
Abstract
Release of the protons from the substrate water molecules is prerequisite for O2 evolution in photosystem II (PSII). Proton-releasing water molecules with low pKa values at the catalytic moiety can be the substrate water molecules. In some studies, one of the ligand water molecules, W2, is regarded as OH−. However, the PSII crystal structure shows neither proton acceptor nor proton-transfer pathway for W2, which is not consistent with the assumption of W2 = OH−. Here we report the pKa values of the four ligand water molecules, W1 and W2 at Mn4 and W3 and W4 at Ca2+, of the Mn4CaO5 cluster. pKa(W1) ≈ pKa(W2) << pKa(W3) ≈ pKa(W4) in the Mn4CaO5 cluster in water. However, pKa(W1) ≈ pKa(D1-Asp61) << pKa(W2) in the PSII protein environment. These results suggest that in PSII, deprotonation of W2 is energetically disfavored as far as W1 exists.
Similar content being viewed by others

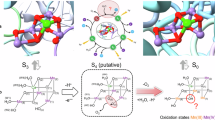
Introduction
In the water-splitting enzyme, photosystem II (PSII), oxygen evolution proceeds, removing four protons (H+) and four electrons from two substrate water molecules at the oxygen-evolving complex, Mn4CaO5 (Fig. 1)1,2. The Mn4CaO5 cluster has two ligand water molecules, W1 and W2, at the dangling Mn4 site and another two ligand water molecules, W3 and W4, at the Ca2+ site. These bound water molecules are candidates as potential substrates for water oxidation. As electron transfer occurs, the oxidation state of the oxygen-evolving complex, Sn, increases. Release of protons is observed with the typical stoichiometry of 1:0:1:2 for S0 → S1 → S2 → S3 → S0, and O2 is evolved in the S3 to S0 transition. After O2 evolution, the first proton-releasing step is the S0 to S1 transition. The Mn4CaO5 cluster has a chain of strongly H-bonded 8 water molecules (O4-water chain) directly linked to O4 (linking Mn4 and Mn3 in the Mn3CaO4-cubane) and the release of the proton occurs along the O4-water chain in the S0 to S1 transition3,4,5. In S0 and S16, the ligand water molecules W1–W4 are H2O in quantum mechanical/molecular mechanical (QM/MM) models (i.e., in the presence of the PSII protein environment)3,7,8, whereas W2 is assumed to be OH− in simplified QM models (i.e., in the absence of the PSII protein environment)9,10,11. The release of the proton is also observed in the S2 to S3 transition. Based on the observations of the recent radiation-damage-free structures obtained using the X-ray free electron laser (XFEL), the sixth O site, O6, may be incorporated into the Mn1 and O5 moieties of the Mn4CaO5cluster in the S2 to S3 transition12,13,14.

The second sphere ligand residues (D1-Asp61 and CP43-Arg357) are not shown. Dotted lines indicate ligations of the ligand water molecules to the Mn4 and Ca2+ sites.
During the water incorporation process, deprotonation of the water molecule may occur as proposed (e.g., water incorporation into the Mn1 moiety15,16, water incorporation from the Ca2+ moiety17,18,19). In theoretical models by Shoji et al.17 and Isobe et al.20, it was assumed the presence of OH− at W2 in S2 to facilitate the release of the proton from H2O at W3 to OH− at W2, and proposed that deprotonation of W3 at the Ca2+ moiety occurs in the S2 to S3 transition. If OH− needed to be located at either W2 or W3, placing OH− at W2 and H2O at W3 would be consistent with the experimentally measured values, pKa(Ca2+) = 12.8 >> pKa(Mn3+) = 0.721 in water. In theoretical models by Ames et al.22, Rapatskiy et al.23, Pérez-Navarro et al.24, and Capone et al.25, W2 was also assumed to be OH–.
If the Mn4CaO5 cluster were isolated from the protein environment, placing OH− at W2, not at W3, might be explained by pKa(Ca2+) >> pKa(Mn3+). However, placing OH− at W2 is not consistent with the PSII crystal structures. The PSII crystal structures show that W2 has no strong H-bond acceptor, whereas W1 has a strong H-bond acceptor, D1-Asp61. Quantum mechanical/ molecular mechanical calculations show that H2O at W1 forms a low-barrier H-bond with D1-Asp61 and is ready for proton transfer in S226. This may correspond to the significant changes in the H-bond properties between D1-Asp61 and a water molecule in the S1 to S2 transition observed in Fourier transform infrared (FTIR) spectroscopy27. Consistently, FTIR spectra suggested that W2 is H2O in S1 and S28.
As far as we are aware, the pKa values of the four water molecules at the Mn4CaO5 moiety, even those for the ligand water molecules W1–W4 are not reported. Robertazzi et al. reported that in S1, pKa(W2) = 6.1 for the isolated Mn4CaO5 cluster with deprotonated D1-His337 and 7.8 for the isolated Mn4CaO5 cluster with protonated D1-His337 in water, based on quantum chemical calculations28. However, the pKa values for W1, W3, and W4 are not reported, which prevent from identifying the deprotonation sites even in the isolated Mn4CaO5 cluster in water. It should also be noted that the definition of the Mn4CaO5 cluster is vague. It can be comprised of Mn4CaO5, four ligand water molecules (W1–W4), and seven ligand residues (D1-Asp170, D1-Glu189, D1-His332, D1-Glu333, D1-Asp342, D1-Ala344, and CP43-Glu354, Fig. 1). It can also include the second sphere ligand residues, D1-Asp61, and CP43-Arg357, or the O4-water chain that forms an H-bond with O4. D1-Asp61 serves as an H-bond acceptor for W1 and is likely to facilitate proton transfer in the S2 to S3 transition26. The O4-water chain forms a significantly short H-bond with O4 (O…O < 2.5 Å) in the crystal structures in S1 (or a slightly lower S-state)29,30 and facilitates the release of the proton from O4 in the S0 to S1 transition3,4,5. The involvement of these proton acceptor groups (i.e., proton transfer pathways) facilitates deprotonation of the H-bond donor sites of the Mn4CaO5 cluster and decreases the pKa values. This fact already implies that the pKa values of the isolated Mn4CaO5 cluster in water are far from the relevant pKa values in the PSII protein environment.
Here we report the pKa values of the W1–W4 sites in the isolated Mn4CaO5 cluster in water, using quantum chemical approaches. To investigate the energetics of release of the proton towards the proton-transfer pathways in PSII, we analyze the potential-energy profiles of the H-bonds between the ligand water molecules and the H-bond acceptor (i.e., the proton acceptor) groups in the PSII protein environment. The results show that pKa(W1) ≈ pKa(W2) << pKa(W3) ≈ pKa(W4) in the Mn4CaO5 cluster in water (i.e., in the absence of the PSII protein environment), whereas pKa(W1) ≈ pKa(D1-Asp61) << pKa(W2) in PSII.
Results and discussion
We calculate the energy difference (ΔEwater) between the protonated and deprotonated states of hexa-aqua metal complexes in water (Fig. 2). The calculated ΔEwater values of hexa-aqua metal complexes with the valences of II, III, and IV show a correlation with the experimentally measured pKa values (Fig. 3) and are best fitted to the following equation:

a Protonated state. b Deprotonated state. Magenta balls indicate metal ions. Dotted lines indicate ligations of the ligand water molecules to metal ions.

Blue open circles for divalent (II) metals, green open squares for trivalent (III) metals, and red closed diamond for the tetravalent (IV) metal.
The calculated pKa values of hexa-aqua metal complexes obtained using Eq. 1 are listed in Table 1. Note that in vacuum, the experimentally measured pKa values cannot be reproduced using a single equation (Supplementary Fig. 1, Supplementary Table 1), because the electrostatic influence between the cationic metal and anionic OH− in the deprotonated state is overestimated in vacuum. Thus, pKa predominantly depends on the metal valence in vacuum.
The isolated Mn4CaO5 cluster is comprised of Mn4CaO5, four ligand water molecules (W1–W4), and seven ligand residues (D1-Asp170, D1-Glu189, D1-His332, D1-Glu333, D1-Asp342, D1-Ala344, and CP43-Glu354, Fig. 1). Using Eq. (1), the pKa values for W1–W4 are calculated at the isolated Mn4CaO5 cluster in water (in the absence of the PSII protein environment, Fig. 1). pKa(W1) and pKa(W2) on the dangling Mn4 site are 7–11, whereas pKa(W3) and pKa(W4) on Ca2+ site are 14–18 (Table 2). pKa(W1) and pKa(W2) are higher than pKa of 0.5 for Mn3+ (Table 1), because Mn4 has two ligand acidic residues, D1-Asp170 and D1-Glu333, and two μ-oxo O atoms, O4 and O5 (Fig. 1). The difference in the pKa of >5 between the Mn4 and Ca2+ sites (Table 2) indicate that the Ca2+ site is originally disadvantageous for H2O deprotonation with respect to the Mn4 site. In particular in the PSII protein environment, W1 at Mn4 has D1-Asp61 as an H-bond acceptor, whereas W3 and W4 at Ca2+ do not have the corresponding acidic residues (see below). Thus, deprotonation of H2O and incorporation of the generated OH– into the Mn4CaO5 cluster occurring at the Ca2+ moiety in the S2 to S3 transition (e.g., refs. 17,18,19) needs to overcome the energetic disadvantage.
As far as the ligand coordination in the PSII protein structure is maintained (i.e., the torsion angles are fixed), pKa(W2) is only marginally (~1 pKa unit) lower than pKa(W1) in the absence of the PSII protein environment (Table 2). When the geometry is fully relaxed (i.e., the torsion angles are not fixed) and OH− is initially placed at W4 [to calculate pKa(W4)], proton transfer occurs from W2 via W3 to W4 occurs and OH− is finally stabilized at W2, not at W1 (Supplementary Fig. 2). Deprotonation of W2 instead of W1 is just an artifact as W2, W3, and W4 form the H-bond network, pushing W1 and W3 away from Mn4 and Ca2+, respectively (W1...Mn4 = 3.8 Å and W3...Ca2+ = 3.6 Å). These observations may be a basis of why electron paramagnetic resonance (EPR) signals were often interpreted based on theoretical models, in which W2 was assumed to be OH− in QM-based models (i.e., in the absence of the PSII protein environment, e.g., refs. 10,22,23). Interestingly, W2 = OH− are also assumed in other QM-based models (e.g., by Siegbahn9 and Retegan et al.11), without using QM/MM approaches. It should be noted that W2, W3, and W4 never form the H-bond network as far as the PSII protein environment exists.
The marginally low pKa(W2) with respect to pKa(W1) (Table 2) were the case only when the Mn4CaO5 cluster could be ideally isolated from the PSII protein environment. In such a model system, pKa(W1) and pKa(W2) can change easily, depending on even the definition of the Mn4CaO5 cluster. When the second sphere ligand residues (D1-Asp61 and CP43-Arg357) are included in the model system of the Mn4CaO5 cluster in water, H2O at W1 is not stable, releasing the proton, and is stabilized as OH– at W1 in the presence of protonated D1-Asp61 (Fig. 4a), i.e., pKa(W1) << pKa(W2) (Table 3). The absence of the corresponding acidic residue as an H-bond acceptor for W2 and the proceeding proton transfer pathway (e.g., the D1-Asp61 pathway for W126) contribute to an increase in pKa(W2) with respect to pKa(W1).

Dotted lines indicate ligations of the ligand water molecules to metal ions. a Quantum-chemically optimized structure in the absence of the PSII protein environment. W1 is stabilized as OH− in the presence of protonated D1-Asp61, as the release of the proton occurs from H2O at W1 to ionized D1-Asp61. b QM/MM-optimized Mn4CaO5 structure in the PSII protein environment.
In the isolated Mn4CaO5 cluster in water (in the absence of the PSII protein environment), proton release occurs along the transiently formed H-bond between the ligand water molecule and a bulk water molecule (i.e., mobile water molecule with a high dielectric constant ≈ 80). The acceptor water molecule is best represented implicitly using the polarizable continuum model (PCM) method; in this case, the pKa value of the deprotonation site of the Mn4CaO5 cluster can be calculated, whereas the pKa difference between the deprotonation site and the adjacent proton-acceptor water molecule cannot be calculated directly. On the other hand, in the presence of the PSII protein environment, proton release occurs along the H-bond between the ligand water molecule and the fixed acceptor group (i.e., fixed dipole with a low dielectric constant << 80) (Fig. 4b). The acceptor group is represented explicitly based on the crystal structure; in this case, the energy barrier, which is associated with the pKa difference between the deprotonation site and the acceptor group31, can be calculated based on the potential-energy profile of the H-bond, whereas the pKa value of the deprotonation site of the Mn4CaO5 cluster cannot be calculated directly. Note that only when the acceptor groups are always the same for all deprotonation sites (e.g., H2O), the pKa values may be calculated from the pKa difference [e.g., the difference from pKa(H2O/H3O+)]31. However, this is not the case for W1–W4 in the PSII protein environment, where the individual explicit acceptor groups already exist (e.g., D1-Asp61 for W1 and W446 for W2).
QM/MM calculations show that H2O at W1 forms a low-barrier H-bond with D1-Asp61 and the proton migrates towards the D1-Asp61 moiety in S2 (Fig. 5a), whereas H2O at W2 forms a standard H-bond with an adjacent water molecule (W446, Fig. 4b) and the proton is localized at the W2 moiety, i.e., proton transfer from W2 to the acceptor H2O is energetically uphill (Fig. 5b). This suggests that pKa(W1) is significantly lower than pKa(W2) in the PSII protein electrostatic environment and W2 cannot release the proton as more deprotonatable W1 exists at the Mn4CaO5 moiety in the PSII protein environment32. The results are consistent with W2 being H2O in S1 and S2 based on FTIR spectra and theoretical calculations by Nakamura and Noguchi8.

a W1 and D1-Asp61. The isoenergetic proton transfer from W1 to D1-Asp61 indicates that pKa(W1) ≈ pKa(D1-Asp61). b W2 and the H-bond acceptor water molecule, W446. The energetically uphill proton transfer from W2 to W446 indicates pKa(W2) >> pKa(W1), i.e., deprotonation of W2 is energetically less favorable than deprotonation of W1.
In summary, pKa(W1) ≈ pKa(W2) << pKa(W3) ≈ pKa(W4) in the Mn4CaO5 cluster in water (Table 2). pKa(W2) is only marginally (~1 pKa unit) lower than pKa(W1) in the absence of the PSII protein environment (Table 2) if the Mn4CaO5 cluster is defined as shown in Fig. 1, which may be a basis of why electron paramagnetic resonance (EPR) signals were often interpreted based on simplified theoretical models with OH− at W2 (e.g., refs. 10,22,23). pKa(W1) is significantly lower than pKa(W2) if the Mn4CaO5 cluster includes the second sphere ligand residues, D1-Asp61 and CP43-Arg357 (Table 3, Fig. 4a). Thus, as pKa(W1) and pKa(W2) depend strongly on the definition of the Mn4CaO5 region, pKa(W1) and pKa(W2) in water (in the absence of the protein environment) do not provide any clue to understanding the deprotonation sites in the physiological S-state transitions. The potential energy profiles of the H-bonds show that in the presence of the PSII protein environment in S2 Fig. 4b), H2O at W1 forms a low-barrier H-bond with D1-Asp61 and proton transfer is barrier-less (Fig. 5a)26, whereas H2O at W2 forms a standard H-bond with the adjacent H2O and proton transfer is energetically uphill (Fig. 5b)32. These suggest that pKa(W1) ≈ pKa(D1-Asp61) << pKa(W2) in PSII. As far as W1 exists, W2 can never release the proton (i.e., not OH−) in PSII.
Methods
pK a calculation
In the deprotonation reaction of the protonated state (AH) to deprotonated state (A–) in water, pKa is defined as
where ΔGaq is the free energy difference between (AH) and (A– + H+) (i.e., ΔGwater = Gwater(A–) – Gwater(AH) + Gwater(H+)), R is the gas constant, and T is the temperature. ΔGwater can also be approximated as
where k is the scaling factor, ΔEwater is the energy difference between AH and A–, which can be calculated using a quantum chemical approach with the PCM method, and C is the constant (simple pKa estimation with energy of the optimized geometry scheme33). If the pKa values of molecules are obtained at the same temperature, Eq. (2) can be written into Eq. (4) using the Eq. (3) as
where k′ is the scaling factor and C′ is constant. To determine k′ and C′, we calculated ΔEwater for 23 hexa-aqua metal complexes whose experimentally measured pKa values are reported21,34.
Hexa-aqua metal complex
The optimized geometry of the protonated hexa-aqua metal complex was obtained, using the restricted or unrestricted density functional theory with the B3LYP functional. The CSDZ* basis set was used for lanthanides except for La, the ERMLER2* basis set for actinides, and the LACVP* basis set for all other atoms. The spin states were consistent with previous studies by Galstyan et al.34. The optimized geometries of the deprotonated hexa-aqua metal complexes were obtained, fixing the torsion angles to prevent OH− from forming an H-bond with other ligand H2O molecules. However, the H-bond formation between the ligand OH− and H2O molecules could not be avoided for Ca2+, Zn2+, Cd2+, Dy3+, Th4+, Pa4+, U4+, Np4+, and Pu4+, which were excluded from the present study. It should be noted that by adding a few external H2O molecules to the ligand OH− moiety, the H-bond formation between the ligand OH− and H2O molecules could be avoided without fixing the torsion angles. It was reported that the pKa values for hexa-aqua metal complexes in water did not differ significantly when calculated by fixing the torsion angles or adding a few external H2O molecules34, probably because the shape of the hexa-aqua metal complex is symmetrical. However, this does not hold true for W1–W4 in the Mn4CaO5 cluster whose shape is not symmetric. Adding a few external H2O molecules to the ligand OH− moiety causes structural changes with respect to the original coordination geometry of the PSII crystal structure. In addition, explicit H2O water molecules (i.e., fixed dipole) form a specific H2O cluster (i.e., the dielectric constant << 80) at the deprotonatable ligand moiety, which does neither represent bulk water (i.e., the dielectric constant ≈ 80) nor provide the relevant pKa values. Based on these, the torsion angles were fixed to obtain the optimized geometry for pKa calculations in the present study. Using the optimized geometries, the energy difference (ΔEwater) between the protonated and deprotonated states of hexa-aqua metal complexes were calculated with PCM method, using the Jaguar program35.
Mn4CaO5 cluster
The optimized geometry of the Mn4CaO5 cluster in the PSII protein environment was obtained as follows: the atomic coordinates of PSII were taken from the X-ray structure of PSII monomer unit “A” of the PSII complexes from Thermosynechococcus vulcanus at a resolution of 1.9 Å (PDB code, 3ARC)29. Atomic partial charges of the amino acids were adopted from the all-atom CHARMM2236 parameter set, respectively. D1-His337 was considered to be protonated8. We employed the electrostatic embedding QM/MM scheme, in which electrostatic and steric effects created by a protein environment were explicitly considered, and we used the Qsite37 program code. We employed the unrestricted DFT method with the B3LYP functional and LACVP* basis sets. To analyze the Mn4CaO5 geometries and the H-bond potential-energy profiles, the QM region was defined as the Mn4CaO5 cluster (including the ligand side-chains of D1-Asp170, D1-Glu189, D1-His332, D1-Glu333, D1-Asp342, CP43-Glu354, the ligand carboxy-terminal group of D1-Ala344, and the ligand water molecules, W1–W4), the Cl-1 binding site (Cl-1, W442, W446, and the side-chains of D1-Asn181 and D2-Lys317), and the second-sphere ligands (side-chains of D1-Asp61 and CP43-Arg357). Specifically, the coordinates of the heavy atoms in the surrounding MM region were fixed at their original X-ray coordinates, while those of the H atoms in the MM region were optimized using the OPLS2005 force field. All of the atomic coordinates in the QM region were fully relaxed (i.e., not fixed) in the QM/MM calculation. All of the H-bond partners were included in the QM region. The cluster was considered to comprise ferromagnetically coupled Mn atoms, where the total spin S = 15/2 in S0, 14/2 in S1, and 13/2 in S2. The resulting Mn oxidation states (Mn1, Mn2, Mn3, Mn4) were (III, IV, III, III) in S0, (III, IV, IV, III) in S1, (III, IV, IV, IV) in open-cubane S2, and (IV, IV, IV, III) in closed-cubane S2. It should be noted that the difference in S (e.g., S = 1/2 in S238, high, low, ferromagnetic, and antiferromagnetic) did not affect the values; e.g., (i) the resulting geometry39,40, (ii) the potential energy profile of proton transfer26, (iii) the redox potential of each Mn site41, and (iv) the pKa values for the ligand water molecules W1–W4 in the absence of the protein environment (see Supplementary Table 2). To obtain the potential energy profiles of the O…H+…O bond, the QM/MM optimized geometry was used as the initial geometry. The H atom under investigation was moved between the two O moieties by 0.05 Å, after which the geometry was optimized by constraining the distance between O–H+ and H+–O distances, and the energy was calculated. This procedure was repeated until the H atom reached the O moieties.
In the absence of the PSII protein environment (i.e., in vacuum), the QM/MM-optimized geometry was re-optimized, using the unrestricted density functional theory with the B3LYP functional and LACVP* basis sets and fixing the torsion angles to maintain the overall shape of the less stable complex. The QM region was defined as either the Mn4CaO5 cluster (including the ligand side-chains of D1-Asp170, D1-Glu189, D1-His332, D1-Glu333, D1-Asp342, CP43-Glu354, the ligand carboxy-terminal group of D1-Ala344, and the ligand water molecules, W1–W4) or the Mn4CaO5 cluster and the second-sphere ligands (side-chains of D1-Asp61 and CP43-Arg357). Using the optimized geometries, the energy difference (ΔEwater) between the protonated and deprotonated states of the Mn4CaO5 cluster were calculated with the PCM method, using the Jaguar program35.
Data availability
All data generated or analyzed during this study are included in this article (and its Supplementary Information files).
References
Shen, J. R. The structure of photosystem II and the mechanism of water oxidation in photosynthesis. Annu. Rev. Plant. Biol. 66, 23–48 (2015).
Cardona, T. & Rutherford, A. W. Evolution of photochemical reaction centres: more twists? Trends Plant Sci. 24, 1008–1021 (2019).
Saito, K., Rutherford, A. W. & Ishikita, H. Energetics of proton release on the first oxidation step in the water-oxidizing enzyme. Nat. Commun. 6, 8488 (2015).
Takaoka, T., Sakashita, N., Saito, K. & Ishikita, H. pKa of a proton-conducting water chain in photosystem II. J. Phys. Chem. Lett. 7, 1925–1932 (2016).
Shimizu, T., Sugiura, M. & Noguchi, T. Mechanism of proton-coupled electron transfer in the S0-to-S1 transition of photosynthetic water oxidation as revealed by time-resolved infrared spectroscopy. J. Phys. Chem. B 122, 9460–9470 (2018).
Pantazis, D. A. Missing pieces in the puzzle of biological water oxidation. ACS Catal. 8, 9477–9507 (2018).
Pal, R. et al. S-state model of the oxygen-evolving complex of photosystem II. Biochemistry 52, 7703–7706 (2013).
Nakamura, S. & Noguchi, T. Infrared determination of the protonation state of a key histidine residue in the photosynthetic water oxidizing center. J. Am. Chem. Soc. 139, 9364–9375 (2017).
Siegbahn, P. E. Mechanisms for proton release during water oxidation in the S2 to S3 and S3 to S4 transitions in photosystem II. Phys. Chem. Chem. Phys. 14, 4849–4856 (2012).
Krewald, V. et al. Metal oxidation states in biological water splitting. Chem. Sci. 6, 1676–1695 (2015).
Retegan, M. et al. A five-coordinate Mn(IV) intermediate in biological water oxidation: spectroscopic signature and a pivot mechanism for water binding. Chem. Sci. 7, 72–84 (2016).
Suga, M. et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 543, 131–135 (2017).
Kern, J. et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 563, 421–425 (2018).
Suga, M. et al. An oxyl/oxo mechanism for oxygen-oxygen coupling in PSII revealed by an x-ray free-electron laser. Science 366, 334–338 (2019).
Siegbahn, P. E. Water oxidation mechanism in photosystem II, including oxidations, proton release pathways, O–O bond formation and O2 release. Biochim. Biophys. Acta 1827, 1003–1019 (2013).
Siegbahn, P. E. M. Computational investigations of S3 structures related to a recent X-ray free electron laser study. Chem. Phys. Lett. 690, 172–176 (2017).
Shoji, M., Isobe, H. & Yamaguchi, K. QM/MM study of the S2 to S3 transition reaction in the oxygen-evolving complex of photosystem II. Chem. Phys. Lett. 636, 172–179 (2015).
Ugur, I., Rutherford, A. W. & Kaila, V. R. I. Redox-coupled substrate water reorganization in the active site of Photosystem II—the role of calcium in substrate water delivery. Biochim. Biophys. Acta 1857, 740–748 (2016).
Kim, C. J. & Debus, R. J. Evidence from FTIR difference spectroscopy that a substrate H2O molecule for O2 formation in photosystem II is provided by the Ca ion of the catalytic Mn4CaO5 cluster. Biochemistry 56, 2558–2570 (2017).
Isobe, H., Shoji, M., Shen, J.-R. & Yamaguchi, K. Strong coupling between the hydrogen bonding environment and redox chemistry during the S2 to S3 transition in the oxygen-evolving complex of photosystem II. J. Phys. Chem. B 119, 13922–13933 (2015).
Dean, J.A. in Lange’s Handbook of Chemistry (ed J. A. Dean) (McGraw-Hill Book Co, 1985).
Ames, W. et al. Theoretical evaluation of structural models of the S2 state in the oxygen evolving complex of photosystem II: protonation states and magnetic interactions. J. Am. Chem. Soc. 133, 19743–19757 (2011).
Rapatskiy, L. et al. Detection of the water-binding sites of the oxygen-evolving complex of photosystem II using W-band 17O electron-electron double resonance-detected NMR spectroscopy. J. Am. Chem. Soc. 134, 16619–16634 (2012).
Pérez Navarro, M. et al. Ammonia binding to the oxygen-evolving complex of photosystem II identifies the solvent-exchangeable oxygen bridge (μ-oxo) of the manganese tetramer. Proc. Natl Acad. Sci. USA 110, 15561–15566 (2013).
Capone, M., Narzi, D., Bovi, D. & Guidoni, L. Mechanism of water delivery to the active site of photosystem II along the S2 to S3 transition. J. Phys. Chem. Lett. 7, 592–596 (2016).
Kawashima, K., Takaoka, T., Kimura, H., Saito, K. & Ishikita, H. O2 evolution and recovery of the water-oxidizing enzyme. Nat. Commun. 9, 1247 (2018).
Debus, R. J. Evidence from FTIR difference spectroscopy that D1-Asp61 influences the water reactions of the oxygen-evolving Mn4CaO5 cluster of photosystem II. Biochemistry 53, 2941–2955 (2014).
Robertazzi, A., Galstyan, A. & Knapp, E. W. Reprint of PSII Manganese Cluster: Protonation of W2, O5, O4 and His337 in the S1 state explored by combined quantum chemical and electrostatic energy computations. Biochim. Biophys. Acta 1837, 1389–1394 (2014).
Umena, Y., Kawakami, K., Shen, J.-R. & Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473, 55–60 (2011).
Suga, M. et al. Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses. Nature 517, 99–103 (2015).
Ikeda, T., Saito, K., Hasegawa, R. & Ishikita, H. The existence of an isolated hydronium ion in the interior of proteins. Angew. Chem. Int. Ed. 56, 9151–9154 (2017).
Saito, K., Mandal, M. & Ishikita, H. Energetics of ionized water molecules in the H-bond network near the Ca2+ and Cl− binding sites in photosystem II. Biochemistry, doi: 10.1021/acs.biochem.0c00177 (2020).
Matsui, T., Baba, T., Kamiya, K. & Shigeta, Y. An accurate density functional theory based estimation of pKa values of polar residues combined with experimental data: from amino acids to minimal proteins. Phys. Chem. Chem. Phys. 14, 4181–4187 (2012).
Galstyan, G. & Knapp, E. W. Computing pKA values of hexa-aqua transition metal complexes. J. Comput Chem. 36, 69–78 (2015).
Jaguar. version 7.9, Schrödinger, LLC, New York, NY (2012).
MacKerell, A. D. Jr. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
QSite. version 5.8, Schrödinger, LLC, New York, NY (2012).
Zimmermann, J. L. & Rutherford, A. W. Electron paramagnetic resonance properties of the S2 state of the oxygen-evolving complex of photosystem II. Biochemistry 25, 4609–4615 (1986).
Ames, W. et al. Theoretical evaluation of structural models of the S2 state in the oxygen evolving complex of photosystem II: protonation states and magnetic interactions. J. Am. Chem. Soc. 133, 19743–19757 (2011).
Isobe, H. et al. Theoretical illumination of water-inserted structures of the CaMn4O5 cluster in the S2 and S3 states of oxygen-evolving complex of photosystem II: full geometry optimizations by B3LYP hybrid density functional. Dalton Trans. 41, 13727–13740 (2012).
Mandal, M., Kawashima, K., Saito, K. & Ishikita, H. Redox potential of the oxygen-evolving complex in the electron transfer cascade of photosystem II. J. Phys. Chem. Lett. 11, 249–255 (2020).
Acknowledgements
This research was supported by JST CREST (JPMJCR1656 to H.I.), JSPS KAKENHI (18H05155, 18H01937, 20H03217, and 20H05090 to H.I., 16H06560 and 18H01186 to K.S.), and the Interdisciplinary Computational Science Program in CCS, University of Tsukuba (K.S.).
Author information
Authors and Affiliations
Contributions
H.I. designed research; K.S., M.N., and H.I. performed research; K.S., M.N., and H.I. analyzed data; and H.I. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saito, K., Nakagawa, M. & Ishikita, H. pKa of the ligand water molecules in the oxygen-evolving Mn4CaO5 cluster in photosystem II. Commun Chem 3, 89 (2020). https://doi.org/10.1038/s42004-020-00336-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-020-00336-7
This article is cited by
-
Binding and functions of the two chloride ions in the oxygen-evolving center of photosystem II
Photosynthesis Research (2022)
-
Glycerol binding at the narrow channel of photosystem II stabilizes the low-spin S2 state of the oxygen-evolving complex
Photosynthesis Research (2022)
-
Role of redox-inactive metals in controlling the redox potential of heterometallic manganese–oxido clusters
Photosynthesis Research (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
