
Abstract
Axially chiral heterobiaryls play a vital role in asymmetric synthesis and drug discovery. However, there are few reports on the synthesis of atropisomeric heterobiaryls compared with axially chiral biaryls. Thus, the rapid enantioselective construction of optically active heterobiaryls and their analogues remains an attractive challenge. Here, we report a concise chiral amine-catalyzed atroposelective heterocycloaddition reaction of alkynes with ortho-aminoarylaldehydes, and obtain a new class of axially chiral 2-arylquinoline skeletons with high yields and excellent enantioselectivities. In addition, the axially chiral 2-arylquinoline framework with different substituents is expected to be widely used in enantioselective synthesis.
Similar content being viewed by others
Introduction
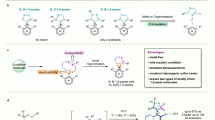
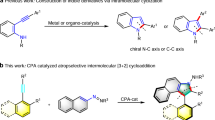
Axially chiral biaryl scaffold is one of the most important structural units, which is widely found in many natural products1,2, bioactive molecules3,4,5, and functional materials6,7. Therefore, the study of axially chiral compounds has attracted extensive attention, and plays an important role in the development of chiral ligands8,9 and organic catalysts10,11,12. In particular, axially chiral 1,1′-bi-2-naphthol (BINOL) (Fig. 1a, R1 = OH) and its derivatives, as the most successful catalysts and ligands in enantioselective synthesis, have achieved great progress13,14,15. Despite advance has been made in the study of thesis axially chiral biaryls, there are still some limitations in the preparation of axially chiral heterobiaryls16,17,18,19,20,21,22. Atropisomeric isoquinoline derivatives (1-(isoquinolin-1-yl)naphthalen-2-ol, Fig. 1a, R2 = OH) has emerged as a unique backbone of several famous catalysts and ligands (e.g., N,O-ligand, QUINOX) in asymmetric catalysis23,24. Especially, Noyori’s BINAP catalyst25 is widely used in industrial and pharmaceutical production. In sharp contrast to axially chiral BINOLs, the atroposelective synthesis of axially chiral isoquinoline derivatives has not been greatly explored. To date, there are few methods for assembling axially chiral isoquinoline derivatives, most of the thesis reports are focus on transition-metal-catalyzed direct cross-coupling reaction26,27, [2 + 2 + 2] cycloaddition28,29 and recently reported (dynamic) kinetic resolution/transformation strategy30,31,32,33,34. However, building upon the increasing demand for this type of catalysts and ligands, a versatile, practical, and scalable method for the synthesis of optically pure axial chiral isoquinoline and corresponding analogues is undoubtedly an urgent need in this field. In addition, from the perspective of structural diversity, nonclassical isoquinoline-type analogues will offer more possibilities for catalyst/ligand development and drug discovery.

a Current research status on axially chiral biaryls. b Organocatalytic synthesis of 4-arylquinolines via central-to-axial chirality conversion (previous work). c Direct to 2-arylquinolines via atroposelective cycloaddition (this work).
Although quinoline skeleton widely exists in bioactive compounds35, in sharp contrast to atropisomeric 2-arylisoquinoline, the research on axial chiral quinoline skeleton is very limited34. Meanwhile, most of the reports focused on the atroposelective synthesis of aryl-C4-36,37, C5-38, or C8-quinoline39 skeletons, in which the distant from the chiral axis of the decisive nitrogen atom for coordination with metal center leading to the difficulty in stereo-induction, which sometimes restrict their further applications in the area of asymmetric synthesis greatly. To face this issue, Tan and coworkers40 uncovered a chiral phosphoric acid-catalyzed atroposelective [4 + 2] cycloaddition to synthesize IAN analogs via the intermediate of vinylidene ortho-quinone methide. This is the only report on the atroposelective synthesis of 2-arylquinoline skeletons. Overall, the catalytic atroposelective synthesis of axially chiral 2-arylquinolines in a highly atroposelective manner remains an attractive challenge.
In the past two decades, chiral amine catalyst has attracted considerable attention in asymmetric catalysis due to its advantages of operational simplicity, low toxicity, and minimal impact on the environment41,42,43,44,45,46. However, most of the reports solely focus on the assembly of central chirality, and the amine-catalyzed enantioselective construction of axial chirality is still in its infancy47,48,49,50. The Sparr group constructed a series of axially chiral biaryl skeletons via chiral amine-catalyzed asymmetric aldol condensation47,48,49. Recently, Cheng20 and Wang21 reported the synthesis of axially chiral 4-arylquinoline skeletons via amine-catalyzed asymmetric heterocycloaddition of ynals with 2-(tosylamino)aryl ketones, followed by aromatization and central-to-axial conversion (Fig. 1b). As part of our group ongoing efforts on organocatalytic synthesis of axially chiral molecules51,52,53, we have successfully reported carbene-catalyzed atroposelective desymmetrization of biphenols, the [3 + 3] annulation of cyclic 1,3-diones with ynals and the kinetic resolution of anilides, resulting in valuable axially chiral biaryl amino alcohols (NOBIN analogues), α-pyrone-aryls, and isoindolinones, respectively. Despite the aforementioned achievements, unsolved challenges and the continuing demands for atropoisomers continue to drive us to develop more revolutionary protocols. We herein firstly report a chiral amine-catalyzed atroposelective heterocycloaddition to offer a class of axially chiral 2-arylquinolines (nonclassical isoquinoline-type analogues) with high yields and excellent enantioselectivities.
Results
Optimization of the reaction conditions
We commenced our study with the model reaction of 3-(2-methoxynaphthalen-1-yl)propiolaldehyde 1a and N-(2-formylphenyl)-4-methylbenzenesulfonamide 2a. The key results of reaction optimization are summarized in Table 1. First, the chiral secondary amine catalysts A–C derived from L-proline with different steric size on the silyl groups were tested. As a result, these catalysts provided the target product 3a in high yields but with very low enantioselectivities (Table 1, entries 1–3). Inspired by the elegant work reported by the Wang group54,55, the catalyst D was selected for initial test under the model reaction. Pleasingly, the desired axially chiral 2-arylquinoline 3a was separated with high yield (89%) and moderate enantiomeric ratio (er) (75:25) (Table 1, entry 4). To further improve the enantiocontrol, we investigated the effect of different O/N-protecting groups on substrates (1b, 1c, and 2b). It is surprising that the enantiomeric ratio of the reaction was arised to 96:4 and the yield was still kept high (91%) (Table 1, entries 5-7). In the case of the best catalyst (D) and suitable substrate (2b), the influences of solvents and catalyst loading were then examined (Table 1, entries 8–10). Finally, the optimal condition and procedure were obtained as follows: adding 1c (1.2 equiv.) and 2b (1.0 equiv.) to the mixture of catalyst D (10 mol%) and CHCl3 (0.1 M) at room temperture and giving corresponding reaction time, the axially chiral 3c was obtained with 91% yield and 96:4 er (Table 1, entry 7).
Substrate scope
After obtaining the optimal conditions, we turned our attention to the substrate scope of ynals. An array of naphthalen-based propiolaldehydes bearing various R1 were tested (Fig. 2). The results show that the electronic and steric effects of the substituents at different positions on the aromatic rings have little impact on the reaction, and the corresponding products were produced in high yields and good to excellent enantioselectivities (3c–3k). Moreover, the quinoline-based ynal provided the coresponding product 3l in 84% yield with 99:1 er. Notably, phenylpropiolaldehyde 1m generated the corresponding biaryl (quinoline-phenyl) product 3m with 92% yield and 96:4 er. Meanwile, a good level of er (89:11) was achieved for quinoline-pyridine-type biaryl product 3n when pyridine-based propiolaldehyde 1n was used.

Reaction conditions: a mixture of 1c (0.11 mmol), 2 (0.1 mmol) and catalyst D (10 mol%) in CHCl3 (1.0 mL) was stirred at room temperature for 24–36 h. After the reaction was complete, the reaction was cooled to 0 °C, then MeOH (0.5 mL) and NaBH4 (0.2 mmol) were added to the mixture and stirred for another 0.5 h at room temperature. At last, HOAc (5.0 equiv) was added to the mixture and stirred for another 2 h.
Encouraged by these results, we next examined the generality of o-aminoarylaldehyde 2. As shown in Fig. 3, substrates bearing either electron-rich groups or electron-deficient groups reacted smoothly with 1c affording the corresponding axially chiral products (4c–4k) in excellent yields (90–96%) and excellent er (96:4–>99:1). When steric hindrance was introduced at the ortho position of the substrate reaction site, the corresponding high yields (84–85%), and the high er (all 94:6) for product 4a, 4b, and 4l were observed. The absolute configuration of 3k was determined by X-ray crystallography (Fig. 2), and other products were assigned by analogy.

Reaction conditions: a mixture of 1c (0.11 mmol), 2 (0.1 mmol) and catalyst D (10 mol%) in CHCl3 (1.0 mL) was stirred at room temperature for 24–36 h. After the reaction was completed, the reaction was cooled to 0°C, then MeOH (0.5 mL) and NaBH4 (0.2 mmol) were added to the mixture and stirred for another 0.5 h at room temperature. At last, HOAc (5.0 equiv) was added to the mixture and stirred for another 2 h.
Gram-scale synthesis and synthetic transformations
To evaluate the practicality of this protocol, a gram-scale reaction of 1c with 2b was carried out under standard condition (Fig. 4, 89% yield, 96:4 er), which indicated that the large-scale synthesis of enantioenriched 2-arylquinolines can be achieved. Synthetic transformations of 3c were also illustrated in Fig. 4b. On the basis of methylation and hydrogenolysis, 3c was easily converted into a versatile intermediatex axially chiral isoquinoline analogue 5 with a yield of 71%, and the er value is completely maintained. After methylation and oxidation, the axially chiral QUINOX analogue 6 (a potential Lewis base organocatalyst) was obtained in good yield without loss of enantiopurity.

a The gram-scale synthesis of 3c. b The synthetic transformations of 3c. c Plausible mechanism.
A postulated reaction pathway is proposed in Fig. 4c. Initially, chiral secondary amine catalyst D added to alkynaldehyde 3c and subsequently dehydrated to produce alkynylamine cationic intermediate I. Then 2b reacted with I via aza-Michael addition to give axially chiral allenamine intermediate II, which underwent an intramolecular aldol reaction to give the chiral styrene intermediate III. Catalyst D was then released from intermediate III to produce chiral compound IV. Finally, compound IV was reduced by NaBH4 and then dehydrated in the present of acid in one pot to deliever the desired product 3c.
Discussion
In summary, we have successfully synthesized the axially chiral 2-arylquinoline analogues via catalytic heterocycloaddition of alkynaldehydes with N-protected o-aminoarylaldehyde via the formation of a critical intermediate (axially chiral styrene). In the presence of commercially available amine catalyst, this conversion can deliver a variety of axially chiral 1-aryl isoquinoline analogues with high yields and excellent er’s. The synthetic utility of this methodology is illustrated by further conversion to axially chiral QUINOX and isoquinoline analogues. Further studies on the application of the axially chiral 2-arylquinoline skeletons in asymmetric synthesis are currently underway in our laboratory.
Methods
Procedure for enantioselective syntheses of compound 3c
To a flame-dried Schlenk reaction tube equipped with a magnetic stir bar, was added the catalyst D (2.2 mg, 0.01 mmol), 1c (39.4 mg, 0.12 mmol), and 2b (0.10 mmol). The Schlenk tube was closed with a septum, CHCl3 (2.0 mL) was added. The mixture was then stirred at room temperature and monitored by TLC until 2b was full consumed. Then the mixture was cooled to 0 °C and MeOH (1.0 mL) was added, subsequently, NaBH4 (11.3 mg, 0.3 mmol) was added slowly to the mixture, and stirred for 2.0 h at this temperature. After the completion of the reaction, as monitored by TLC, HOAc (5.0 equiv) was added to the mixture and then the mixture was warmed to room temperature and stirred for another 6 h. After the reaction was completed, as monitored by TLC, saturated NaHCO3 was added and stirred for another 0.5 h. Then the mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by a silica gel flash chromatography (Hexane/EtOAc = 5:1) to afford the desired product 3c.
Procedure for enantioselective synthesis of compound rac-3c
To a flame-dried Schlenk reaction tube equipped with a magnetic stir bar, was added the catalyst rac-A (2.2 mg, 0.01 mmol), 1c (39.4 mg, 0.12 mmol) and 2b (0.10 mmol). The Schlenk tube was closed with a septum, CHCl3 (2.0 mL) was added. The mixture was then stirred at room temperature and monitored by TLC until 2b was full consumed. Then the mixture was cooled to 0 °C and MeOH (1.0 mL) was added, subsequently, NaBH4 (11.3 mg, 0.3 mmol) was added slowly to the mixture and stirred for 2.0 h at this temperature. After the completion of the reaction, as monitored by TLC, HOAc (5.0 equiv) was added to the mixture and then the mixture was warmed to room temperature and stirred for another 6 h. After the reaction was completed, as monitored by TLC, saturated NaHCO3 was added and stirred for another 0.5 h. Then the mixture was extracted with EtOAc. The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by a silica gel flash chromatography (Hexane/EtOAc = 5:1) to afford the desired product 3c.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and Supplementary information file, or from the corresponding author upon reasonable request. The supplementary crystallographic data for this paper could be obtained free of charge from The Cambridge Crystallographic Data Centre (3k: CCDC 2070558) via www.ccdc.cam.ac.uk/data_request/cif.
References
Kozlowski, M. C., Morgan, B. J. & Linton, E. C. Total synthesis of chiral biaryl natural products by asymmetric biaryl coupling. Chem. Soc. Rev. 38, 3193–3207 (2009).
Bringmann, G., Gulder, T., Gulder, T. A. M. & Breuning, M. Atroposelective total synthesis of axially chiral biaryl natural products. Chem. Rev. 111, 563–639 (2011).
Clayden, J., Moran, W. J., Edwards, P. J. & LaPlante, S. R. The challenge of atropisomerism in drug discovery. Angew. Chem. Int. Ed. 48, 6398–6401 (2009).
LaPlante, S. R. et al. Assessing atropisomer axial chirality in drug dicovery and development. J. Med. Chem. 54, 7005–7022 (2011).
Smyth, J. E., Butler, N. M. & Keller, P. A. A twist of nature-the significatnce of atropisomers in biological systems. Nat. Prod. Rep. 32, 1562–1583 (2015).
Pu, L. 1, 1′-Binaphthyl dimers, oligomers, and polymers: molecular recognition, asymmetric catalysis, and new materials. Chem. Rev. 98, 2405–2494 (1998).
Hembury, G. A., Borovkov, V. V. & Inoue, Y. Chirality-sensing supramolecular systems. Chem. Rev. 108, 1–73 (2008).
Noyori, R. & Takaya, H. BINAP: an efficient chiral element for asymmetric catalysis. Acc. Chem. Res. 23, 345–350 (1990).
Carroll, M. P. & Guiry, P. J. P,N ligands in asymmetric catalysis. Chem. Soc. Rev. 43, 819–833 (2014).
Kumarasamy, E., Raghunathan, R., Sibi, M. P. & Sivaguru, J. Nonbiaryl and heterobiaryl atropisomers: molecular templates with promise for atropselective chemical tranformations. Chem. Rev. 115, 11239–11300 (2015).
Min, C. & Seidel, D. Asymmetric Brønsted acid catalysis with chiral carboxylic acids. Chem. Soc. Rev. 46, 5889–5902 (2017).
Wang, Q., Gu, Q. & You, S.-L. Enantioselective carbonyl catalysis enabled by chiral aldehydes. Angew. Chem. Int. Ed. 58, 6818–6825 (2019).
Brunel, J. M. BINOL: A versatile chiral reagent. Chem. Rev. 105, 857–897 (2005).
Parmar, D., Sugiono, E., Raja, S & Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: history and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. M. Chem. Rev. 114, 9047–9153 (2014).
Cheng, D.-J. & Shao, Y.-D. Advances in the catalytic asymmetric synthesis of atropisomeric hexatomic N-heterobiaryls. Adv. Synth. Catal. 362, 3081–3099 (2020).
Gao, D. W., Gu, Q. & You, S.-L. Pd(II)-Catalyzed intermolecular direct C–H bond iodination: an efficient approach toward the synthesis of axially chiral compounds via kinetic resolution. ACS Catal. 4, 2741–2745 (2014).
Miyaji, R., Asano, K. & Matsubara, S. Bifunctional organocatalysts for the enantioselective synthesis of axially chiral isoquinoline N-oxides. J. Am. Chem. Soc. 137, 6766–6769 (2015).
Quinonero, O. et al. Combining organocatalysis with central-to-axial chirality conversion: atroposelective Hantzsch-Type synthesis of 4-arylpyridines. Angew. Chem. Int. Ed. 55, 1401–1405 (2016).
Shao, Y.-D. et al. Organocatalytic atroposelective Friedländer quinoline heteroannulation. Org. Lett. 21, 4831–4836 (2019).
Shao, Y.-D., Han, D.-D., Dong, M.-M., Yang, X.-R. & Cheng, D.-J. A one-pot stepwise approach to axially chiral quinoline-3-carbaldehydes enabled by iminium–allenamine cascade catalysis. Org. Chem. Front. 8, 605–612 (2021).
Zhang, J., Xu, Y., Wang, Z., Zhong, R. & Wang, Y. Organocatalyzed cascade Aza-Michael/Aldol reaction for atroposelective construction of 4-Naphthylquinoline-3-carbaldehydes. J. Org. Chem. 86, 4262–4273 (2021).
Jiang, P.-Y., Fan, K.-F., Li, S.-Y., Xiang, S.-H. & Tan, B. Metal-free oxidative cross-coupling enabled practical synthesis of atropisomeric QUINOL and its derivatives. Nat. Commun. 12, 2384 (2021).
Malkov, A. V. & Kočovský, P. Chiral N-oxides in asymmetric catalysis. Eur. J. Org. Chem. 1, 29–36 (2007).
Sakiyama, N., Hojo, D., Noguchi, K. & Tanaka, K. Enantioselective synthesis of axially chiral 1-arylisoquinolines by Rhodium-catalyzed [2+2+2] cycloaddition. Chem. Eur. J. 17, 1428–1432 (2011).
Rokade, B. V. & Guiry, P. J. Axially chiral P,N-ligands: some recent twists and turns. ACS Catal. 8, 624–643 (2018).
Mosquera, A., Pena, M. A., Sestelo, J. P. & Sarandeses, L. A. Synthesis of axially chiral 1,1′-binaphthalenes by Palladium-catalysed cross-coupling reactions of triorganoindium reagents. Eur. J. Org. Chem. 13, 2555–2562 (2013).
Shen, D., Xu, Y. & Shi, S.-L. A bulky chiral N-heterocyclic carbene Palladium catalyst enables highly enantioselective Suzuki–Miyaura cross-coupling reactions for the synthesis of biaryl atropisomers. J. Am. Chem. Soc. 141, 14938–14945 (2019).
Gutnov, A. et al. Cobalt(I)-catalyzed asymmetric [2+2+2] cycloaddition of alkynes and nitriles: synthesis of enantiomerically enriched atropoisomers of 2-arylpyridines. Angew. Chem. Int. Ed. 43, 3795–3797 (2004).
Hapke, M. et al. Asymmetric synthesis of axially chiral 1-aryl-5,6,7,8-tetrahydroquinolines by Cobalt-catalyzed [2 + 2 + 2] cycloaddition reaction of 1-aryl-1,7-octadiynes and nitriles. J. Org. Chem. 75, 3993–4003 (2010).
Ros, A. et al. Dynamic kinetic cross-coupling strategy for the asymmetric synthesis of axially chiral heterobiaryls. J. Am. Chem. Soc. 135, 15730–15733 (2013).
Bhat, V., Wang, S., Stoltz, B. M. & Virgil, S. C. Asymmetric synthesis of QUINAP via dynamic kinetic resolution. J. Am. Chem. Soc. 135, 16829–16832 (2013).
Zheng, J., Cui, W.-J., Zheng, C. & You, S.-L. Synthesis and application of chiral spiro Cp ligands in Rhodium-catalyzed asymmetric oxidative coupling of biaryl compounds with alkenes. J. Am. Chem. Soc. 138, 5242–5245 (2016).
Wang, Q., Cai, Z.-J., Liu, C.-X., Gu, Q. & You, S.-L. Rhodium-catalyzed atroposelective C–H arylation: efficient synthesis of axially chiral heterobiaryls. J. Am. Chem. Soc. 141, 9504–9510 (2019).
Romero-Arenas, A. et al. Ir-Catalyzed atroposelective desymmetrization of heterobiaryls: hydroarylation of vinyl ethers and bicycloalkenes. J. Am. Chem. Soc. 142, 2628–2639 (2020).
Wang, N. et al. Synthesis and in vitro cytotoxic effect of 6-amino-substituted 11H- and 11Me-indolo[3,2-c]quinolones. Eur. J. Med. Chem. 78, 314–323 (2014).
Wang, J. et al. Catalytic asymmetric synthesis of atropisomeric quinolines through the Friedländer Reaction. Synlett 30, 2198–2202 (2019).
Wang, S.-J., Wang, Z., Tang, Y., Chen, J. & Zhou, L. Asymmetric synthesis of quinoline-naphthalene atropisomers by central-to-axial chirality conversion. Org. Lett. 22, 8894–8898 (2020).
Shen, D., Xu, Y. & Shi, S.-L. A bulky chiral N-heterocyclic carbene palladium catalyst enables highly enantioselective Suzuki−Miyaura cross coupling reactions for the synthesis of biaryl atropisomers. J. Am. Chem. Soc. 141, 14938–14945 (2019).
Wang, J., Chen, M.-W., Ji, Y., Hu, S.-B. & Zhou, Y.-G. Kinetic resolution of axially chiral 5- or 8-substituted quinolines via asymmetric transfer hydrogenation. J. Am. Chem. Soc. 138, 10412–10416 (2016).
Zhang, L. et al. Design and atroposelective construction of IAN analogues by organocatalytic ssymmetric heteroannulation of alkynes. Angew. Chem. Int. Ed. 59, 23077–23082 (2020).
MacMillan, D. W. C. The advent and development of organocatalysis. Nature 455, 304–308 (2008).
Dondoni, A. & Massi, A. Asymmetric Organocatalysis: From Infancy to Adolescence. Angew. Chem. Int. Ed. 47, 4638–4660 (2008).
Melchiorre, P., Marigo, M., Carlone, A. & Bartoli, G. Asymmetric aminocatalysis-gold rush in organic chemistry. Angew. Chem. Int. Ed. 47, 6138–6171 (2008).
Bertelsen, S. & Jørgensen, K. A. Organocatalysis-after the gold rush. Chem. Soc. Rev. 38, 2178–2189 (2009).
Moyano, A. & Rios, R. Asymmetric organocatalytic cyclozation and cycloaddition reactions. Chem. Rev. 111, 4703–4732 (2011).
Donslund, B. S., Johansen, T. K., Poulsen, P. H., Halskov, K. S. & Jørgensen, K. A. The diarylprolinol silyl ethers: ten years after. Angew. Chem. Int. Ed. 54, 13860–13874 (2015).
Link, A. & Sparr, C. Organocatalytic atroposelective Aldol condensation: synthesis of axially chiral biaryls by arene formation. Angew. Chem. Int. Ed. 53, 5458–5461 (2014).
Lotter, D., Neuburger, M., Rickhaus, M., Häussinger, D. & Sparr, C. Stereoselective arene-forming Aldol condensation: synthesis of configurationally stable oligo-1,2-naphthylenes. Angew. Chem. Int. Ed. 55, 2920–2923 (2016).
Witzig, R. M., Fäseke, V. C., Häussinger, D. & Sparr, C. Atroposelective synthesis of tetra-ortho-substituted biaryls by catalyst-controlled non-canonical polyketide cyclizations. Nat. Catal. 2, 925–930 (2019).
Hayashi, Y., Takikawa, A., Koshino, S. & Ishida, K. Asymmetric synthesis of biaryl atropisomers using an organocatalyst-mediated domino reaction as the key step. Chem. Eur. J. 25, 10319–10322 (2019).
Zhao, C.-G., et al. Enantioselective [3 + 3] atroposelective annulation catalysed by N-heterocyclic carbenes. Nat. Commun. 9, 611 (2018).
Bie, J.-B., Lang, M. & Wang, J. Enantioselective N-heterocyclic carbene-catalyzed kinetic resolution of anilides. Org. Lett. 20, 5866–5871 (2018).
Yang, G.-M., Guo, D.-H., Meng, D. & Wang, J. NHC-catalyzed atropoenantioselective synthesis of axially chiral biaryl amino alcohols via a cooperative strategy. Nat. Commun. 10, 3062 (2019).
Zhang, X.-S., Zhang, S.-L. & Wang, W. Iminium-allenamine cascade catalysis: one-pot access to chiral 4H-chromenes by a highly enantioselective Michael–Michael sequence. Angew. Chem. Int. Ed. 49, 1481–1484 (2010).
Zhang, X.-H., et al. An organocatalytic cascade approach toward polysubstituted quinolines and chiral 1,4-dihydroquinolines-unanticipated effect of N-protecting groups. Angew. Chem. Int. Ed. 51, 7282–7286 (2012).
Acknowledgements
Generous financial supports for this work are provided by: the National Natural Science Foundation of China (21871160, 21672121, 22071130), the Bayer Investigator fellow, the fellowship of Tsinghua-Peking centre for life sciences (CLS).
Author information
Authors and Affiliations
Contributions
G.M.Y. conducted the main experiments; Z.P.L., Y.H.L., and X.J.S. prepared several starting materials. J.W. and S.F.S. conceptualized and directed the project, and drafted the paper with the assistance from co-authors. All authors contributed to the discussions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Communications Chemistry thanks Dao-Juan Cheng and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, G., Sun, S., Li, Z. et al. Organocatalytic atroposelective heterocycloaddition to access axially chiral 2-arylquinolines. Commun Chem 4, 144 (2021). https://doi.org/10.1038/s42004-021-00580-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-021-00580-5
This article is cited by
-
Syntheses of quinolines and their derivatives from α,β-unsaturated aldehydes
Chemistry of Heterocyclic Compounds (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
