
Abstract
Carbohydrate-protein interactions are key for cell-cell and host-pathogen recognition and thus, emerged as viable therapeutic targets. However, their hydrophilic nature poses major limitations to the conventional development of drug-like inhibitors. To address this shortcoming, four fragment libraries were screened to identify metal-binding pharmacophores (MBPs) as novel scaffolds for inhibition of Ca2+-dependent carbohydrate-protein interactions. Here, we show the effect of MBPs on the clinically relevant lectins DC-SIGN, Langerin, LecA and LecB. Detailed structural and biochemical investigations revealed the specificity of MBPs for different Ca2+-dependent lectins. Exploring the structure-activity relationships of several fragments uncovered the functional groups in the MBPs suitable for modification to further improve lectin binding and selectivity. Selected inhibitors bound efficiently to DC-SIGN-expressing cells. Altogether, the discovery of MBPs as a promising class of Ca2+-dependent lectin inhibitors creates a foundation for fragment-based ligand design for future drug discovery campaigns.
Similar content being viewed by others

Introduction
All cells are covered in a complex matrix of carbohydrates (glycans) with established roles in health and disease1,2. The mammalian immune system employs glycan-binding proteins (GBPs) for various processes during pathogen and tumor recognition, as well as the embryonic development3,4. However, many viruses (e.g. influenza5 and SARS-CoV-26), ESKAPE pathogens (e.g. Pseudomonas aeruginosa7) and tumors8 exploit GBPs for the host invasion and to suppress the immune cell responses. Consequently, GBPs evolved as promising therapeutic targets for antimicrobials, vaccines, and as drug delivery systems to combat autoimmune diseases9. For example, the neuraminidase inhibitors Zanamivir (Relenza®) and Oseltamivir (Tamiflu®) are currently on the market for the treatment of influenza virus infections, which demonstrate the utility of GBP’s inhibitors in anti-infectious therapy10. Moreover, lectins can be used as selective drug-delivery systems. This approach has been successful for Givosiran targeting hepatocytes through the asialoglycoprotein receptor (ASGPR)11 and the development of dendritic cell-based vaccines as in the case of Dec-205 (CD205)12. Cumulatively, such examples increase the therapeutic interest in other clinically relevant GBPs.
In recent years, the Ca2+-dependent C-type lectin receptors (CLRs) have emerged as therapeutic targets due to their cell-specific expression in immune cells and roles in cell signaling, self- or pathogen recognition, and antigen presentation13. In particular, dendritic cell-specific ICAM-3 grabbing non-integrin (DC-SIGN, CD209) and Langerin (CD207) are expressed in macrophages or dendritic cells (DCs), and a subset of skin cells called Langerhans cells, respectively14,15. Pathogens such as Mycobacterium tuberculosis16 or viruses (e.g. HIV type-117, Ebola18, SARS-CoV19, and SARS-CoV-220) use their D-mannose (Kd of 3.5–6.1 mM21) branched carbohydrates to attach to Langerin and DC-SIGN even promoting an immune escape in some cases. Therefore, reports showing the role of DC-SIGN in promoting HIV trans-infection of T cells22 or the involvement in SARS-CoV-2 uptake23 have drawn a lot of attention to it as a potential target for antiviral therapy. Aside from pathogen recognition, Langerin is an efficient endocytic recycling receptor with established roles in immunity and tolerance24. Thus, both CLRs evolved as attractive targets for the design of anti-infectives, as well as for vaccine and antigen delivery to dendritic cells in immunotherapy25.
Besides mammalian CLRs, bacterial lectins have also been proposed as viable targets for drug development, such as the opportunistic pathogens Burkholderia ambifaria and P. aeruginosa expressing BambL and LecA (PA-IL) / LecB (PA-IIL), respectively26. LecA and LecB attach with a remarkably high affinity to α-D-galactose (Kd = 50 µM27) and α-L-fucose (Kd = 3 µM28) containing glycans on the surface of mammalian cells causing bacterial biofilms in the lungs of immunocompromised patients. Therefore, blocking these interactions may replace or complement the antibiotic treatment of ESKAPE pathogens29.
Developing the lectin-directed therapeutics is desirable to interfere with the bacterial30 and viral31 infections, as well as for cancer32 and autoimmune diseases33. However, inhibitors for the orthosteric site of Ca2+-dependent lectins have been a bottleneck for drug discovery34. This is not surprising given its hydrophilic nature and the high affinity for a metal cofactor such as Ca2+ (e.g. Langerin: Kd = 130 µM35, LecA: Kd = 60 µM36), which directly determines the affinity and the specificity of the carbohydrate-lectin interactions37. Therefore, the flexibility and electronic structure of the metal-centered complexes further complicate shaping metal-fragment interactions in ‘classical’ drug design38. This overall rendered this target class challenging and contributed to the vast underrepresentation of lectin inhibitors in the drug space39.
Recent advances towards the development of lectin inhibitors focus on mimicking the natural ligands of lectins (glycomimetics) through the design of mono- and multivalent oligosaccharides with various conjugates (glycopeptides, glycoclusters, and glycopolymers)40. However, they pose a pharmaceutical challenge such as routes of administration and possible side effects39. Several carbohydrate-based glycomimetics are available for CLRs41,42 and bacterial lectins29,43,44, but only a few non-carbohydrate drug-like lectin inhibitors are reported45,46. Previously, fragment-based drug design (FBDD) was successfully applied for assessing the druggability of CLRs47. However, fragments likely to interfere with primary in vitro screening assays (PAINS) were excluded from the general fragment libraries as their presence can partially or fully contradict the screen48. Therefore, a target-directed FBDD campaign using metal-binding pharmacophores (MBPs) has been proposed49. Certainly, drugs on the market as captopril50 and suberanilohydroxamic acid (Zolinza)51 demonstrate the success of this strategy. Interestingly, many clinically relevant proteins in the genome as lectins require a metal ion to maintain their stability and activity, but MBPs have not been considered for non-metalloenzymes49.
Here, we show that metal-dependent lectins are more druggable than previously anticipated. To address this, we screened four fragment libraries identifying MBPs as promising scaffolds. To demonstrate the potential of MBPs in inhibition of carbohydrate-protein interactions, we studied their effect on clinically relevant Ca2+-binding lectins (DC-SIGN, Langerin, LecA, and LecB), whereas a metal-independent lectin BambL served as a control (Fig. 1a). Employing NMR52, surface plasmon resonance (SPR), X-ray crystallography, as well as fluorescence polarization (FP53) assays, we explored the structure-activity relationships of several MBPs to further improve lectin binding and selectivity. Finally, the activity of MBPs in a physiologically relevant cellular environment was assessed in a cell-based fragment screen assay (CellFy)54.

a Cartoon representation of monomer crystal structures showing the carbohydrate-recognition domain (CRD) bound to the monosaccharides (orange) in a Ca2+-dependent manner (green spheres): LecA (D-galactose, PDB: 4CP9), Langerin (D-mannose, PDB: 3P5D), DC-SIGN (D-mannose, PDB: 1SL4), LecB (L-fucose, PDB: 5A70). BambL does not require Ca2+ for carbohydrate binding (L-fucose, PDB: 3ZZV). b All targets were screened computationally and experimentally using 3F Fsp3, general and MBP libraries. c Shown are 19F T2-filtered NMR spectra of a fragment mixture of the MBP library in the presence of 5 mM EDTA alone or with 20 µM LecA followed by competition with 30 mM CaCl2. The fragments undergoing a chemical shift perturbation (CSP) above 0.01 ppm (violet arrow) in the presence of LecA and CaCl2 indicated a Ca2+-dependent binding. d Heat map shows the percentage values (%) of total hit rates of four libraries screened against five lectins revealing that MBP library improved the hit rates for Ca2+-binding lectins (37–50%).
Results
Metal-binding pharmacophores target Ca2+-dependent lectins
The druggability of four Ca2+-dependent lectins was assessed and compared to a metal-independent lectin BambL using a virtual and two experimental: 3F Fsp3-rich and diversity-oriented (general), libraries (Fig. 1b), as described in the Supplementary Results and Discussion (Figure S1). Despite the limitations of in silico approaches, 19F NMR screening of the general library revealed the druggability of Ca2+-dependent lectins LecA and LecB, where MBPs were identified as the most potent drug-like molecules (Figures S2 and S3).
To demonstrate the potential of MBPs in targeting the carbohydrate-binding site of Ca2+-dependent lectins, we subjected 142 commercial MBP fragments for the binding studies with one non-Ca2+-(BambL) and four Ca2+-dependent lectins (LecA, LecB, Langerin and DC-SIGN). In the 19F NMR screening of the MBP library, the chemical shifts of many 19F-labeled fragments were perturbed in the presence of CaCl2 alone, whereas even larger CSPs were observed in the presence of both CaCl2 and the Ca2+-dependent lectins as shown on the example of LecA binding to 1s-1v (Fig. 1c), which allowed us to exclude direct fragment-metal interactions as a major contribution. Notably, the MBP library improved the hit rates for lectins with one (LecA: 54.2%, Langerin: 62.8%), two (LecB: 42.8%) and three Ca2+ ions (DC-SIGN: 42.5%), but not for the metal-independent lectin BambL (7.7%) compared to 3F Fsp3-rich and general libraries (Fig. 1d).
Next, we validated 19F NMR hits from the general and MBP libraries for binding to 15N-labeled LecA, LecB and the carbohydrate recognition domain (CRD) of DC-SIGN in 1H-15N HSQC/TROSY NMR experiments, identifying hydroxamic (1) and malonic (58) acids as potent scaffolds for targeting Ca2+-dependent lectins (LecA LecB and DC-SIGN). Further, both scaffolds were subjected to explorative structure-activity relationship (SAR) studies aiming to demonstrate the Ca2+-dependency and selectivity of the scaffolds for the Ca2+-dependent lectins.
Hydroxamates as Ca2+-dependent inhibitors of LecA
To characterize the interaction of LecA with the hydroxamate 1 and to enhance its potency, we initiated a SAR study employing five biophysical assays. A full discussion can be found in the Supplementary Results and Discussion. Briefly, we ranked 49 analogs of 1 using TROSY NMR, where we quantified the changes in the chemical shift perturbations (CSPs) of 15N LecA in the presence of the 1 analogs and MeGal as a positive control (Figure S4a). The derivatives promoted CSPs in 15N LecA similar to MeGal and 1 as shown in the case of 35 (Fig. 2a, b), suggesting that hydroxamates targeted the orthosteric site of LecA. Moreover, none of the marketed metalloproteinase inhibitors (47, 49, 50) bound to 15N LecA (Figures S4b-c), which indicated the presence of functional groups in the marketed drugs that sterically prevented a beneficial coordination of Ca2+ in LecA. Cumulatively, we identified 18 analogs of hydroxamate 1 targeting LecA with the potentially higher affinities.

a Examples of the residues in the carbohydrate-binding site of 15N LecA in TROSY NMR in the presence of DMSO (gray), 3 mM 1 (blue), 35 (red) or 1 mM methyl α-D-galactoside (MeGal, black). b Quantitative analysis of the chemical shift perturbations (CSPs > 0.01 ppm, dashed line) and changes in the peak intensity (dotted line) promoted by 1, 35 and MeGal. The resonances 16 and 68 were highlighted (violet) showing hydroxamates perturbed the residues similarly to MeGal. c Shown is a Ca2+-dependent binding of 0.01 mM LecA to 5 (dashed line) in the competitive 19F T2-filtered NMR, as evident by a decrease and recovery of the spy 5 with Ca2+ (10 mM CaCl2) and without (2 mM EDTA), respectively. Moreover, 5-LecA interaction showed a higher selectivity over other non- and Ca2+-dependent lectins. d Competitive binding assay based on fluorescence polarization with MeGal and GalNAc (left) as positive controls for strong and weak ligands, respectively. The hydroxamates 6, 20, 35, 36 and 39 (right) demonstrated LecA-dependent inhibitory properties. Inhibition values of compounds were derived at 10 mM and referenced to MeGal (16 h). e Shown is PrOF NMR of 0.15 mM 5FW LecA with 3 mM 35 (upper panel) binding to the carbohydrate pocket as evident by a CSP of W42. One-site binding model of 35 (bottom panel) used to derive the affinity (Kd) by following CSPs of W42. The error bars are showing the mean (n = 3). The Kd values were used to calculate LE. f Crystal structures show the interactions between LecA and 35 (PDB: 7FJH) and (g) galactose (PDB: 1OKO).
In parallel, we validated TROSY NMR results using SPR, competitive 19F NMR, FP assay and protein-observed 19F (PrOF) NMR. Given a weak affinity of hydroxamates towards LecA, SPR did not allow ranking of the derivatives (Figure S5). Therefore, we designed a competitive T2-filtered 19F NMR experiment using the hydroxamate derivative (5) as a 19F spy molecule. To prove the utility of this assay, we evaluated the Ca2+-dependency and selectivity of the hydroxamate-LecA interaction and compared it to other metal-dependent lectins, i.e. LecB, DC-SIGN and Langerin (Fig. 2c). To test the Ca2+-dependency of the interactions of 5 with lectins, a T2-filtered 19F NMR spectrum of 5 was recorded with 2 mM EDTA, 10 mM CaCl2 alone and with 10 µM lectins added. Both Langerin and DC-SIGN showed a Ca2+-independent binding to hydroxamates (Figure S6), whereas only 5-LecA interactions required Ca2+, demonstrating the Ca2+-dependency and early selectivity of the hydroxamate-LecA interaction. Consequently, we used this assay to rank the derivatives as discussed in the Supporting Information. Briefly, we observed a higher competition of 35 over the reporter 5 and other analogs suggesting 35 as the most potent hydroxamate derivative in this assay (Figure S7).
Next, we investigated the inhibitory properties and derived the affinities (Kd) of hydroxamates, which were active in NMR. In the FP assay the strongest inhibition compared to natural ligands of LecA, MeGal and GalNAc, was observed with linear 35 and cyclic 20 (Fig. 2d, Table 1)53. Following, PrOF NMR using LecA labeled with 5-fluorotryptophans (5FW) was employed. Since W42 is located in the orthosteric site of LecA, the binding and affinities of weak ligands could be determined36. As result, the perturbation of W42 in the presence of hydroxamates confirmed its binding to the orthosteric site of LecA (Fig. 2e and S8a, Table S4). Moreover, changes in W42 were used to estimate the affinities (Kd) of hydroxamates for LecA, as shown on example of 35 (Kd (35) = 4.6 ± 0.9 mM, Fig. 2e and S8b-c, Table S3). The analogs showed a similarly poor affinity compared to 35. Due to the small size of the fragments, there are only a limited number of the interactions they can engage in. However, they may bind tightly enough relative to their size and number of heavy atoms (HAs)55. Ligand efficiency (LE) is the key measure relating the potency of a fragment to the number of HAs and thus, would allow the development of drug-like molecules with enhanced binding modes for LecA56. Therefore, we selected only fragments with the highest affinity, LE and the % of inhibition values (Table 1).
Crystal structure of LecA in complex with 35 at 1.8 Å resolution in space group P212121 provided the first evidence for the interaction between a hydroxamate and a nonenzyme metal-binding protein. The electron density corresponding to the full structural model for 35 was detected at the carbohydrate-binding sites in subunits A and D of LecA (Fig. 2f and S9a-b). Comparing the structures of LecA-35 and LecA-galactose complexes illustrated the galactose-mimicking properties of 35 (Fig. 2g). In the LecA-35 complex, two oxygen atoms in the hydroxamic acid functional group coordinate the Ca2+ ion via bidentate chelation, which is also typical for interactions between hydroxamate-based inhibitors and metal ions in metalloenzymes49. The two oxygen atoms form hydrogen bonds with N107 and D100, mimicking OH3 and OH4 of galactose. A water molecule (WAT2) is in contact with the nitrogen atom in the hydroxamic acid via a hydrogen bond. WAT2 mimics the role of galactose OH6 by forming hydrogen bonds with the side chains of H50, Q53, the main chain oxygen atom of P51 and water WAT1. Notably, the terminal benzyl ring forms CH-π interactions with P38, an interaction not observed for other LecA-targeting glycomimetics (Figure S9c)57.
Taken together, we report the first evidence of hydroxamates targeting carbohydrate-protein interactions. Moreover, we conducted several experiments highlighting the selectivity of hydroxamates towards LecA over LecB and mammalian Ca2+-dependent lectins (Langerin/DC-SIGN). Therefore, we believe hydroxamates are promising molecules for the design of drug-like inhibitors against bacterial infections.
Malonates target lectins with multiple Ca2+ ions
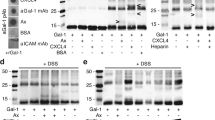
In the 19F and 1H-15N HSQC/TROSY NMR screenings of the MBP library, we discovered malonate 58 as a potent scaffold for targeting the Ca2+-dependent lectins LecA, LecB and DC-SIGN. To prove the utility of a target-oriented MBP-FBDD approach, we aimed to unravel its interactions with lectins having one, two, and three Ca2+ ions in or close to the orthosteric site. Here, BambL served as control for a metal-independent lectin expected not to interact with MBPs such as malonates. For this, the malonate 58 was subjected to docking and an initial SAR study using commercial analogs (Fig. 3a, Table S6). The results of both studies are discussed in detail in the Supplementary Results and Discussion. Briefly, docking simulation proposed that the malonate 58 could coordinate one or multiple Ca2+ ions in LecA, DC-SIGN and LecB, respectively (Fig. 3a, S10–S12). Therefore, we investigated the Ca2+-dependency and selectivity of malonate-lectin interactions. Taking advantage of a C-F bond in α-position of the malonic acid group of 61, we used it similar to 5 as a 19F spy in a competitive 19F T2-filtered NMR experiment (Fig. 3b). We observed 61 binding to all lectins except for Langerin in the presence of 10 mM CaCl2 suggesting that malonates target the orthosteric sites of lectins in a Ca2+-dependent manner. To rule out an off-target effect, we added a competitor (LecA: 10 mM MeGal, LecB/BambL: 10 mM MeFuc, and DC-SIGN/Langerin: 30 mM D-mannose) expecting the 19F peaks to recover if malonates target the orthosteric site of Ca2+-dependent lectins. Indeed, the competitors displaced 61 in LecA, LecB and DC-SIGN. On the other hand, the 19F peak intensities remained unchanged in Langerin and BambL, which was probably due to 61 binding to the secondary druggable sites58,59,60. Cumulatively, our results proposed that malonates interact with the orthosteric sites of metal-dependent lectins LecA, LecB and DC-SIGN in a Ca2+-dependent manner.

a Docking poses of 58 with one and two Ca2+ in or near the carbohydrate-binding site of LecA and LecB (PDB: 4CP9, 1OXC). The residues proposed to interact with lectins were highlighted (violet). b 19F T2-filtered NMR using the analog of 58 (61) was used to assess the Ca2+-dependency and selectivity of the malonate-lectin interaction, where 61 bound to 10 mM CaCl2 given a perturbation of fluorine resonance. All lectins interacted with 61 in a Ca2+-dependent manner. The presence of competitors: 10 mM MeGal (LecA), 10 mM MeFuc (LecB/BambL) and 30 mM D-mannose (DC-SIGN/Langerin) displaced 61 in LecA, LecB and DC-SIGN ECD verifying that malonates target the carbohydrate-binding site. c Shown is PrOF NMR of 0.15 mM 5FW LecA bound to 2 mM 58 or 61 as evident by CSPs of W42 and W33 (arrow). DMSO and MeGal served as negative and positive controls, respectively. (d) Examples of 15N LecB residues in TROSY NMR alone (black) and with 2 mM 58 (orange) or MeFuc (gray). 58 perturbed the resonances 85, 46, 29 and 79 similarly to MeFuc. e and f One-site binding model was applied to derive the Kd values of 58 and 64 by following CSPs of the resonances perturbed similar to MeFuc.
Next, we aimed to confirm our observations for LecA and LecB and investigated whether the number of Ca2+ ions in the orthosteric site plays a role for the interaction with malonates. For LecA, PrOF NMR with 5FW-labeled lectin was used to gain information on the binding site of 58 analogs. Similar to hydroxamates, 58 perturbed W42 in PrOF NMR demonstrating its binding to the orthosteric site of 5FW LecA (Fig. 3c). Subsequently, we aimed to rank the impact of 13 analogs of 58 on W42 to prioritize scaffolds for future fragment evolution. The compounds with an acetic acid group (63 and 70, Table S6) did not interact with 5FW LecA, which supported our docking result (Figures S10a-b), whereas the scaffold 61 promoted the strongest perturbation of W42 (Figures S10c-d). Altogether, malonates (58 and 61) interact with the orthosteric site of LecA bearing one Ca2+ ion, similar to hydroxamates.
For LecB as a lectin with two Ca2+ ions, a similar tendency in malonate binding to the orthosteric site was observed in TROSY NMR. Here, we observed similar changes in residues of 15N LecB resonances with 58 compared to the positive control MeFuc, whereas the binding was in a Ca2+-dependent manner (Fig. 3d, S13–S14). Therefore, we determined the affinities (Kd) and LE of 58 analogs using TROSY NMR, which were in a low mM range (58: Kd = 1.2 ± 0.4 mM, Fig. 3e). Interestingly, all structural derivatives of 58 gave a rather flat SAR and showed comparable affinities for LecB (Table 2, Figure S15), which is discussed in detail in the Supporting Information. Notably, only 64 had a decrease in affinity (64: Kd = 2.6 ± 0.6 mM, Fig. 3f) indicating the role of an electronegative group in binding to LecB. This observation supported our docking study, where CF2 group of 58 was predicted to interact with the protein surface through T98 using a hydrogen bond (Figures S11a-b). However, due to the lack of 15N LecB protein backbone assignment, co-crystallization studies are ongoing to support it.
Taken together, malonates interacted with lectins with one (LecA) and two (LecB) Ca2+ ions in the orthosteric site unlike hydroxamates coordinating only one Ca2+ ion in the orthosteric site of LecA. Moreover, our SAR study with LecA and LecB suggested that the selectivity of malonates is tunable as 61 had a stronger tendency to bind LecA, whereas 58 showed the preference for LecB. Finally, malonates have a higher tendency to bind to secondary sites in allosteric lectins, such as Langerin and BambL. Cumulatively, malonates offer a potential scaffold for design of orthosteric and allosteric inhibitors.
Malonates to design inhibitors of DC-SIGN (CD209)
To investigate the ability of malonates to bind lectins with three Ca2+ ions, we probed malonate—DC-SIGN interaction using protein-observed 15N HSQC NMR and a cell-based assay (cellFy)54. A full discussion on the SAR study of 58 and DC-SIGN can be found in the Supplementary Results and Discussion (Figure S16). Briefly, malonate 58 perturbed the resonances in the EPN motif coordinating Ca2+ (site 1) and D367, whereas L321 and E324 near Ca2+ (site 2) and Ca2+ (site 3) showed weaker effects (Fig. 4a, b). Quantitative analyses of the chemical shift perturbations (CSPs) caused by 58 revealed similar CSPs compared to the positive control D-mannose, suggesting that 58 mimics the carbohydrate binding to DC-SIGN (Fig. 4c, d). In particular, the interaction of 58 with the Ca2+(site 1)-binding site of DC-SIGN occurred through N365, N344 and F313 being in agreement with our docking predictions. We then derived the affinities (Kd) and LE values of 58 analogs for DC-SIGN CRD using 15N HSQC NMR. The analogs of 58 showed a similar affinity (58: Kd = 1.2 ± 0.5 mM, Table 2, Fig. 4e and S17) and thus, three scaffold groups were defined as interchangeable (58, 62 and 69). Similar to LecB, the compounds with an electronegative group on the ring (58, 62, 63 and 67) were predominant and thus, in agreement with the predicted F313 interaction of DC-SIGN with CF2 group in 58. Compared to LecA and LecB, a methyl group in 59 was well tolerated in DC-SIGN CRD (Fig. 4f). Therefore, this position is potentially suitable for future fragment growing to gain malonate specificity towards DC-SIGN. Together, both computational and experimental data demonstrated that malonates could target the Ca2+ (site 1) binding site of DC-SIGN similarly to D-mannose.

a Protein fingerprint of 15N DC-SIGN CRD in 15N HSQC NMR alone (black) and with 2 mM 58 (orange) or 30 mM D-mannose (gray). Similar to D-mannose, 58 perturbed the resonances D367, G346 and L321, but not E324 being in the Ca2+ (site 1) and Ca2+(site 1)/Ca2+(site 3)-binding sites, respectively. These residues are highlighted in (b), which shows a docked 58 in the Ca2+ site 1 of DC-SIGN CRD (PDB: 2XR5). c Quantitative analysis of chemical shift perturbation (CSP) of 15N DC-SIGN CRD in the presence of 58 and D-mannose. Resonances D367, G346, L321 and E324 are highlighted (violet). A schematic representation of DC-SIGN CRD is shown on top. Malonates perturbed resonances similarly to D-mannose, i.e. in the long loop locating the EPN motif (violet). d The CSPs promoted in 15N DC-SIGN CRD by 58 were transferred to DC-SIGN CRD (PDB: 2XR5). e One-site binding model of 58 to DC-SIGN CRD derived by following CSPs of the resonances perturbed in the long loop and β4 sheet. f Heat map shows the 1:0 hit rate of 58 analogs for LecA, LecB and DC-SIGN normalized to the effect of MeGal, MeFuc and D-mannose, respectively. g Shown are flow cytometry histograms of DC-SIGN+ and Langerin+ Raji cells bound to FITC-dextran in the presence of DMSO or D-mannose used as a negative and positive controls in a cell-based (cellFy) assay, respectively. 58 inhibited FITC-dextran binding to DC-SIGN similarly to D-mannose, but not to Langerin.
To assess the activity of 58 in a physiologically more relevant cellular environment, we explored the ability of malonates to inhibit the carbohydrate binding in a cell-based system (cellFy)54. For this, we assessed the binding of 58 to DC-SIGN+ and Langerin+ Raji cells by flow cytometry (Fig. 4g). Indeed, we observed dose- and DC-SIGN-dependent binding of 58 to DC-SIGN+ cells as well as negligible cytotoxicity at low concentrations. Most importantly, malonate 58 specifically bound to DC-SIGN+ cells and not to Langerin-expressing cells, although both lectins share a common recognition motif called EPN. Together, our cellFy data demonstrated that malonates specifically inhibit DC-SIGN-carbohydrate interactions in the cellular context similar to D-mannose.
Together, our SAR study demonstrated that the selectivity of malonates is tunable. Moreover, the malonate scaffold can inspire the design of lectin inhibitors with one, two and three Ca2+ ions, as shown exemplarily for LecA, LecB and DC-SIGN, respectively. Thereby, 61 had a stronger tendency to interact with LecA, whereas LecB and DC-SIGN preferred 58 and 62 scaffolds, and the observation with 59 is valuable to gain malonate specificity towards DC-SIGN.
Discussion
Interfering with GBP interactions has a high therapeutic potential. However, identifying a suitable starting point for the development of lectin inhibitors is challenging. Despite that several mono- and multivalent carbohydrate-based glycomimetics are available for lectins40, poor pharmacokinetic properties of the most potent multivalent inhibitors are a large drawback for the clinical approval. This further underscores the attractiveness of drug-like noncarbohydrate lectin inhibitors. Applying fragment screening, we approached this challenge focusing on metal-dependent lectins. As result, we identified metal-binding pharmacophores (MBPs) as novel scaffolds for targeting Ca2+-dependent carbohydrate-protein interactions.
Recently, we reported the molecular basis of a catechol binding to LecA, where two hydroxyls coordinated a Ca2+ ion in the orthosteric site of LecA and Langerin45. Here, we identified hydroxamic acids as additional drug-like molecules to target the Ca2+-bearing orthosteric site of LecA. As hydroxamates are the most widely used inhibitors of various metalloenzymes, to the best of our knowledge this is the first report demonstrating their interaction with non-metalloenzymes. The SAR study revealed a sterically optimal presentation of the hydroxamic acid group. However, the potencies of hydroxamates are certainly lower than those observed for metalloenzymes, and still have to be improved. Nevertheless, hydroxamates offer a promising selectivity towards LecA over other Ca2+-dependent lectins. This knowledge will contribute to the design of specific hydroxamate-based inhibitors for LecA, which have the potential to interfere with P. aeruginosa biofilm integrity and increase susceptibility to antibiotic treatment of immunocompromised patients.
The improved druggability rates for the orthosteric site of the Ca2+-dependent lectins with catechols and hydroxamates encouraged us to initiate a target-oriented MBP-FBDD campaign. Employing MBP library, we have shown a fourfold improved hit rates for lectins with one (LecA and Langerin) and multiple Ca2+ ions (DC-SIGN and LecB) in the orthosteric site. Hereby, a malonate scaffold 58 was identified as a potent drug-like molecule for targeting lectins with one or multiple Ca2+-ions. Notably, this malonate showed low cytotoxicity (up to 10 mM) and selectivity for DC-SIGN+, but not Langerin+ Raji cells in vitro using a cell-based assay (cellFy). Moreover, using commercially available analogs of malonate 58, we demonstrated that tuning the selectivity of malonates towards lectins with one to multiple Ca2+ ions was possible. Aside from the Ca2+-dependent binding of a common malonic acid group, we discovered an additional interaction of 58’s electronegative CF2 group with the protein surfaces of DC-SIGN and LecB in HSQC/TROSY NMR. This secondary interaction was not observed for LecA, which demonstrated a preference for a malonate with a different scaffold (61). We believe our discovery will assist the design of malonate-based lectin inhibitors.
Taken together, metal-coordinating fragments are the most prominent drug-like molecules for targeting the metal-dependent lectins. Since the most potent hydroxamate (35) and malonate (58) scaffolds are still small with 179 and 208 Da, respectively, both serve as suitable starting points for the fragment evolution, which could lead to the first drug-like inhibitors for the orthosteric sites of mammalian and bacterial Ca2+-dependent lectins.
Methods
Materials
The carbohydrates D-mannose (CAS: 3458-28-4) and methyl α-D-galactoside (MeGal, CAS: 3396-99-4) were purchased from Sigma-Aldrich Chemie GmbH, whereas methyl α-L-fucoside (MeFuc, CAS: 14687-15-1) was from Biosynth-Carbosynth (UK). Commercial analogs (Table S3, compounds 3-5, 9-35, 46-50) of hydroxamic and malonic acids (Table S6) were purchased from Life Chemicals Europe GmbH (Germany), Otava Chemicals (Lithuania) or KeyOrganics (UK). The non-labeled or 15N-labeled LecA61, LecB62 and BambL63 were purified in soluble form as reported previously. Human 15N-labeled Langerin CRD and non-labeled ECD or DC-SIGN CRD and non-labeled ECD constructs were expressed in inclusion bodies and prepared as described before35,58.
Virtual screening
The detailed protocol including the list of analyzed LecA (Table S1) and LecB (Table S2) crystal structures used for virtual screening was described in Supplementary Materials and Methods.
Fragment libraries
The 3F and general 19F fragment libraries were prepared as reported previously47,64. MBP library of 142 commercially available fragments purchased from Otava Chemicals (Lithuania) was prepared. MBP fragments were chosen from the ‘Chelator Fragment Library’ based on the presence of a fluorine atom and various chelating groups represented by picolinic acids, pyrimidines, hydroxypyrones, hydroxypyridinones, sulfonamides, β-diketones, salicylic and hydroxamic acids. Fragments were subjected to a quality control for solubility and purity in 1H and 19F NMR yielding 98 fluorinated and 9 nonfluorinated fragments. Following, we combined fluorinated MBP fragments in mixtures of 30 to 32 fragments at 100 µM in a two-fold concentrated 19F screening buffer (25 mM in Tris-HCl pH 7.8, 150 mM NaCl, 100 µM TFA and 20% D2O) and stored at −20 °C upon use.
Chemical synthesis
Hydroxamate derivatives were prepared as well as analyzed for the identity and purity by NMR as described in Supplementary Materials and Methods and Supplementary Note 1, respectively.
NMR studies
All NMR measurements were performed on a Bruker Ascend™700 (AvanceIII HD) spectrometer equipped with a 5 mm TCI700 CryoProbe™ in 3 mm tubes (Norell S-3-800-7).
Fragment screening
NMR screening of fluorinated and non-fluorinated fragments from the 3F, general and MBP libraries was performed in 19F NMR and 1H-15N HSQC/TROSY NMR, respectively. For 19F NMR, two samples containing 1:1 fragment mixture and 10 µM protein in TBS/EDTA buffer (25 mM in Tris-HCl pH 7.8, 150 mM NaCl, 5 mM EDTA) or TBS/EDTA buffer alone were prepared and followed by addition of 10 mM CaCl2 and 1 mM MeGal (LecA), 1 mM MeFuc (LecB/BambL) and 30 mM D-mannose (DC-SIGN/Langerin). Two separate 19F NMR spectra were recorded at 298 K for CF3 and CF groups with 32 and 64 scans, a spectral width of 100 ppm, a transmitter offset at −50 and −150 ppm, acquisition time of 2 s and 1 s relaxation time, respectively. 19F T2-filtered (CPMG) spectra were recorded using a CPMG pulse sequence with a 180° pulse repetition rate of 384 ms using the same acquisition and relaxation times with 64 and 256 scans for CF3 and CF compounds, respectively. Data was acquired without proton decoupling. All spectra were referenced to the internal standard trifluoroacetic acid (TFA) at −75.6 ppm and analyzed in MestReNova 11.0.0 (Mestrelab Research SL) for the changes in peak intensity and chemical shift perturbations in the presence of protein. For 19F NMR spectra, chemical shift changes of 0.01 ppm or intensity changes between 25 and 50 % were defined as ‘high’ and ‘low’ confidence hits, respectively. For 19F T2-filtered NMR spectra, intensity changes of 20 and 50% or more than 50% were defined as ‘low’ and ‘high’ confidence hits, respectively. Fragments that bound proteins in the presence of 10 mM CaCl2 were used to derive total hit rates.
1H-15N HSQC and TROSY
The 1H-15N TROSY pulse sequence trosyf3gpphsi19 with 128 increments and 32 scans per increment was applied for 0.15 mM 15N LecA and 0.07 mM LecB in HBS/Ca2+ buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 10 mM CaCl2, 10% D2O and 100 µM sodium trimethylsilylpropanesulfonate (DSS) as internal reference) at 310 K. The 1H-15N HSQC pulse sequence hsqcf3gpph19 with 128 increments and 8 scans per increment was applied for 0.1 mM 15N DC-SIGN and Langerin CRD in HBS/Ca2+ buffer at 298 K. For screening of 650 non-fluorinated fragments of the general library against 15N LecA, a mix of 10 compounds at 1 mM each was combined with 0.15 mM 15N LecA in HBS/Ca2+ buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 10 mM CaCl2, 10% D2O and 100 µM sodium trimethylsilylpropanesulfonate (DSS) as internal reference, whereas the fragments with the MBP scaffolds were validated separately at 1 mM. Similarly, 9 nonfluorinated fragments from the MBP library were screened against 15N-labeled DC-SIGN, LecA and LecB. To validate the fragment binding to proteins, we recorded 1H-15N HSQC and TROSY NMR of proteins in the presence of DMSO as negative control and 2 mM fragments in HBS/Ca2+ buffer. Natural ligands were used as positive controls at 1 mM MeGal, MeFuc and 30 mM D-mannose to LecA/LecB and DC-SIGN CRD/Langerin CRD, respectively. Titration experiments with 15N-labeled DC-SIGN CRD or LecB were recorded in HBS/Ca2+ buffer with 30 mM CaCl2 and 0.25 to 4–8 mM malonic acid derivatives. The Kd values were calculated according to the one-site-binding model in Origin(Pro) 2020b (OriginLab Corp., USA). Ligand efficiency calculations were done according to Eq. (1) for a temperature of 298 K and 310 K for DC-SIGN CRD and LecB, respectively.
with the temperature T [K], the gas constant R [kcal mol−1 K−1], and the number of non-hydrogen atoms HA56.
All data were processed with NMRpipe65 and analyzed with CcpNmr analysis66. For data analysis, the protein fingerprint of 15N DC-SIGN (Table S7) or Langerin CRD was assigned as reported previously58. The 1H-15N TROSY resonances of 15N LecA and 15N LecB were indexed with IDs due to a lack of protein backbone resonance assignment (Tables S8-S9). Next, resonance IDs from protein spectra were transferred to the spectra obtained in the presence of compounds in order to compare the changes in chemical shift perturbations for the fast exchange peaks. The changes in chemical shift perturbations (CSPs) were calculated according to Eq. (2):
in which \(\delta\) is the difference in chemical shift (in ppm) and \(\alpha\) is an empirical weighting factor of 0.14 for all amino acid backbone resonances67. The threshold value was set based on three independent measurements of reference spectra to 0.01 ppm for LecA, LecB, DC-SIGN CRD and Langerin CRD.
Reporter-based 19F NMR
The analogs of 1 (5) and 58 (61) were used as fluorinated spy molecules in 19F T2-filtered NMR experiments. Two samples containing 0.1 mM 5 or 61 alone or a 10 µM protein in TBS/EDTA buffer followed by addition of 10 mM CaCl2 and the carbohydrates: 1 mM MeGal (LecA), 1 mM MeFuc (LecB/BambL) and 30 mM D-mannose (DC-SIGN/Langerin). Similarly, 0.1 mM 5 was used to prioritize hydroxamate derivatives, where 3 mM analogs of 1 were added to the samples instead of the carbohydrates. The 19F and 19F T2-filtered spectra were recorded with 64 scans, a spectral width of 5 ppm, a transmitter offset at −155.5 ppm (5) and −155.5 ppm (61), the acquisition time of 0.8 s, 2 s relaxation time applying a T2-filter of 384 ms. All spectra were analyzed as described above.
PrOF NMR
PrOF NMR was performed as reported previously36. Briefly, 150 µM 5FW LecA was recorded alone and in the presence of DMSO or 2–4 mM compounds in TBS/Ca2+ buffer. The difference in chemical shift perturbations of W42 in 5FW LecA free vs bound forms was followed to determine Kd values of compounds. The Kd values were calculated according to the one-site-binding model in Origin(Pro) 2020b (OriginLab Corp., USA) from two or three independent titrations. Ligand efficiency values were calculated according to Eq. (1) for a temperature of 310 K.
SPR
All experiments were performed on a BIACORE X100 instrument (GE Healthcare) at 25 °C in phosphate-buffered saline (10 mM phosphate buffer pH 7.4, 2.7 mM KCl, 137 mM NaCl, 0.05% Tween-20, 100 µM CaCl2, 5% DMSO). LecA was immobilized onto a CM7 chip (BIACORE) following standard amine coupling procedures. The CM7 chip was activated by three injections of a NHS/EDC mixture with a contact time of 540 s at a flow rate of 10 µL min−1 until the response exceeded 800 RU, followed by multiple injections of LecA dissolved in 10 mM sodium acetate pH 4.5 (100 µg/mL) onto channel 2 (contact time of 540 s at a flow rate of 10 µL min−1). A minimum of 10,000 RU of LecA was captured onto the chip. Initial binding screens were performed with the injections of 0.2 and 1 mM of each hydroxamate (association 30 s, dissociation 60 s, 30 µL min−1 flow rate), to identify the initial binders eliciting dose-response behaviors. Injection of the positive control (0.1 mM 4-Nitrophenyl-β-d-galactoside) was included after every forth injection cycle to monitor the activity of immobilized LecA throughout the binding screen experiments. All data evaluation was performed using BIACORE X100 evaluation software (v.2.0).
Competitive fluorescence polarization (FP) assay
The FP assay was performed in a black 384-well plate (Greiner Bio-One, Germany, 781900) with the final volume of 20 µL as described previously53. Briefly, 10 µL of a sample solution series (10–0.078 mM) in TBS buffer containing 10 mM Ca2+ (TBS/Ca2+-buffer) and 20% DMSO were added in technical triplicates to 10 µL LecA (40 µM) preincubated with a galactose-based Cy5 conjugate68 (20 nM) in TBS/Ca2+-buffer. Two positive controls (MeGal, IC50 = 140 ± 30 µM and GalNAc, IC50 = 1230 ± 20045), a blank (20 µM LecA, 10% DMSO in TBS/Ca2+-buffer), and a negative control (20 µM LecA, 10% DMSO, 10 nM galactose-based Cy5 conjugate in TBS/Ca2+-buffer) were included. The plate was sealed (EASYseal, Greiner Bio-One, 676,001), centrifuged (1 min, 1500 rpm, 25 °C) and incubated in a humidity chamber for 16 h. The fluorescence was measured with an excitation 590 nm and emission 675 nm filter68 on a PheraStar FS plate reader (BMG Labtech GmbH, Germany). The blank signal of TBS/Ca2+-buffer containing 10% DMSO was subtracted and the compounds were analyzed with the MARS Data Analysis Software (BMG Labtech GmbH Germany) using the four-parameter variable slope model. The top and bottom plateaus were defined according to the positive controls. The graphs were visualized using Graphpad Prism 5. The percentage (%) of inhibition was calculated at the highest concentration compared with 10 mM MeGal set to 100%.
Crystallography
Lyophilized powder of recombinant LecA was dissolved in MilliQ water containing 1 mM CaCl2 to the final protein concentration of 11.7 mg mL−1. 100 mM hydroxamate 1 stock solution in DMSO was diluted in the LecA solution to the final concentration of 20 mM and incubated at 25 °C to allow hydroxamate 1 to interact with LecA. 1.2 µL of the protein solution containing hydroxamate 1 was then mixed with 0.3 μL of LecA seed solution containing LecA microcrystals. 1.5 μL of reservoir solution (20% PEG6K, 1 M LiCl, 0.1 M sodium acetate pH 4) was added to the mixture. The entire mixture (total volume of 3 µL) was deposited on a siliconized glass slide. Crystallization was performed by the hanging drop vapor diffusion method on a 24-well plate with sealant (Hampton Research) at 19 °C. Crystals were cryo-protected in 30% PEG6000, 1 M LiCl, 100 mM sodium acetate pH 4, supplemented with 10 mM hydroxamate 1 and flash cooled in liquid nitrogen. X-ray diffraction data were collected at SOLEIL-PROXIMA2 (Saint Aubin, France) using EIGER X 9 M (Dectris) detector and analyzed as described in the Supporting Information (Table S5).
CellFy
The cellFy experiments using Langerin+ and DC-SIGN+ Raji cell lines were performed as described before54. Briefly, 50k cells were plated in a 96-well plate (clear, round bottom; Greiner Bio-One and mixed with varying concentrations of malonates 58, D-mannose and 0.025 mg mL−1 FITC-conjugated dextran (500 kDa, Sigma Aldrich) in a final volume of 50 µL following incubation for 30 min on ice. After centrifugation at 500 g for 3 min at 4 °C, the supernatant was discarded. After washing cells were treated with 50 µL 4 % paraformaldehyde (Roti-Histofix, Carl Roth) for 20 min on ice and resuspended in 100 µL fresh culture medium. The fluorescence of cells was measured by flow cytometry (MACSQuant Analyzer 16). Data was analyzed in FlowJo.
Docking
Docking poses were obtained by docking compound 58 in MOE69 using the Triangle Matcher and the Rigid Receptor as placement and refinement methods, respectively. For LecA and DC-SIGN CRD, only the highest-ranking pose was taken into account, whereas for LecB the best two ones were considered. Interaction maps were also retrieved from MOE.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Relevant data are available from the corresponding author on reasonable request. Atomic coordinates for LecA bound to hydroxamic acid 35 crystal structure has been deposited in the Protein Data Bank under accession numbers 7FJH.
References
Freeze, H. H., Baum, L., Varki, A., Glycans in Systemic Physiology. In Essentials of Glycobiology (eds Varki, A., Cummings, R. D., Esko, J. D., Stanley, P., Hart, G. W., Aebi, M., Darvill, A. G., Kinoshita, T., Packer, N. H., Prestegard, J. H., Schnaar, R. L., Seeberger, P. H.). Cold Spring Harbor Laboratory Press Copyright 2015-2017 by The Consortium of Glycobiology Editors, La Jolla, California. 521–516 (Cold Spring Harbor NY, 2015).
Freeze, H. H., Kinoshita, T., Varki, A., Glycans in Acquired Human Diseases. In Essentials of Glycobiology, (eds Varki, A., Cummings, R. D., Esko, J. D., Stanley, P., Hart, G. W., Aebi, M., Darvill, A. G., Kinoshita, T., Packer, N. H., Prestegard, J. H., Schnaar, R. L., Seeberger, P. H.) 583–595. Cold Spring Harbor Laboratory Press Copyright 2015-2017 by The Consortium of Glycobiology Editors, La Jolla, California. (Cold Spring Harbor NY, 2015).
van Kooyk, Y. & Rabinovich, G. A. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat. Immunol. 9, 593–601 (2008).
Fuster, M. M. & Esko, J. D. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat. Rev. Cancer 5, 526–542 (2005).
Ng, W. C., Tate, M. D., Brooks, A. G. & Reading, P. C. Soluble Host Defense Lectins in Innate Immunity to Influenza Virus. J. Biomedicine Biotechnol. 2012, 732191 (2012).
Gao, C. et al. SARS-CoV-2 Spike Protein Interacts with Multiple Innate Immune Receptors. bioRxiv 2020.07.29.227462, 2020.
Heggelund, J. E., Varrot, A., Imberty, A. & Krengel, U. Histo-blood group antigens as mediators of infections. Curr. Opin. Struct. Biol. 44, 190–200 (2017).
Munkley, J. & Elliott, D. J. Hallmarks of glycosylation in cancer. Oncotarget 7, 35478–35489 (2016).
Smith, B. A. H. & Bertozzi, C. R. The clinical impact of glycobiology: targeting selectins, Siglecs and mammalian glycans. Nat. Rev. Drug Discov. 20, 217–243 (2021).
Rewar, S., Mirdha, D. & Rewar, P. Treatment and Prevention of Pandemic H1N1 Influenza. Ann. Glob. health 81, 645–653 (2015).
Springer, A. D. & Dowdy, S. F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 28, 109–118 (2018).
Dhodapkar, M. V. et al. Induction of Antigen-Specific Immunity with a Vaccine Targeting NY-ESO-1 to the Dendritic Cell Receptor DEC-205. Sci. Transl. Med. 6, 232ra51–232ra51 (2014).
Geijtenbeek, T. B. & Gringhuis, S. I. Signalling through C-type lectin receptors: shaping immune responses. Nat. Rev. Immunol. 9, 465–479 (2009).
Geijtenbeek, T. B. et al. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell 100, 575–585 (2000).
Romani, N., Clausen, B. E. & Stoitzner, P. Langerhans cells and more: langerin-expressing dendritic cell subsets in the skin. Immunological Rev. 234, 120–141 (2010).
Geijtenbeek, T. B. H. et al. Mycobacteria Target DC-SIGN to Suppress Dendritic Cell Function. J. Exp. Med. 197, 7–17 (2002).
van den Berg, L. M. & Geijtenbeek, T. B. Antiviral immune responses by human langerhans cells and dendritic cells in HIV-1 infection. Adv. Exp. Med Biol. 762, 45–70 (2013).
Alvarez, C. P. et al. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 76, 6841–6844 (2002).
Yang, Z.-Y. et al. pH-Dependent Entry of Severe Acute Respiratory Syndrome Coronavirus Is Mediated by the Spike Glycoprotein and Enhanced by Dendritic Cell Transfer through DC-SIGN. J. Virol. 78, 5642–5650 (2004).
Lenza, M. P. et al. Structural Characterization of N-Linked Glycans in the Receptor Binding Domain of the SARS-CoV-2 Spike Protein and their Interactions with Human Lectins. Angew. Chem. (Int. ed. Engl.) 59, 23763–23771 (2020).
Holla, A. & Skerra, A. Comparative analysis reveals selective recognition of glycans by the dendritic cell receptors DC-SIGN and Langerin. Protein Eng. Des. Sel. 24, 659–669 (2011).
Geijtenbeek, T. B. et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100, 587–597 (2000).
Lempp, F. A. et al. Lectins enhance SARS-CoV-2 infection and influence neutralizing antibodies. Nature 598, 342–347 (2021).
Clausen, B. E. & Kel, J. M. Langerhans cells: critical regulators of skin immunity? Immunol. Cell Biol. 88, 351–360 (2010).
Engering, A., Geijtenbeek, T. B. H. & van Kooyk, Y. Immune escape through C-type lectins on dendritic cells. Trends Immunol. 23, 480–485 (2002).
Imberty, A., Wimmerova, M., Mitchell, E. P. & Gilboa-Garber, N. Structures of the lectins from Pseudomonas aeruginosa: insight into the molecular basis for host glycan recognition. Microbes Infect. 6, 221–228 (2004).
Rodrigue, J. et al. Aromatic thioglycoside inhibitors against the virulence factor LecA from Pseudomonas aeruginosa. Org. Biomol. Chem. 11, 6906–6918 (2013).
Sabin, C. et al. Binding of different monosaccharides by lectin PA-IIL from Pseudomonas aeruginosa: Thermodynamics data correlated with X-ray structures. FEBS Lett. 580, 982–987 (2006).
Meiers, J., Siebs, E., Zahorska, E. & Titz, A. Lectin antagonists in infection, immunity, and inflammation. Curr. Opin. Chem. Biol. 53, 51–67 (2019).
Poole, J., Day, C. J., von Itzstein, M., Paton, J. C. & Jennings, M. P. Glycointeractions in bacterial pathogenesis. Nat. Rev. Microbiol. 16, 440–452 (2018).
Van Breedam, W., Pöhlmann, S., Favoreel, H. W., de Groot, R. J. & Nauwynck, H. J. Bitter-sweet symphony: glycan–lectin interactions in virus biology. FEMS Microbiol. Rev. 38, 598–632 (2014).
Liu, Z., Zhang, Q., Peng, H. & Zhang, W.Z. Animal Lectins: Potential Antitumor Therapeutic Targets in Apoptosis. Appl. Biochem. Biotechnol. 168, 629–637 (2012).
Buzás, E. I. et al. Carbohydrate recognition systems in autoimmunity. Autoimmunity 39, 691–704 (2006).
Hopkins, A. L. & Groom, C. R. The druggable genome. Nat. Rev. Drug Discov. 1, 727–730 (2002).
Hanske, J. et al. Intradomain Allosteric Network Modulates Calcium Affinity of the C-Type Lectin Receptor Langerin. J. Am. Chem. Soc. 138, 12176–12186 (2016).
Shanina, E. et al. Protein-observed 19F NMR of LecA from Pseudomonas aeruginosa. Glycobiology 31, 159–165 (2021).
Imberty, A. & Prestegard, J. H., Structural Biology of Glycan Recognition. In Essentials of Glycobiology (eds Varki, A. et al.) 387–400. Cold Spring Harbor Laboratory Press Copyright 2015-2017 by The Consortium of Glycobiology Editors, La Jolla, California. (Cold Spring Harbor NY, 2015).
Riccardi, L., Genna, V. & De Vivo, M. Metal–ligand interactions in drug design. Nat. Rev. Chem. 2, 100–112 (2018).
Ernst, B. & Magnani, J. L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov. 8, 661–677 (2009).
Cecioni, S., Imberty, A. & Vidal, S. Glycomimetics versus multivalent glycoconjugates for the design of high affinity lectin ligands. Chem. Rev. 115, 525–561 (2015).
Wamhoff, E.-C. et al. 19F NMR-Guided Design of Glycomimetic Langerin Ligands. ACS Chem. Biol. 11, 2407–2413 (2016).
Thépaut, M. et al. Structure of a Glycomimetic Ligand in the Carbohydrate Recognition Domain of C-type Lectin DC-SIGN. Structural Requirements for Selectivity and Ligand Design. J. Am. Chem. Soc. 135, 2518–2529 (2013).
Sommer, R. et al. Glycomimetic, Orally Bioavailable LecB Inhibitors Block Biofilm Formation of Pseudomonas aeruginosa. J. Am. Chem. Soc. 140, 2537–2545 (2018).
Sommer, R. et al. Anti-biofilm Agents against Pseudomonas aeruginosa: A Structure-Activity Relationship Study of C-Glycosidic LecB Inhibitors. J. Med Chem. 62, 9201–9216 (2019).
Kuhaudomlarp, S. et al. Non-Carbohydrate Glycomimetics as Inhibitors of Calcium(II)-binding Lectins. Angew. Chem. (Int. ed. Engl.). 60, 8104–8114 (2020).
Sethi, A., Sanam, S. & Alvala, M. Non-carbohydrate strategies to inhibit lectin proteins with special emphasis on galectins. Eur. J. Med Chem. 222, 113561 (2021).
Aretz, J., Wamhoff, E.-C. Hanske, J., Heymann, D. & Rademacher, C. Computational and Experimental Prediction of Human C-Type Lectin Receptor Druggability. Front. Immunol. 5, 323 (2014).
Baell, J. B. & Nissink, J. W. M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 13, 36–44 (2018).
Chen, A. Y. et al. Targeting Metalloenzymes for Therapeutic Intervention. Chem. Rev. 119, 1323–1455 (2019).
Marte, F., Sankar, P., Cassagnol, M., Captopril. In StatPearls, StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC (Treasure Island FL, 2021).
Marks, P. A. & Breslow, R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 25, 84–90 (2007).
Erlanson, D. A. Introduction to Fragment-Based Drug Discovery. In Fragment-Based Drug Discovery and X-Ray Crystallography (eds Davies, T. G., Hyvönen, M.) 1–32. (Springer Berlin Heidelberg: Berlin, Heidelberg, 2012).
Joachim, I. et al. Development and optimization of a competitive binding assay for the galactophilic low affinity lectin LecA from Pseudomonas aeruginosa. Org. Biomol. Chem. 14, 7933–7948 (2016).
Schulze, J. et al. CellFy: A Cell-Based Fragment Screen against C-Type Lectins. ACS Chem. Biol. 13, 3229–3235 (2018).
Hann, M. M., Leach, A. R. & Harper, G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. computer Sci. 41, 856–864 (2001).
Hopkins, A. L., Groom, C. R. & Alex, A. Ligand efficiency: a useful metric for lead selection. Drug Discov. Today 9, 430–431 (2004).
Kadam, R. U. et al. CH-pi “T-shape” interaction with histidine explains binding of aromatic galactosides to Pseudomonas aeruginosa lectin LecA. ACS Chem. Biol. 8, 1925–1930 (2013).
Aretz, J. et al. Identification of Multiple Druggable Secondary Sites by Fragment Screening against DC-SIGN. Angew. Chem. Int. Ed. 56, 7292–7296 (2017).
Aretz, J. et al. Allosteric Inhibition of a Mammalian Lectin. J. Am. Chem. Soc. 140, 14915–14925 (2018).
Shanina, E. et al. Druggable Allosteric Sites in β-Propeller Lectins. Angew. Chem. Int. Ed. 61, e202109339 (2022).
Denavit, V. et al. Stereoselective Synthesis of Fluorinated Galactopyranosides as Potential Molecular Probes for Galactophilic Proteins: Assessment of Monofluorogalactoside-LecA Interactions. Chemistry 25, 4478–4490 (2019).
Sommer, R. et al. Cinnamide Derivatives of d-Mannose as Inhibitors of the Bacterial Virulence Factor LecB from Pseudomonas aeruginosa. ChemistryOpen 4, 756–767 (2015).
Audfray, A. et al. Fucose-binding lectin from opportunistic pathogen Burkholderia ambifaria binds to both plant and human oligosaccharidic epitopes. J. Biol. Chem. 287, 4335–4347 (2012).
Troelsen, N. S. et al. The 3F Library: Fluorinated Fsp3-Rich Fragments for Expeditious 19F NMR Based Screening. Angew. Chem. Int. Ed. 59, 2204–2210 (2020).
Delaglio, F. et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Vranken, W. F. et al. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins: Struct. Funct. Bioinf. 59, 687–696 (2005).
Williamson, M. P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson Spectrosc. 73, 1–16 (2013).
Gasteiger, E. et al. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788 (2003).
Molecular Operating Environment (MOE), C. C. G. U., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2021.
Acknowledgements
German Research Foundation (DFG) [Ti756/5-1, RA1944/7-1] funded this work, which was in the scope of German Research Foundation and French National Research Agency [ANR-17-CE11-0048] project ‘Glycomime’. We thank Max Planck Society and Olaf Niemeyer for support. Moreover, ES and CR thank a DAAD Rise Germany student J. Johnson for her contribution to screening of non-fluorinated fragments for LecA. SK and AI thank synchrotron SOLEIL (Saint Aubin, France) for access and technical support at beamline PROXIMA-2. SK thanks Annabelle Varrot for her contribution to data collection for crystallographic experiment. AI acknowledges support from Glyco@Alps (ANR-15-IDEX-02) and Labex Arcane/CBH-EUR-GS (ANR-17-EURE-0003). MHC acknowledges funding for DK-OPENSCREEN from the Ministry of Higher Education and Science (grant case no. 5072-00019B), the Technical University of Denmark, and other contributing universities.
Author information
Authors and Affiliations
Contributions
C.R. and E.S. initiated this study. E.S. performed NMR experiments. A.T. and E.S. performed the chemical synthesis and FP assay. A.I. and S.K. performed X-ray and SPR studies. D.R. and P.G. performed virtual screening. F.F. and M.D. performed CellFy and docking experiments. P.H.S. provided the infrastructure to support this study. M.H.C. supported the study with providing the 3F library. E.S. and C.R. wrote the manuscript with assistance from A.I., A.T., D.R., E.S., M.D., P.G., and S.K. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Jesús Jiménez-Barbero, Sébastien Vidal and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shanina, E., Kuhaudomlarp, S., Siebs, E. et al. Targeting undruggable carbohydrate recognition sites through focused fragment library design. Commun Chem 5, 64 (2022). https://doi.org/10.1038/s42004-022-00679-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-022-00679-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
