro (Ki: 1–10 μM) good anti-SARS-CoV-2 infection activity in the low micromolar range (EC50: 1–12 μM) without significant toxicity. Additional kinetic studies of compounds FGA145, FGA146 and FGA147 show that all three compounds inhibit cathepsin L, denoting a possible multitarget effect of these compounds in the antiviral activity. Structural analysis shows the binding mode of FGA146 and FGA147 to the active site of the protein. Furthermore, our results illustrate that peptidyl nitroalkenes are effective covalent reversible inhibitors of the Mpro and cathepsin L, and that inhibitors FGA145, FGA146 and FGA147 prevent infection against SARS-CoV-2.
Similar content being viewed by others

Introduction
The impact of SARS-CoV-2 pandemic (COVID-19) has made the search for new therapies against coronaviruses urgent. The pandemic has resulted so far in over 600 million infections and over 6 million deaths worldwide, according to World Health Organization1. The evolution of SARS-CoV-2 virus has resulted in several highly contagious SARS-CoV2 strains that evade antibodies targeting the receptor binding domain and threaten the effectiveness of the current vaccines2,3,4.
Among approved antivirals for the treatment of COVID-19 are nucleoside derivatives remdesivir4,5 and molnupiravir5,6, with uncertain efficacy for certain types of patients, and Paxlovid6,7, a combination of the protease inhibitor nirmatrelvir and ritonavir, a metabolic booster that increase the effectiveness of the protease. Despite the remarkable efficacy of Paxlovid, it cannot be administered to patients with liver or kidney dysfunction. Furthermore, ritonavir, which blocks the rapid metabolism of nirmatrelvir by CYP3A, interacts in turn with other drugs limiting its use. These issues demonstrate that there is an urgent need to find new antivirals for SARS-CoV-2 and for other coronavirus outbreaks in the future.
During the replication cycle the coronavirus expresses two overlapping polyproteins, (pp1a and pp1b) and four structural proteins from the viral RNA8. In order to liberate the mature viral proteins required for replication, these polyproteins must be properly processed. There are two proteases coded in the viral genome, known as 3-chymotrypsin-like protease (3CLpro, 3CLP or nsp5, also termed main protease Mpro) and papain-like protease (PLpro or nsp3). Most of the cleavages are carried out by Mpro9,10. Both Mpro and PLpro are cysteine proteases with different site specificities.
Mpro is a three-domain cysteine protease essential for most maturation events within the precursor polyprotein9,10,11. The active protease is a homodimer. The active site is made up by a non-canonical Cys-His dyad located in the cleft between domains I and II10,11,12. Mpro hydrolyzes proteins predominantly between a P1 glutamine and a small P1′ amino acid, such as alanine, serine or glycine. For the P2 position, leucine is the most common amino acid in the sequence specificity for coronaviruses. There is no human homolog of Mpro which makes it an ideal antiviral drug target13,14,15,16.
Peptidyl compounds have been shown to inhibit Mpro in in vitro assays and to prevent infection by SARS-CoV-2 in cell culture17. The warhead of these reported inhibitors are carbonyl groups [aldehydes18, ketoamides19, ketones20], nitriles (as marketed inhibitor nirmatrelvir)21 and enoates22. Mpro inhibitors with new reactive warheads, chlorofluoroacetamide23, dichloroacetamide24, and nirmatrelvir analogs25, have been designed with potent enzymatic inhibition and antiviral activity. In this context, the nitroalkene moiety has been previously reported by us as a valid warhead for inhibitors against cysteine proteases belonging to the papain family26. Irreversible inhibitors such as enoates can give rise to undesired side reactions. Alternatively, nitroalkenes represent Michael acceptor inhibitors following a reversible mode-of-action due to the low basicity of the nitronate intermediate27. Computational studies previously reported by us pointed to nitroalkenes as promising inhibitors of SARS-CoV-2 Mpro28. Interestingly, peptidyl nitroalkenes have been shown to act aspotent inhibitors of human cathepsin L (CatL) as we previously reported27. CatL has been also recognized as a potential target for the search of drugs against COVID-19 as it is found to enable viral cell entry by activating the SARS-CoV-2 spike protein by cleavage29,30,31,32. Dual inhibition against SARS-CoV-2 Mpro and human Cathepsin-L has been reported33,34.
Analysis of interaction energies between the substrate (the peptide in the proteolysis reaction or the inhibitor in the case of the inhibition reaction) and the different binding pockets of SARS-CoV-2 Mpro based on multiscale quantum mechanics/molecular mechanics (QM/MM) studies, indicated that they are dominated by those in the P1:::S1 site28,35,36. However, the recognition portion dictates how the inhibitor is accommodated in the active site, which in turn affects the subsequent chemical reaction step. Consequently, the reactivity of the warhead and the favorable interactions between the recognition portion and the active site of the enzyme must be considered to design an efficient inhibitor36. In all, the experience accumulated based on the results derived from previous studies on this and other cysteine proteases can be used to guide the design of new compounds, and QM/MM simulations can be considered a useful tool to get a detailed description of the chemical steps of the inhibition of protein targets by covalent inhibitors.
Based on our previous studies on proteolysis reaction of the SARS-CoV-2 Mpro35 and its inhibition mechanisms by peptidyl inhibitors with different warheads28,35,36, we have designed and synthesized six peptidyl inhibitors with a nitroalkene warhead. These six inhibitors were able to inhibit the Mpro in vitro activity in the low micromolar range, and three of them were found to prevent SARS-CoV-2 infection in cell culture in the low micromolar range. In addition, these compounds were also tested against cathepsin L, a key enzyme for the viral entry into the cells. The crystal structures of Mpro in complex with these two most active inhibitors were solved to provide detailed information about the binding to SARS-CoV-2 Mpro. Molecular dynamics (MD) simulations with multiscale QM/MM potentials calculations were carried out to obtain the full free energy landscape of the inhibition reaction with the two most active inhibitors, confirming the interactions established with the active site residues of SARS-CoV-2 Mpro as well as their mechanism of action for the enzyme-inhibitor covalent complexes formation.
Results
Design and synthesis of the peptidyl nitroalkenes inhibitors
Six inhibitors were designed and synthesized: three of them having the typical coronaviral protease glutamine surrogate (gamma-lactam) at the P1 site and a L-leucine at the P2 site (FGA145, FGA146 and FGA147); the other three inhibitors display the typical cathepsin like inhibitors backbone: two having a L-homophenylalanine at the P1 site and a L-leucine at the P2 site (FGA159 and FGA177), and one having a L-homophenylalanine at the P1 site and a L-phenylalanine at the P2 site (FGA86). The design of the inhibitors was based upon nitroalkene inhibitors reported by us26,27 and previous Mpro inhibitors: compound FGA146 is a direct analog of the Pfizer human SARS Mpro inhibitor PF-0083523120, which is the progenitor for the clinically used inhibitor nirmatrelvir and FGA147 is a direct nitroalkene analog of the well-established feline coronavirus protease inhibitor GC37637.
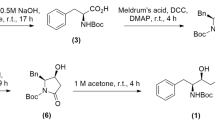
For the synthesis of the ones having the glutamine surrogate at P1, the N-Boc protected amino alcohol 1 was prepared first as previously reported starting from L-glutamic acid38. The alcohol was then submitted to oxidation followed by a nitroaldol reaction with nitromethane in a one-pot procedure. The mixture of nitroaldols was then transformed into the corresponding inhibitors following a three-step sequence: Boc deprotection, peptide coupling and then dehydration through mesylate activation (Fig. 1). For the synthesis of the other three inhibitors having a homophenylalanine at the P1 site, N-Boc protected homophenyl alaninal was reacted with nitromethane and the nitroaldols were coupled with the corresponding peptide with free carboxylic terminus (Fig. 1, Table 1) (see Supplementary Experimental procedures for the preparation of the compounds).

Inhibitors FGA145, FGA146 and FGA147 were prepared through a common synthetic route starting from Boc-L-glutamic acid. FGA77, FGA86 and FGA159 were prepared through a synthetic route starting from Boc-L-homophenyl alaninal.
Inhibition of the Mpro activity by the peptidyl nitroalkenes
The molecular structures and the Ki values obtained for the inhibition of the Mpro activity by the six peptidyl nitroalkene compounds are summarized in Table 1, respectively. For testing the inhibitory effect of the compounds, enzymes from two different expression systems were used (see Supplementary Enzymatic assays). An example of the enzymatic activity inhibition curves obtained from the enzyme using the pMal-Mpro vector is shown in Supplementary Fig. 1. In Supplementary Fig. 2 the inhibition profiles for the six compounds obtained using the Mpro from the expression using the pET21-Mpro vector are shown. Compounds FGA145, FGA146 and FGA147 with a glutamate surrogate at the P1 site, leucine at P2 and an aromatic residue at P3 displayed inhibition in the low micromolar range, less than 10 μM. The N-terminal substitution of the aromatic residue is well tolerated which is shown through the substitution of the benzyloxycarbonyl (Cbz) group of FGA147 by a 4-methoxy-1H-indole-2-carbonyl residue in FGA146 leads to similar values of inhibition. Compounds FGA86 with homophenylalanine at P1 and phenylalanine at the P2 sites, and an aromatic residue at P3; FGA159 with a homophenylalanine at the P1 site, leucine at P2, and three more residues at P3, P4 and P5; and FGA177 with a homophenylalanine at the P1 site, leucine at P2, and an aromatic residue at P3, also showed inhibition in the low micromolar range. No irreversible character was observed over a 10-min period (Supplementary Fig. 1) denoting the compounds to be non-time dependent inhibitors as it was predicted by us27,28. The values of Ki obtained using the plate reader assay and the continuous fluorometric assay were very similar, and the small non-significant differences might be due to differences in the enzymatic assay conditions, especially the different amount of the organic solvent DMSO (Table 1) (Supplementary Enzymatic assays).
Cellular antiviral activity and cytotoxicity
Three compounds (FGA145, FGA146 and FGA147) were selected for the antiviral assay with infectious SARS-CoV-2. Huh-7-ACE2 cells were used for this antiviral assay. The antiviral activity and cytotoxicity assays are shown in Fig. 2. Compounds FGA146 and FGA147 showed potent antiviral activity against SARS-CoV-2 with EC50 values in the low micromolar range (0.9 and 1.9 μM, respectively). FGA145 showed less potent activity (EC50 = 11.7 μM), in line with the enzymatic activity inhibition results (Supplementary Methods and Supplementary Data 4). The cytotoxicity of these compounds was very low with CC50 values over 100 μM. To validate the therapeutic potential of the designed compounds, antiviral assays in suitable, primary cell systems (e.g., human bronchial epithelial cells) need to be done in the future.

Cytotoxicity assays for the three compounds (left column), all of them presented a CC50 greater than 100 μM. Effect of the three compounds on the virus titer (center and right columns); FGA146 was the most potent inhibitor with an EC50 of 0.9 μM, followed by FGA147 and FGA145 with EC50 of 1.9 and 11.7 μM, respectively. All graphs show means ± s.d.; asterisks indicate p values (*p ≤ 0.05) obtained by two-tailed unpaired t tests.
Inhibition of other proteases
While the cysteine protease Mpro is inhibited with high potency by inhibitors FGA145, FGA146 and FGA147, no inhibitory activity against the serine proteases: human matriptase (membrane-type serine protease 1, MT-SP1, prostamin) and bivalent expressed Zika Virus NS2B/NS3 (bZiPro) was observed for these compounds (Table 2) (Supplementary Methods). Besides inhibition of Mpro, compound FGA145 was found to be a very potent inhibitor of cysteine proteases rhodesain (RhD), cruzain (CRZ), cathepsin L (CatL) and cathepsin B (CatB) with decreasing potencies from RhD to CatB (1.63 nM, 12.6 nM, 53.0 nM, 206 nM, respectively, Table 2). This finding is in line with previous reports of nitroalkenes as potent reversible inhibitors of these proteases26. Interestingly, all three compounds inhibit CatL, especially compound FGA145, denoting a possible multitarget effect of these compounds in the antiviral activity.
Crystal structure of SARS-CoV-2 Mpro in the apo form and in complex with FGA146 and FGA147
The SARS-CoV-2 Mpro in complex with FGA146 crystallized in the P21 space group and diffracted up to 1.98 Å resolution (Supplementary Table 2) with one biological dimer in the asymmetric unit (Supplementary Data 1), and the complex with FGA147 crystallized in the orthorhombic P21212 space group and diffracted up to 1.62 Å resolution (Supplementary Table 2) (Supplementary Data 2) with one monomer per asymmetric unit that forms the biological dimer with a crystallographic-symmetry related neighboring molecule (Supplementary Fig. 4). The protein structure can be subdivided into three domains (as shown in Supplementary Fig. 4), domain I and domain II containing the active site and domain III as the dimerization domain.
After the protein structures of the respective complexes were solved, significant electron density was found at the active site. This electron density could be unequivocally assigned to the corresponding inhibitors FGA146 (Fig. 3a) and FGA147 (Fig. 3b). Both inhibitors are covalently bound to the catalytic Cys145. A comparison of both inhibitors bound to the active site is shown in Fig. 3c.

Mpro is shown in ribbon and the inhibitors in ball and stick representation. 2Fo-Fc electron density map contoured at 1σ (shown in gray mesh) of FGA146 (a) and FGA147 (b) bound covalently to the catalytic cysteine (Cys145). c Electrostatic surface representation of the active site of Mpro with bound FGA146 (violet) and FGA147 (light blue). Red indicates negative charge and blue positive charge.
The bound inhibitors, that mimic the natural peptide substrate, show a good geometric complementarity within the active site for subsites S1, S2 and S3 with the warhead located at the S1’ subsite (Supplementary Fig. 5a–c). The nitro group of the warhead occupies, in both complexes, the “oxyanion hole” formed by the backbone amides of Gly143, Ser144, and Cys145 (Supplementary Fig. 5). The Sγ atom of the nucleophilic Cys145 forms a covalent bond to carbon C19 of the nitroalkene warhead of the inhibitor through a Michael addition (for numbering of the compounds see Supplementary Fig. 3). The S1 subsite is occupied by the glutamine surrogate γ-lactam ring that forms hydrogen bonds with the main chain of Phe140 and the side chain of Glu166 through the nitrogen atom (N16) of the ring, and to the side chain of His163 through the oxygen atom (O18) of the ring (Supplementary Fig. 5a–c and Fig. 6b, d). The carbon atoms of the side chain of these residues lie in a hydrophobic cavity defining the S1 subsite (Supplementary Fig. 5a–c and Fig. 6b, d). The second residue of both inhibitors is a Leu that is inserted into the hydrophobic S2 subsite made up by His41, Met49, and Met169 (Supplementary Fig. 5a–c). There are three hydrogen bonds between the main chain of the peptidyl inhibitor and the protein. They involve interactions between atoms N10 from the inhibitors and the backbone carbonyl of His164, N3 and OE1 from Gln189, and O1 and the backbone nitrogen from Glu166. The P3 residue side chain of the inhibitors, a methoxy indole carbonyl group in FGA146 and a Cbz group in FGA147, shows different conformations when bound to the protein (Fig. 3c). In the FGA147, the Cbz group is oriented towards the solvent lacking interactions with the protein residues (Figs. 3c and S5a). However, the methoxy indole carbonyl group of FGA146 is occupying the S4 subsite (Figs. 3c and S5a). This group forms a hydrogen bond between the N35 atom and the carbonyl atom from Glu166 (Supplementary Fig. 5b–e). This side chain is encased inside the S4 subsite formed by residues Glu166, Leu167, Pro168, Gln189, Thr190 and Ala191 (Supplementary Fig. 5d, e). The methoxy group is surrounded by the side and main chain atoms of residue Gln189, and by the main chain atoms of residues Thr190 and Ala191 (Supplementary Fig. 5d, e). The distance between the O atom of this methoxy group and the potential hydrogen bond partners is too far away and/or without a favorable geometry for this type of interaction (Supplementary Table 2), but it is enough to fix the position of this group and to orient the methyl group towards the solvent. Interactions between the protein and inhibitor atoms with the distances between them are summarized in Supplementary Table 3. Besides the extensive hydrogen bond network, there are numerous non-polar interactions that contribute to the tight binding of the inhibitor.
The position of the nitro group of the warhead in the structure of Mpro in complex with FGA146 is not fixed by the interactions with the residues that form the “oxyanion hole” (Gly143, Ser144 and Cys145, Supplementary Fig. 7). In one of the monomers (mon. B), this nitro group forms hydrogen interactions with the N atoms from Gly143 and Cys145 (Supplementary Table 3, and Supplementary Fig. 7, protein in green and ligand in purple); while in the other monomer (mon. A), the nitro group moves away from the “oxyanion hole” and interacts with His41 (Supplementary Table 3, and Supplementary Fig. 7, protein in cyan and ligand in brown); showing a certain degree of flexibility upon binding to this S1 site. In the case of the inhibitor FGA147, the nitro group interacts with “oxyanion hole” (Supplementary Table 3) and, because there is only one monomer in the asymmetric unit, we observe only one conformation.
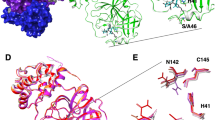
Concerning the protein, there are different conformations of some residues forming the active site. The most significant changes are located in the P2 helix, Ser46 to Asn51 (Fig. 4a) and in the P5 loop, Asp187 to Ala193 (Fig. 4a). The first segment is the α-helix that takes part in the formation of the S2 binding subsite. In the structure of the FGA146 complex, this segment is displaced towards the inside of the active site with respect to the structure of the FGA147 complex, consequently widening the S2 subsite (Fig. 4a). The second segment is the P5 loop, at this subsite only the inhibitor FGA146 was observed to be bound, while the P3 side chain of FGA147 is pointing towards the solvent. This segment is tightly packed around FGA146 fixing its conformation. FGA147 does not bind to this subsite in our structure, and the residues are positioned further away (Fig. 4a). Also, there is a small difference at the P4 β-hairpin flap. There is one loop close to the active site with a different conformation (Fig. 4a, loop I), and another loop further away from the active site also with a different conformation (Fig. 4a, loop II).

a Most significant changes in the active site and domain I are located at P2 helix and the P5 loop. Smaller changes can be observed at the P4 β-hairpin flap, loop I and loop II. b Superposition of Mpro in complex with FGA146 and FGA147. Monomer A and B from the complex with FGA146 are shown in light green and yellow orange, respectively, and from the complex with FGA147 are shown in salmon and light blue, respectively. Significant displacements of some of the helices from the dimerization domain (Domain III, circles) of monomer A can be observed, while the same domain from monomer B does not show these displacements. c Superposition of monomer A (light green) and B (yellow orange) of the Mpro in complex with FGA146. The helical dimerization domain (Domain III) shows differences in the relative positions of some of the helices. Four of them show significant displacements (α-6, α-7, α-8 and α-9) while the las helix (α-10) does not show any significant displacement.
Besides the differences observed for the active site located in domains I and II, there are additional differences in domain III between the structures of the Mpro in complex with FGA146 and FGA147 (Fig. 4b). The superposition of these structures shows clear displacements in the positions of four of the helices (α-6, α-7, α-8 and α-9) in monomer A, as a consequence the loops linking those helices are also displaced.
These changes could not be observed in monomer B where all the helices from this domain are very well aligned with only small non-significant differences (Fig. 4b). Upon observing these differences in only one monomer we superposed monomer A and monomer B from the Mpro in complex with FGA146, shown in Fig. 4c. Here, we were able to observe the same changes that occur in monomer A between the FGA146 and FGA147 structures (Fig. 4b). This indicates a great flexibility of the Mpro with no coordinated changes in both monomers. These changes could be due to the different space groups in which the protein crystallized, as stated previously39 or just might indicate the flexibility of this protein which has to accommodate itself to be able to catalyze the proteolysis of the polyprotein to liberate the mature proteins essential for the virus replication.
Computational study of the SARS-CoV-2 Mpro inhibition by FGA146 and FGA147
Based on the results derived from the kinetic studies presented in previous section, the inhibition reaction was studied according to the general mechanism proposed in Fig. 5 with the two most promising inhibitors: FGA146 and FGA147. After the binding between the inhibitor and the enzyme to form the non-covalent E:I complex, the first chemical step would involve the activation of Cys145 by a proton transfer to His41 which can take place concomitantly with the nucleophilic attack of the sulfur atom of Cys145 to the C19 atom of the inhibitor to form an intermediate, E-I(-). Then, the reaction is completed by the transfer of the proton from the protonated His41 to the C20 atom of the inhibitor to render the final E-I covalent adduct.

The mechanism has three steps: entry of the inhibitor into the active site, then addition of the cysteine thiolate, and finally protonation of the nitronate.
The free energy profiles derived from free energy surfaces (FESs) of the SARS-CoV-2 Mpro inhibition with both FGA146 and FGA147 obtained by M06-2X/6-31 + G(d,p)/MM MD simulations (see Supplementary Figs. 12, 13; Tables 4, 5, 6 and Computational Methods) confirm that the activation of Cys145 takes place concertedly with the inhibitor-enzyme covalent bond formation, E:I to E-I(-) step. Interestingly, we already observed this concerted activation and nucleophilic attack of Cys145 when exploring the acylation step of the proteolysis reaction35, but previous inhibition processes explored in our laboratory have rendered stepwise processes where the activation of the Cys145 precedes the covalent formation between the sulfur atom of Cys145 and the different tested warheads of the inhibitors28,36. The last step of the inhibition reaction corresponds to the proton transfer from His41 to the C20 atom of the inhibitor, E-I(-) to E-I step.
The chemical steps of the inhibition process are exergonic in both cases (−15.6 and −9.8 kcal·mol−1 with FGA146 and FGA147, respectively), and the activation of free energies, determined by the formation of the intermediate covalent intermediate, E-I(-), are almost equivalent (15.3 and 15.6 kcal·mol−1 with FGA146 and FGA147, respectively).
Structures of the different states appearing along the reaction optimized at M06-2X/MM level are shown in Fig. 6, while a list of key inter-atomic distances obtained on the representative stable states is listed in Supplementary Tables 7 and 8. The estimation of the main interaction energies between residues of SARS-CoV-2 Mpro and the inhibitors FGA146 and FGA147 computed in the E:I and in the E-I states are shown in Supplementary Fig. 15.

Carbon atoms of the inhibitor are shown in yellow, and those of the catalytic residues C145 and H41 are shown in cyan. Key distances are in Å.
Thermal stability of Mpro in the absence and presence of the inhibitors
We have examined the thermal stability of the protease in the absence and in the presence of the inhibitors using circular dichroism (CD). The Tm value for Mpro did not change, significantly, in the presence of FGA177 (0.1 ± 0.2 °C in the range of 25 to 100 μM, Figs. 7c and S8d). In the presence of FGA86 (Figs. 5c and S8a) at the lower concentration (25 μM) there was a slight increase in stability (0.9 ± 0.4 °C) and at the higher concentration (100 μM) there was a slight decrease in stability (−1.7 ± 0.4 °C). The presence of the inhibitors FGA145 (−1.1 ± 0.2 °C at 25 μM and −3.7 ± 0.1 °C at 100 μM, Figs. 5c and S8b) and, while FGA146 (−4.2 ± 0.2 °C at 25 μM and −6.4 ± 0.1 °C at 100 μM, Fig. 7a, c) and FGA147 (−3.0 ± 0.1 °C at 25 μM and −4.2 ± 0.2 °C at 100 μM, Fig. 7b, c) lead to a significant decrease of the Tm value. It has been reported that the association of covalently-bound compounds induce shifts of Tm values to lower temperatures with an apparent destabilization of the protein40,41,42. Thus, these data agree with the formation of a covalent bond between the protein and compounds FGA86, FGA145, FGA146 and FGA147. Based on the X-ray crystallographic analyses and thermal stability data, we can conclude that these last four compounds bind covalently to the protein. The inhibitor FGA159 showed an increase in stability of 2.5 ± 0.2 °C at a concentration range between 5 and 100 μM (Figs. 5c and S8c).

a Thermal stability of Mpro in the presence of FGA146 using circular dichroism. b Thermal stability of Mpro in the presence of FGA147 using circular dichroism. The Tm value of Mpro in the absence of inhibitors (black squares) was 49.2 °C, while in the presence of 25 (blue circles) and 100 μM (green triangles) of FGA146, the values decreased to 45.0 and 42.8 °C, respectively. In the case of FGA147, these values decreased to 46.2 and 45.0 °C, respectively. c Change of the Tm in the presence of the inhibitors. FGA86 shows a slight increase in stability at the lower concentration (25 μM) and a decrease in stability at the higher concentration (100 μM). There is no significant change in stability in the presence of FGA177; while in the presence of FGA145, FGA146 and FGA147 shows a significant decrease in stability, suggesting that they could bind covalently to the protein. The presence of FGA159 increased the stability of the protein. The error bars represent the fitting error of the denaturation data.
Binding of inhibitors to Mpro
Figure 8a shows the binding isotherms of FGA145 and FGA146 to Mpro by ultracentrifugation, and Fig. 8b shows the binding of FGA147 to Mpro measured by ITC (Supplementary Data 5). The measured binding dissociation constants for these compounds were in the low micromolar range, the Kd for FGA145 was 11.8 ± 1.05 μM, the Kd for FGA146 was 7.28 ± 0.58 μM (the average obtained from using both, absorption and fluorescence, data), and the Kd for FGA147 was 2.86 ± 0.25 μM. For all three inhibitors we obtained a stoichiometry of one, which is compatible with binding of one molecule of the inhibitor to the active site.

a Binding isotherm of FGA145 (black squares) and FGA146 (blue circles and green triangles) to Mpro. The concentration of FGA145 was measured by absorption, and that of FGA146 was measured by absorption (blue circles) and fluorescence (green triangles). All data are mean values ± standard deviation of two technical replicates. b ITC binding profile of FGA147 to Mpro.
Reversibility
The results of the experiments carried out by dilution to check for the reversibility of the binding of the inhibitor FGA146 to Mpro are shown in Fig. 9 (Supplementary Data 6). The activity of the Mpro preincubated with FGA146 seems to recover over time (shown in blue in Fig. 9), which makes it likely that the compound inhibits its target reversibly. For the reversible control Nirmatrelvir (nitrile warhead) there is only partial recovery of activity and for the irreversible control (vinyl sulfone warhead) there is no visible recovery of activity.

In black a DMSO control with no inhibitor added is shown, in green Nirmatrelvir as a reversible control and in blue the compound FGA146. All samples were measured as triplicates. Due to substrate depletion over time only the first 1000 s are shown.
Pharmacokinetic assays
A pharmacokinetic profile has been performed for most active inhibitor FGA146 including physicochemical parameters (Supplementary Table 9), cysteine reactivity (Supplementary Figs. 20 and 21), stability (Supplementary Table 10 and Figs. 22, 23 and 24), dilution assay, inhibition assay (Supplementary Fig. 25), DNMT2 MST-displacement assay (Supplementary Fig. 26), logP determination by HPLC (Supplementary Table 11, Figs. 27, 28) and permeability (Supplementary Figs. 29 and 30).
Discussion
We have designed, synthesized and measured the inhibitory effect of a series of peptidomimetic compounds with a nitroalkene warhead on the enzymatic activity of Mpro and cell infection. We have also examined the possibility of using a nitroalkene warhead that due to its reversible binding26 should decrease the possibility of side effects due to unwanted reactions with other cellular components.
Six compounds (FGA86, FGA145, FGA146, FGA147, FGA159 and FGA177) were prepared in good yields via a short and straightforward synthetic route. Among the inhibitors we have synthesized there are three similar to inhibitors already reported, they have the same peptidomimetic moiety but they differ in the warhead. FGA145 is similar to two previously reported inhibitors43. FGA146 is similar to PF-0083523120 and FGA147 is similar to GC37644, the rest of the inhibitors present a new molecular structure. All of them exhibited enzyme inhibitory activity (Ki: 110 μM) and three of them (FGA145, FGA146 and FGA147), having the typical coronaviral protease glutamine surrogate (gamma-lactam) at P1 site and a L-leucine at P2 site, gave good anti-SARS-CoV-2 infection activity in the low micromolar range (EC50: 112 μM) without significant toxicity. Additional kinetic studies of the selectivity of FGA145, FGA146 and FGA147 show that they are also potent inhibitors of cathepsin L (CatL), revealing a multitarget effect.
The crystal structures of the Mpro in complex with FGA146 and FGA147 were solved and confirmed the binding modes. The covalent inhibitory character of these inhibitors is similar to other peptidomimetic inhibitors18,19,22. Our crystal structures, that virtually overlap with the structures derived from the computer simulations that were initiated from a previously crystallized Mpro in complex with a different inhibitor (N3), the structures corroborate the great conformational flexibility of the dimer of Mpro. Flexibility of the active site has been reported to be needed to accommodate the different natural cleavage sites present in the polyprotein of SARS-CoV-2, as well as some conformational flexibility of other regions of the protein (Domain II and III)39. The crystal structures of Mpro in complex with FGA146 and FGA147 show a very similar binding mode to that of the similar inhibitors PF-00835231 and GC376. The only significant difference is the warhead and its binding to the “oxyanion hole” (Supplementary Fig. 9).
Through thermal denaturation we have been able to observe that the inhibitors might stabilize or destabilize the protein, in some cases a destabilization of more than 6 °C was found. The analysis of the crystal structures together with these thermal denaturation data shows the influence of the inhibitors on the whole structure of Mpro. This instability induced by active site inhibitors might be exploited to increase their potency against the virus replication if they could be combined with inhibitors that bind to sites other than the active site to further disrupt the activity of this essential protease for the virus.
Finally, QM/MM computer simulations assisted in elucidating the way of action of the most promising compounds, FGA146 and FGA147, by generating the complete free energy landscape of the inhibitor-enzyme covalent complex formation. The results of the inhibitory mechanism, that appear to be equivalent in both cases, suggest that, after the non-covalent enzyme:inhibitor binding step, the E:I formation, the activation of Cys145 takes place concertedly with the inhibitor-enzyme covalent bond formation. In the second chemical step, the final proton transfer takes place from His41 to the Cα atom of the inhibitors, E-I(-) to E-I step. The resulting free energy profiles for the covalent inhibition of SARS-CoV-2 Mpro with FGA146 and FGA147 show how the processes are exergonic in both tested inhibitors, which determined not only by the favorable binding step but by the stability of the final adduct, E-I, with respect to the initial E:I complex (see Supplementary Computational Methods).
In summary, we have designed and synthesized six compounds as covalent reversible inhibitors of the SARS-Cov-2 Mpro with the nitroalkene warhead. The three most active inhibitors were active at low micromolar concentrations against SARS-CoV-2 and did not show significant toxicity. These compounds were also active against human Cathepsin L in the nanomolar range, denoting a dual activity. The crystal structures, aided by computer simulations, of the two most promising inhibitors in complex with Mpro show the mechanism of action of these inhibitors and the interactions established between the inhibitor and the protein.
Methods
General procedure for the preparation of nitroalkenes
To a stirred solution of alcohol 1 (1.05 g, 4 mmol) in dichloromethane (32 mL) was added Dess-Martin periodinane (1.82 g, 4.3 mmol) and sodium bicarbonate (361 mg, 4.3 mmol). The resulting mixture was stirred at room temperature for 1 h. Then the reaction mixture was cooled with an ice-bath and triethylamine (0.17 mL, 1.21 mmol) and nitromethane (1.33 mL, 24.4 mmol) were. Then the mixture was stirred for 15 h at room temperature and then was quenched with a saturated aqueous solution of NH4Cl (10 mL), the mixture was extracted with CH2Cl2 (3 × 15 mL) and the combined organic layers were washed with HCl 1 M, then with a saturated aqueous solution of sodium bicarbonate and then dried over Na2SO4. Then the solvent was evaporated and the residue was purified by column chromatography silica gel, CH2Cl2/MeOH (99:1 to 85:15) to afford the desired product as a yellow oil (71%).
The corresponding nitroaldol (0.73 mmol) was dissolved in dichloromethane (2.1 mL) and placed in an ice-bath. Then trifluoroacetic acid (1.1 mL) in dichloromethane (1.1 mL) was added dropwise and the mixture was stirred at room temperature for 3 h. The reaction mixture was evaporated in vacuo to give the product as a colorless solid. The resulting mixture was submitted to the next step without any further purification.
To a solution of the ammonium trifluoroacetate (0.80 mmol) and the carboxylic acid (0.89 mmol) in dichloromethane (8 mL) cold with an ice-bath, HOBt·H20 (121 mg, 0.89 mmol) was added. After 15 min at the same temperature, DIPEA (0.56 mL, 3.23 mmol) was added dropwise. After another 15 min, EDC (186.2 mg, 0.97 mmol) was added and the mixture was stirred for 16 h at room temperature. Then the mixture was quenched with saturated ammonium chloride solution (10 mL) and extracted with dichloromethane (3 × 20 mL). The combined organic layers were washed with HCl 1 M, with a saturated aqueous sodium bicarbonate solution and then dried over Na2SO4. Then the solvent was evaporated and the residue was purified by column chromatography silica gel, CH2Cl2/MeOH (100:0 to 85:15) to afford the desired product (64%, two steps).
To an ice bath cold solution of peptidyl nitroaldol (0.66 mmol) in dichloromethane (6.6 mL) was added DIPEA (0.24 mL, 1.39 mmol), then methanesulfonyl chloride (0.056 mL, 0.73 mmol). The resulting mixture was stirred overnight, then it was quenched with a saturated aqueous solution of NH4Cl (10 mL) and extracted with dichloromethane (3 × 20 mL). The combined organic layers were washed with HCl 1 M then with a saturated aqueous sodium bicarbonate solution and then dried over Na2SO4. Then the solvent was evaporated and the residue was purified by column chromatography silica gel, CH2Cl2/MeOH (99:1 to 9:1) to afford the desired product (68–81%) (NMR Spectra of all compounds in Supplementary Data 3).
For the preparation of all the compounds, the coupling steps and nitroalkene formation were done following the experimental procedure detailed above.
For the preparation of compound FGA159, the hydrolysis and hydrogenation steps were done following standard experimental procedures.
Cloning of Mpro gene
The Mpro gene was cloned in two different vectors with a similar strategy. First, the sequence of the gene coding for Mpro (nsp5) SARS-CoV-2 was optimized for Escherichia coli expression, synthesized and cloned directly into a pUCIDTKan. The Mpro gene was amplified from the vector pUCIDTKan-Mpro and cloned into the vector pET21a (Novagen) named pET21-Mpro. Second, the Mpro gene was inserted into the pMal plasmid harboring the C-terminal hexahistidine-tagged sequence of SARS-CoV-2 Mpro named pMal-Mpro (Prof. John Ziebuhr, Justus Liebig University Gießen, Germany). The sequence contained the native nsp4/nsp5 Mpro cleavage site between MBP and Mpro as well as the native nsp5/nsp6 cleavage site between Mpro and the hexahistidine tag, thus enabling the purification of native Mpro.
Protein expression and purification
SARS-CoV-2 Mpro The vector pET21-Mpro was transformed into E. coli Tuner (DE3) cells (Novagen, Merck, Madrid, Spain). These cells were grown in 2xYT medium supplemented with ampicillin (100 mg/L) at 37 °C. The expression was induced by the addition of 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG), and let to grow for an additional 16 h. The cells were harvested by centrifugation and resuspended in the lysis buffer. The cells were lysed by sonication and the supernatant was then loaded onto a HisTrap FF column (GE Healthcare). The fractions containing the protease were then pooled, and PreScission protease containing a hexahistidine tag was added. The mixture was then dialyzed and the PreScission-treated Mpro solution was applied to a HisTrap FF column to remove the PreScission protease, the C-terminal tag, and Mpro with uncleaved hexahistidine tag. The processed Mpro was collected in the flow-through and concentrated to 10 mg/mL. The expression of SARS-CoV-2 Mpro using the vector pMal-Mpro was performed exactly as described previously45. Human matriptase. Recombinant expression and purification were mainly performed as described previously46. The pQE30 plasmid, containing the human matriptase (membrane-type serine protease 1, MT-SP1, prostamin) was kindly provided by Prof. Torsten Steinmetzer (Philipps University Marburg, Germany). Since MT-SP1 is expressed as inclusion bodies, no leakage suppression was needed and, hence, the plasmid was transformed in E. coli BL21-Gold (DE3) (Agilent Technologies, Santa Clara, CA, USA) cells. After growing them in LB medium supplemented with ampicillin (100 mg/mL) to an OD600 of 0.6–0.8, overexpression was induced by addition of 1 mM IPTG over night (o.n.) at 20 °C. Cells were harvested by centrifugation, flash frozen in liquid N2 and stored at −80 °C until further usage. For protein refolding and purification from inclusion bodies, cell pellets were resuspended in lysis buffer (50 mM TRIS−HCl pH 8.0, 300 mM NaCl, 10% (v/v) glycerol and 1 mM β-ME), supplemented with lysozyme and DNase and stirred for 1 h at room temperature (rt). After that, cells were further lysed by sonication (Sonoplus HD 2200; Bandelin, Berlin, Germany) and again centrifuged. The supernatant was discarded and the pellet was washed with lysis buffer. Proteins were solubilized in a denaturing solubilization buffer (50 mM TRIS−HCl at pH 8.0, 6 M urea, 10% (v/v) glycerol and 1 mM β-ME) by stirring o.n. at rt. The suspension was again centrifuged to remove cell debris. The supernatant was subjected to IMAC on a HisTrap HP 5 ml column (Cytiva Europe GmbH, Freiburg im Breisgau. Germany), using IMAC buffer A (50 mM TRIS−HCl pH 8.0, 6 M urea, 20 mM imidazole and 1 mM β-ME) in a linear gradient with IMAC buffer B (50 mM TRIS−HCl at pH 8.0, 6 M urea, 250 mM imidazole and 1 mM β-ME). The fractions, containing eluted MT-SP1 were refolded by a 2-step dialysis over 12 h each at 4 °C in dialysis buffer A (50 mM TRIS−HCl at pH 9.0, 3 M urea and 1 mM β-ME) and anion exchange (IEX) buffer A (50 mM TRIS−HCl at pH 9.0, 1 mM β-ME) prior to IEX chromatography on a Resource Q 1 ml column (Cytiva Europe GmbH, Freiburg im Breisgau. Germany), using IEX buffer A in a linear gradient with IEX buffer B (50 mM TRIS−HCl at pH 9.0, 1 M NaCl and 1 mM β-ME). Eluted MT-SP1 was flash frozen in liquid N2 and stored at −80 °C. Zika Virus 2 NS2BCF/NS3pro. The bivalently expressed ZIKV protease was expressed and purified as described previously47. Briefly, the pETDUET vector containing bZiPro (purchased from Addgene) was transformed into competent E. coli BL21 Gold (DE3) cells (Agilent Technologies, Santa Clara, CA, USA) and grown in LB medium containing ampicillin at 37 °C until they attained an optical density (OD600) of 0.8. Overexpression was induced o.n. by addition of 1 mM IPTG at 20 °C. After harvesting, cells were flash frozen in liquid N2 and stored at −80 °C until protein purification. Herein, cell pellets were resuspended in lysis buffer (20 mM TRIS−HCl at pH 8.0, 300 mM NaCl, 20 mM imidazole, 0.1% (v/v) Triton X-100, RNase, DNase, lysozyme and 1 mM DTT) and lysed by sonication. After centrifugation, bZiPro from the cleared supernatant was purified by IMAC on a HisTrap HP 5 ml column with a step-gradient of washing buffer (20 mM TRIS−HCl at pH 8.0, 300 mM NaCl and 20 mM imidazole) and elution buffer (20 mM TRIS−HCl at pH 8.0, 300 mM NaCl and 250 mM imidazole). The eluted fractions, containing bZiPro were subjected to a gel filtration step (HiLoad 16/600 Superdex 75; GE Healthcare, Chicago, IL, USA) in SEC buffer (50 mM TRIS−HCl at pH 8.0 and 150 mM NaCl). Eluted bZiPro was flash frozen in liquid N2 and stored at −80 °C. Cruzain. Cruzain (CRZ) was kindly provided by Dr. Avninder S. Bhambra (De Montfort University, Leicester, UK). Cathepsin L, Cathepsin B. Both cathepsin L (CatL) and cathepsin B (CatB) were purchased from Calbiochem (Merck Millipore, Burlington, Massachusetts). Rhodesain. Rhodesain (RhD) was recombinantly expressed and purified as reported previously48.
Enzymatic assays
Proteolytic activity was determined by cleavage of fluorescence resonance energy transfer peptide substrates. The inhibitor was added into the Mpro solution in the reaction buffer, mixing and allowing the mixture to equilibrate for 10 s, and then initiated by adding the Dabcyl-KTSAVLQ ↓ SGFRKME-(Edans)-Amid substrate solution. Compounds in Table 1 were diluted in DMSO (FGA145, FGA146 and FGA147), N,N-dimethylformamide (FGA86 and FGA177) and ethanol (FGA159). Due to the deleterious effect of the solvents on the activity of Mpro and for consistency of the data, the concentration of solvent was kept constant at 1% (v/v) in all experiments. All measurements were made in triplicates. For measurements using the TECAN Infinite F200 PRO plate reader each well was composed of 180 μL buffer, 5 μL enzyme in buffer, 10 μL inhibitor in DMSO or ethanol, and 5 μL substrate in DMSO (measuring conditions for all the proteases are summarized in Supplementary Table 1). The amount of solvent in these experiments was 7.5%.
FGA146, the indole harboring compound, was the only one revealing a strong fluorescence in this assay at higher concentrations. To rule out that bleaching of this fluorescence interferes with our readout by overlaying the fluorescence increase caused by the enzymatic substrate cleavage, control measurements were performed. Therefore, the assay was repeated without addition of substrate, hence, the negative slope due to bleaching of FGA146 was determined. The relative activity values were then corrected for the negative slope of each inhibitor concentration (Fig. S1).
Cell-based antiviral activity and cytotoxicity assays
Huh-7 cells that overexpress human angiotensin-converting enzyme 2 (ACE2) (Huh-7-ACE2; kindly provided by Friedemann Weber (Institute of Virology, Justus Liebig University Giessen)) were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and antibiotics (100 μ/mL penicillin and 100 μg/mL streptomycin) at 37 °C in an atmosphere containing 5% CO2. The SARS-CoV-2 isolate Munich 92931 was kindly provided by Christian Drosten (Institute of Virology, Charité-Universitätsmedizin, Berlin). Cytotoxic concentrations 50% (CC50) of the compounds used in antiviral activity assays were determined using MTT assays as described previously49. To determine effective concentrations 50% (EC50) of the respective compounds, Huh-7-ACE2 cells were inoculated with SARS-CoV-2 at a multiplicity of infection (MOI) of 0.1 plaque-forming units (pfu) per cell. After incubation for 1 h at 33 °C, the virus inoculum was replaced with fresh cell culture medium containing the test compounds at the indicated concentration. After 23 h at 33 °C, the cell culture supernatants were collected and virus titers were determined by virus plaque assay as described previously50.
Crystallization data collection and structure determination
Crystallization trials were performed at 295 K using the sitting-drop vapor-diffusion method with commercial screening solutions using a Cartesian Honeybee System (Genomic Solutions, Irvine, USA) nano-dispenser robot.
For data collection, crystals were cryo-protected with 30% (v/v). X-Ray data collection experiments were performed at the ALBA Synchrotron (Cerdanyola del Vallès, Spain) BL13 XALOC beamline, and at the ESRF Synchrotron (Grenoble, France) ID30B beamline. Data were indexed and integrated, scaled and merged using XDS51. The structures were solved by molecular replacement using the previously reported SARS-CoV2 Mpro structure (PDB: 7K3T) with Molrep52. The inhibitor molecule was added manually. Data processing and refinement statistics are listed in Supplementary Table 2.
Circular dichroism
Circular dichroism measurements were carried out on a JASCO J-720 (Jasco, Tokyo, Japan) spectropolarimeter equipped with a Peltier type temperature controller and a thermostatized cuvette cell linked to a thermostatic bath. Spectra were recorded in 0.1 cm path length quartz cells with a response time of 4 s and a band width of 2 nm. The protein concentration used was 0.15 mg/mL in 20 mM Tris-HCl buffer at pH 7.5 and 100 mM NaCl. The observed ellipticities were converted into the molar ellipticities [θ] based on a mean molecular mass per residue of 110.45 Da. Thermal denaturation experiments were performed by increasing the temperature from 20 to 80 °C at 30 °C/h. Tm represents the temperature at the midpoint of the unfolding transition. The CD signal was followed at 230 nm and the concentration of organic solvent was kept constant at 2.5%. Two concentrations of each compound were used (25 and 100 μM).
Binding measurements
For direct binding measurements we used the sedimentation method previously reported53. A solution of known concentrations of protein and ligand was centrifuged in 1 mL polycarbonate tubes in an MLA-15 rotor in the Optima MAX-XP ultracentrifuge (Beckman) at 150,000 rpm (700,200 x g) for 4 h at 25 °C. At the end of the centrifugation, the lower half of the tube contained the protein in equilibrium with free ligand; the upper half contained only free ligand and essentially no protein, as checked by control measurements. The upper 0.5 mL was withdrawn carefully, and the free ligand was measured. The bound ligand concentration was taken as the difference between total concentration of ligand added to the tube and the measured free concentration. The values of the binding equilibrium constant and the number of sites were obtained by fitting the binding equation to the data. FGA145 and FGA146 were the only two ligands with an absorption spectrum that allowed us to measure its concentration spectrophotometrically. The higher concentrations were measured directly and for the lower concentrations the solution has to be concentrated to obtain absorption spectra with sufficient intensity. Also, FGA146 was measured fluorometrically due to the fact that this compound present intrinsic fluorescence. The protein concentration used in the experiments was 14.8 μM, and the concentration range used for FGA145 was 0.5 to 100 μM, and for FGA146 was 0.1 to 100 μM. The experiments were performed in 20 mM TRIS-HCl buffer at pH7.5 containing 0.1 M NaCl and 1% DMSO. Isothermal Titration Calorimetry (ITC) titrations were performed in a PEAQ-ITC calorimeter (Malvern, Westborough, MA, USA), using a Mpro-containing solution of 250 μL at 29.5 μM in 20 mM TRIS-HCl buffer at pH 7.5 containing 0.1 M NaCl and injections at 150 s intervals of 2 μL ligand-containing solution (up to adding 36.4 μL) at 25 °C and 750 rpm. The ligand solution consisted of a concentration of 1 mM of the compound FGA147 containing 5% of DMSO. A fitted offset parameter was applied to account for potential background. Data processing was performed using the MiroCal PEAQ-ITC Analysis software. None of the other compounds were completely soluble with 5% DMSO at the concentration required for the ITC experiments.
Reversibility experiments. Dilution assays
Buffer: 20 mM Tris, 0.1 mM EDTA, 1 mM DTT, 200 mM NaCl pH = 7.5. For the dilution assay Nirmatrelvir21 was used as a reversible control and LM18854 as an irreversible control (Fig. 10).

Both were used as controls in the reversibility experiments: inhibitor LM188 was used as an irreversible inhibitor and Nirmatrelvir as a reversible inhibitor.
All compounds were preincubated with the enzyme (10 µM) at a concentration of at least 20 times the IC50 (70 µM for LM188 and 76.4 µM for FGA146). For Nirmatrelvir a (relatively) higher concentration (10 µM) was chosen to ensure the full inhibition of the enzyme. The incubation was done at RT for 30 min in assay buffer. DMSO was used as a control. After incubation the Enzyme/Inhibitor (or DMSO) mixtures were diluted 100-fold in assay buffer. The substrate (Dabcyl-KTSAVLQ ↓ SGFRKME-Edans) was added with a final concentration of 25 µM, and the fluorescence was measured over 1 h at 25 °C as triplicates.
QM/MM simulations
After setting up the molecular models, the reaction was studied using a QM/MM approach from the equilibrated structures. The QM subset of atoms includes the P1’ and P1 positions of the inhibitor, together with C145 and H41 residues of the protein. Four link atoms were inserted where the QM/MM boundary intersected covalent bonds in the positions indicated on Supplementary Fig. 11. Thus, QM part consisted of 57 atoms for both inhibitors. All the calculations were performed with the QMCube suite55, for which the combination of the OpenMM and Gaussian0956 programs was used for constructing the potential energy function. The AMBER ff0357 and the TIP3P58 force fields were selected to describe the MM atoms, and the Minnesota functional M06-2X59 with the split-valence 6–31 + G(d,p) basis set60 were used to treat the QM subset of atoms. This functional has been tested and shown to be suitable for modeling this type of reactivity27,28,35,36,61,62,63,64. The position of any atom over 20 Å from the substrate was fixed to speed up the calculations.
The reaction mechanisms for each inhibitor were initially explored using the nudged elastic band65 approach to set up plausible starting geometries for the transition structures. The information obtained in this stage was used in the fine-tuning of the calculation of the FES, in terms of potential of mean force (PMF). The PMF for each chemical step was obtained using the combination of the umbrella sampling (US) approach66 with the weighted histogram analysis method67.
Finally, the protein:inhibitor interaction energy was estimated as a contribution of each residue of the protein, computed as an average along QM/MM MD simulations. The interaction energy is decomposed in a sum over residues provided that the polarized wave function (Ψ) is employed to evaluate this energy contribution, and the QM sub-set of atoms were described by the semiempirical Hamiltonian AM168 in these QM/MM MD calculations. A detailed description of the computational methods can be found in the Supplementary Material.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data are available in the main text, the Supplementary Information, the Supplementary Data files 1–6, and the Zenodo repository at: https://zenodo.org/uploads/10066288. The atomic coordinates and structure factors have been deposited into the Protein Data Bank with accession codes 8BGA and 8BGD. Materials and all other data are available from the corresponding authors on reasonable request.
References
World Health Organization. Weekly epidemiological update on COVID-1919 - 21 December 2022. https://www.who.int/publications/m/item/covid-19-weekly-epidemiological-update---21-december-2022.
Editorial “There’s no room for COVID complacency in 2023”. Nature 613, 7 (2023).
Cameroni, E. et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature 602, 664–670 (2022).
Cao, Y. et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 602, 657–663 (2022).
Gottlieb, R. L. et al. Early Remdesivir to prevent progression to severe Covid-19 in outpatients. N. Engl. J. Med. 386, 305–315 (2022).
Jayk Bernal, A. et al. Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. N. Engl. J. Med. 386, 509–520 (2022).
Dryden-Peterson, S. et al. Nirmatrelvir plus Ritonavir for early COVID-19 in a large U.S. health system : a population-based cohort study. Ann. Intern. Med. M22, 2141 (2022).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Yan, S. & Wu, G. Potential 3-chymotrypsin-like cysteine protease cleavage sites in the coronavirus polyproteins pp1a and pp1ab and their possible relevance to COVID-19 vaccine and drug development. Faseb j. 35, e21573 (2021).
Yang, H. et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 100, 13190–13195 (2003).
Anand, K. et al. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha-helical domain. EMBO J. 21, 3213–3224 (2002).
Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R. & Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300, 1763–1767 (2003).
Hayden, F. G. et al. Phase II, randomized, double-blind, placebo-controlled studies of ruprintrivir nasal spray 2-percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob. Agents Chemother. 47, 3907–3916 (2003).
Kim, Y. et al. Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathog. 12, e1005531 (2016).
Yang, H. et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 3, e324 (2005).
Xiong, M. et al. What coronavirus 3C-like protease tells us: from structure, substrate selectivity, to inhibitor design. Med. Res. Rev. 41, 1965–1998 (2021).
Agost-Beltran, L. et al. Advances in the development of SARS-CoV-2 Mpro inhibitors. Molecules 27, 2523 (2022).
Dai, W. et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368, 1331–1335 (2020).
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 368, 409–412 (2020).
Hoffman, R. L. et al. Discovery of Ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 63, 12725–12747 (2020).
Owen, D. R. et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593 (2021).
Jin, Z. et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020).
Yamane, D. et al. Selective covalent targeting of SARS-CoV-2 main protease by enantiopure chlorofluoroacetamide. Chem. Sci. 13, 3027–3034 (2022).
Ma, C. et al. Discovery of Di- and Trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. 143, 20697–20709 (2021).
Brewitz, L. et al. Alkyne derivatives of SARS-CoV-2 main protease inhibitors including nirmatrelvir inhibit by reacting covalently with the Nucleophilic Cysteine. J. Med. Chem. 66, 2663–2680 (2023).
Latorre, A. et al. Dipeptidyl Nitroalkenes as potent reversible inhibitors of cysteine proteases Rhodesain and Cruzain. ACS Med. Chem. Lett. 7, 1073–1076 (2016).
Arafet, K., González, F. V. & Moliner, V. Quantum mechanics/molecular mechanics studies of the mechanism of cysteine proteases inhibition by Dipeptidyl Nitroalkenes. Chemistry 26, 2002–2012 (2020).
Arafet, K. et al. Mechanism of inhibition of SARS-CoV-2 M(pro) by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem. Sci. 12, 1433–1444 (2020).
Mellott, D. M. et al. A clinical-stage cysteine protease inhibitor blocks SARS-CoV-2 infection of human and monkey cells. ACS Chem. Biol. 16, 642–650 (2021).
Shang, J. et al. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 117, 11727–11734 (2020).
Hoffmann, M. et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020).
Liu, T., Luo, S., Libby, P. & Shi, G. P. Cathepsin L-selective inhibitors: a potentially promising treatment for COVID-19 patients. Pharm. Ther. 213, 107587 (2020).
Ma, X. R. et al. MPI8 is potent against SARS-CoV-2 by inhibiting dually and selectively the SARS-CoV-2 main protease and the host Cathepsin L. ChemMedChem 17, e202100456 (2022).
Sacco, M. D. et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci. Adv. 6, eabe0751 (2020).
Świderek, K. & Moliner, V. Revealing the molecular mechanisms of proteolysis of SARS-CoV-2 M(pro) by QM/MM computational methods. Chem. Sci. 11, 10626–10630 (2020).
Martí, S. et al. Impact of warhead modulations on the covalent inhibition of SARS-CoV-2 M(pro) explored by QM/MM Simulations. ACS Catal. 12, 698–708 (2022).
Kim, Y., Mandadapu, S. R., Groutas, W. C. & Chang, K. O. Potent inhibition of feline coronaviruses with peptidyl compounds targeting coronavirus 3C-like protease. Antivir. Res. 97, 161–168 (2013).
Zhang, L. et al. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J. Med. Chem. 63, 4562–4578 (2020).
Jaskolski, M. et al. Crystallographic models of SARS-CoV-2 3CL(pro): in-depth assessment of structure quality and validation. IUCrJ 8, 238–256 (2021).
Wossner, N. et al. Sirtuin 1 inhibiting Thiocyanates (S1th)-a new class of isotype selective inhibitors of NAD(+) dependent Lysine Deacetylases. Front. Oncol. 10, 657 (2020).
Simeonov, A. Recent developments in the use of differential scanning fluorometry in protein and small molecule discovery and characterization. Expert Opin. Drug Discov. 8, 1071–1082 (2013).
Weber, P. C. & Salemme, F. R. Applications of calorimetric methods to drug discovery and the study of protein interactions. Curr. Opin. Struct. Biol. 13, 115–121 (2003).
Wang, H. et al. The structure-based design of peptidomimetic inhibitors against SARS-CoV-2 3C like protease as Potent anti-viral drug candidate. Eur. J. Med. Chem. 238, 114458 (2022).
Vuong, W. et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 11, 4282 (2020).
Amendola, G. et al. Lead discovery of SARS-CoV-2 main protease inhibitors through covalent docking-based virtual screening. J. Chem. Inf. Model. 61, 2062–2073 (2021).
Désilets, A., Longpre, J. M., Beaulieu, M. E. & Leduc, R. Inhibition of human matriptase by eglin c variants. FEBS Lett. 580, 2227–2232 (2006).
Phoo, W. W. et al. Structure of the NS2B-NS3 protease from Zika virus after self-cleavage. Nat. Commun. 7, 13410 (2016).
Klein, P. et al. Naphthoquinones as covalent reversible inhibitors of cysteine proteases-studies on inhibition mechanism and kinetics. Molecules 25, 2064 (2020).
Ludewig, S., Kossner, M., Schiller, M., Baumann, K. & Schirmeister, T. Enzyme kinetics and hit validation in fluorimetric protease assays. Curr. Top. Med. Chem. 10, 368–382 (2010).
Müller, C. et al. Broad-spectrum antiviral activity of the eIF4A inhibitor silvestrol against corona- and picornaviruses. Antivir. Res. 150, 123–129 (2018).
Kabsch, W. XDS. Acta Crystallogr. D. Biol. Crystallogr. 66, 125–132 (2010).
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D. Biol. Crystallogr. 66, 22–25 (2010).
Medrano, F. J., Andreu, J. M., Gorbunoff, M. J. & Timasheff, S. N. Roles of ring C oxygens in the binding of colchicine to tubulin. Biochemistry 30, 3770–3777 (1991).
Müller, P. et al. Investigation of the compatibility between warheads and peptidomimetic sequences of protease inhibitors-A comprehensive reactivity and selectivity study. Int. J. Mol. Sci. 24, 7226 (2023).
Martí, S. QMCube (QM(3)): an all-purpose suite for multiscale QM/MM calculations. J. Comput Chem. 42, 447–457 (2021).
Frisch, M. J. et al. Gaussian 09, Version A.1. Gaussian Inc. (Wallingford, CT, 2009).
Duan, Y. et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012 (2003).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of Four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2008).
Hehre, W. J., Radom, L., Schleyer, P. V. R. & Pople, J. A. Ab Initio Molecular Orbital Theory, (John Wiley, New York, 1986).
Arafet, K., Ferrer, S. & Moliner, V. Computational study of the michaelis complex formation and the effect on the reaction mechanism of Cruzain Cysteine protease. ACS Catal. 7, 1207–1215 (2017).
Arafet, K., Swiderek, K. & Moliner, V. Computational study of the Michaelis complex formation and the effect on the reaction mechanism of Cruzain Cysteine protease. ACS Omega 3, 18613–18622 (2018).
Arafet, K., Gonzalez, F. V. & Moliner, V. Elucidating the dual mode of action of Dipeptidyl Enoates in the Inhibition of Rhodesain Cysteine Proteases. Chemistry 27, 10142–10150 (2021).
Chan, H. T. H. et al. Discovery of SARS-CoV-2 M(pro) peptide inhibitors from modelling substrate and ligand binding. Chem. Sci. 12, 13686–13703 (2021).
Henkelman, G. & Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 113, 9978–9985 (2000).
Torrie, G. M. & Valleau, J. P. Non-physical sampling distributions in Monte_Carlo Free-Energy estimation-umbrella sampling. J. Comput. Phys. 23, 187–199 (1977).
Kumar, S., Bouzida, D., Swendsen, R. H., Kollman, P. A. & Rosenberg, J. M. The weighted histogram analysis method for free-energy calculations on biomolecules.1. The Method. J. Comput. Chem. 13, 1011–1021 (1992).
Dewar, M. J. S., Zoebisch, E. G., Healy, E. F. & Stewart, J. J. P. The development and use of quantum mechanical molecular-models. 76. AM1: a new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 107, 3902–3909 (1985).
Acknowledgements
This research was funded by the Consejo Superior de Investigaciones Científicas, grant number PIE-202020E224, the Spanish Ministerio de Ciencia e Innovación (ref. PID2021-123332OB-C21 and PID2019-107098RJ-I00), the Generalitat Valenciana (PROMETEO with ref. CIPROM/2021/079, and SEJI/2020/007), Universitat Jaume I (UJI-B2020-03, UJI-B2021-71 and SomUJIcontracovid crowdfunding campaign). K.Ś. thanks to Ministerio de Ciencia e Innovación and Fondo Social Europeo for a Ramon y Cajal contract (Ref. RYC2020-030596-I). The authors wish to thank the staff of beamlines ID30B (ESRF Synchrotron) and BL13-XALOC (ALBA Synchrotron) for their generous and much appreciated support, and the Serveis Centrals d’Instrumentació Científica of Universitat Jaume I for technical support. The work was also supported by a Research Grant of the University Medical Center Giessen and Marburg (UKGM, to C.M.), the von Behring-Röntgen-Stiftung (project 71_0016, to C.M.) the Deutsche Forschungsgemeinschaſt (DFG, project 530813989, to C.M.). Finally, the authors acknowledge the computer resources at Mare Nostrum of the Barcelona Supercomputing Center (QH-2022-2-0004 and QH-2022-3-0008), as well as the local computational resources founded by Generalitat Valenciana - European Regional Development Fund (REF: IDIFEDER/2021/02).
Author information
Authors and Affiliations
Contributions
All authors contributed to this work. A.R. and F.V.G. conceived and designed the study; F.J.M. obtained the crystal structures and performed the CD experiments; A.G.-M. performed the kinetic experiments; E.S. performed the cloning; S.d.l.H.-R. performed the synthesis; S.J.H. performed protein expression, purification, and enzymatic assays of Mpro and off-targets; C.M. and J.Z. performed the virus and cell assays; C.Z., A.W. and R.Z. performed the pharmacokinetic assays; S.M., K.A. and A.L. carried out the computer simulations; All authors participated in the discussion of the results; F.J.M., F.V.G., V.M., A.L., K.Ś., A.R. and T.S. participated in writing the original draft. All authors have read and approved the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Jun Wang and the other anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Medrano, F.J., de la Hoz-Rodríguez, S., Martí, S. et al. Peptidyl nitroalkene inhibitors of main protease rationalized by computational and crystallographic investigations as antivirals against SARS-CoV-2. Commun Chem 7, 15 (2024). https://doi.org/10.1038/s42004-024-01104-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-024-01104-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
