
Abstract
Construction of artificial higher order protein complexes allows sampling of structural architectures and functional features not accessible by classical monomeric proteins. Here, we combine in silico modelling with expanded genetic code facilitated strain promoted azide-alkyne cycloaddition to construct artificial complexes that are structurally integrated protein dimers and demonstrate functional synergy. Using fluorescent proteins sfGFP and Venus as models, homodimers and heterodimers are constructed that switched ON once assembled and display enhanced spectral properties. Symmetrical crosslinks are found to be important for functional enhancement. The determined molecular structure of one artificial dimer shows that a new long-range polar network comprised mostly of organised water molecules links the two chromophores leading to activation and functional enhancement. Single molecule analysis reveals the dimer is more resistant to photobleaching spending longer times in the ON state. Thus, genetically encoded bioorthogonal chemistry can be used to generate truly integrated artificial protein complexes that enhance function.
Similar content being viewed by others


Introduction
The importance of protein oligomerisation to biology is illustrated clearly in nature1,2, with dimers being the most commonly observed final structural state of proteins3,4. Protein oligomerisation is now emerging as an alternative route to construct novel higher-order complexes with a limited repertoire of monomeric building blocks5,6,7. There are challenges as the subunit interfaces normally comprise numerous weak non-covalent interactions1,8. Great strides have been made in generating assembled peptide and protein oligomeric systems using approaches such as helix–helix interactions9,10, metal coordination11,12,13, fusion domains6, disulfide bridging14 and remodelled naturally inspired protein–protein interfaces15,16,17. However, one key aspect is generally missing that is common in natural protein oligomer systems, functional synergy between the individual components, so complexes manifest the properties of the starting components. This is because long-range bond networks beyond the local direct interactions at the interface region (which drive the initial assembly event) need to be considered for connecting functional centres, which is a fundamentally more challenging proposition. For example, an impressive range of Green Fluorescent Protein (GFP) oligomers have been constructed through disulfide or metal-mediated approaches but functional communication was not apparent14.
Here, we demonstrate the construction of protein dimers assembled by structure-guided biorthogonal chemistry using genetically encoded strain promoted azide-alkyne cycloaddition chemistry (SPAAC)18 (Fig. 1a). The benefit of such an approach is that the design can be simpler, linkage position is defined and be easily combined with other protein interfacing approaches noted above if required. Mutually compatible reaction handles can be placed at different sites in each protein monomer; a single covalent crosslink species then acts as a molecular “bolt” so stabilising the complex. Non-canonical amino acids (ncAAs) such as tyrosine or lysine derivatives (Fig. 1a for example) are ideal for such an approach. Their relatively long side chains will reduce steric clashes while maintaining structural intimacy to promote and stabilise favourable non-covalent interactions required for dimerisation and inter-monomer communication. They are also less labile compared with, for example, disulfide bridges. While previous work has generated proteins linked by bioorthogonal crosslinks, normally via extended linker molecules or restricted linkage residues position19,20,21,22,23,24,25,26, the proteins are structurally and functionally distinct so provide little improvement on classical genetic fusions. SPAAC27 is a biocompatible 1-to-1 reaction that does not require toxic catalysts and can occupy either available regioisomer (Fig. 1a) around the linking triazole. It can also be implemented by genetic code expansion using two separate ncAAs (azF and SCO in Fig. 1a) so bypassing the requirement of linking molecules thus enabling an intimate interaction between the monomers.

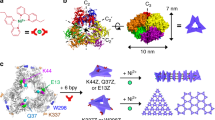
Bioorthogonal-driven protein dimerisation and implementation phases. a Concept. SPAAC reaction between the two genetically encoded ncAAs (azF, green and SCO, blue) intimately links two monomeric proteins via either a syn- or anti-triazole link to promote the formation additional non-covalent interactions. b Structure-guided design. In silico modelling to predict potential dimer interfaces and residues contributing to the interface. The highest ranked sfGFP-sfGFP dimer model (see Supplementary Table 1 for statistics), with residues E132 (magenta), H148 (cyan) and Q204 (yellow), selected for replacement with either azF or SCO is shown. Inset is the structural relationship of H148, Q204 and the structured water molecule W1 with the chromophore (CRO). c, d Construction. Dimerisation as analysed by gel mobility shift; c formation of dimeric sfGFP148x2 from monomers sfGFP148azF and sfGFP148SCO; d formation of dimeric sfGFP204x2 from monomers sfGFP204azF and sfGFP204SCO
Super-folder green fluorescent protein (sfGFP)28 and its yellow relative Venus29 were chosen as target proteins, with sfGFP in particular proving to be an excellent model for investigating and understanding the molecular influence of ncAA incorporation on protein function (see refs. 30,31,32,33 for examples), including BioClick reactions34 and biohybrid assemblies35,36,37. Fluorescent proteins in general are also an excellent system to study important chemical and biological process, such as charge transfer networks and coupled photochemistry38,39.
We show that favourable dimer interface regions can be identified through the use of in silico docking approaches, and that dimerization through symmetrical SPAAC switches ON and enhances function. Non-symmetrically linked dimers do not show functional enhancement. Our experimentally determined structure highlights the formation of a new long-range interaction network between the protein’s functional centres, with organised water molecules playing a key role. Heterodimers are also constructed, which show apparent integrated function combining facets of each individual monomer so the dimer acts as one single functional unit.
Results
Click chemistry interface sites
We surmised that areas of a protein’s surface that are compatible in terms of association are more likely to generate an integrated structure through the formation of mutually compatible non-covalent interactions. As the proteins are monomeric these interactions are at best, weak and transient so will not persist, we reasoned that we needed a molecular “bolt” as part of the interface site to promote and stabilise any new interactions. The first step is to identify regions on the target proteins that have the inherent potential to interact. ClusPro 2.040 (cluspro.org) was used to generate potential dimer configurations. The output sfGFP homodimer models were refined, analysed and ranked using RosettaDock41,42 (Supplementary Table 1). The highest ranked configuration is shown in Fig. 1b (which is the closest model to the determined structure below; vide infra), with the next 4 ranked configurations shown in Supplementary Fig. 1. While different orientations of one sfGFP to the other were observed, docking revealed residues 145–148, 202–207 and 221–224 were routinely found to contribute to the dimer interface. To bolt the two proteins together, genetically encoded bioorthgonal Click chemistry was used (Fig. 1a). The benefit compared with, for example, disulfide linkages include longer side chains to overcome potential steric clashes, improved crosslink stability and the ability to generate non-symmetrical (different linking residues on different monomers) and heterodimers in a designed 1-to-1 manner (vide infra).
Based on the dimer models, three residues were selected for replacement with the two Click compatible ncAAs, SCO43 (strained alkyne) and azF (azide)44,45 (Fig. 1a). H148 and Q204 were chosen based on their location at the putative dimer interface (Fig. 1b and Supplementary Fig. 1). Both residues are known to be readily modified with small molecule cyclooctyne adducts on azF incorporation34,46, and lie close to the functional centre, the sfGFP chromophore (CRO) (Fig. 1b). Gel mobility shift analysis revealed that dimerisation was successful (Fig. 1c, d); this was confirmed by mass spectrometry analysis (Supplementary Fig. 2). Residue 132 was not predicted to be at the dimer interface (Fig. 1b and Supplementary 1) but is known to be compatible with a range of strained alkyne adducts ranging from dyes46 to carbon nanotubes35 to single stranded DNA36. Thus, it acts as a good test of our ability to predict protein–protein interfaces and Click reaction compatibility. Despite residue 132 being surface exposed, no dimer product was observed using sfGFP132azF with SCO containing protein (Supplementary Fig. 3) indicating the importance of surface interface compatibility and the utility of in silico analysis in helping to identify click chemistry compatible sites. Either steric clashes and/or protein–protein interactions that persist for longer at other regions may hinder covalent crosslinking at residue 132.
Positive functional switching on forming sfGFP148x2 dimer
H148 forms a H-bond with CRO (Fig. 1b) and plays an important role in proton shuttling that regulates the population of the neutral A (λmax ~ 400 nm, CRO A) and the anionic B form (λmax ~ 490 nm, CRO B)47 present in the ground state. The B form predominates in sfGFP but on incorporating azF in place of H148 (sfGFP148azF) removal of the H-bond results in the A state now predominating33,34 (Fig. 2a and Table 1). Incorporating SCO at residue 148 (sfGFP148SCO) elicits a similar effect, with CRO A predominating but with a smaller red shift (λmax 492 nm) in the minor CRO B form (Fig. 2a and Table 1).

Spectral properties of sfGFP148 variants before and after dimerisation. a Absorbance and b fluorescence emission (on excitation at 492 nm) of sfGFP148x2 (red), sfGFP148SCO (black dashed) and sfGFP148azF (black). Fluorescence emission was normalised to sfGFPWT. Absorbance peaks due to the neutral CRO A state and phenolate CRO B state are indicated. c Comparison of sfGFPWT absorbance spectra (green) with sfGFP148x2 (red). The green dashed line represents the expected value if ε at λmax is simply doubled for sfGFPWT. d Single molecule fluorescence intensity histogram for sfGFP148x2 dimers (115 trajectories comprised of 1742 spots), with two representative fluorescence time-course traces of individual dimers inset (both with raw and Cheung-Kennedy filtered data). The histogram of observed sfGFP148x2x2 fluorescence intensities is described by a two-component mixed log-normal distribution. Representative fluorescent time-course traces illustrate typically observed fluorescent behaviour of the dimer. With prolonged fluorescence observed at ~80–100 counts corresponding to the first component in the histogram. Some dimers exhibit rapid and brief forays to higher intensity states, giving rise to the second higher intensity peak in the histogram. Additional traces can be found in Supplementary Fig. 5
Dimerisation of sfGFP148azF and sfGFP148SCO produces two significant positive effects: (i) switches ON fluorescence at ~490 nm due to promotion of the CRO B form; (ii) greatly enhanced brightness through increased molar absorbance coefficient at 490 nm (Fig. 2a and Table 1). The major excitation peak is red-shifted on dimerisation (λmax 492 nm) compared with sfGFPWT (λmax 485 nm) (Supplementary Table 3). The 490:400 nm absorbance ratio shifts by an order of magnitude from ~0.5 for the monomers to ~5 for the dimer, with CRO B form dominating the dimer absorbance spectrum (Fig. 2a) despite the apparent absence of a species that can replace the role of the H148 imidazole group. Previous examples of modifying sfGFP148azF with small molecule adducts or photoactivation at best result in partial conversion to CRO B form33,34. The 10-fold switch in absorbance is mirrored in fluorescence emission; excitation at 490 nm results ~20-fold higher emission than either monomer (Fig. 2a). In addition, the dimer shows enhanced function even when compared with the original superfolder sfGFPWT (Fig. 2b and Table 1). Molar absorbance and brightness increased ~320% for sfGFP148x2 (~160% on a per CRO basis) (Fig. 2b) higher than expected for a simple additive increase if monomer units are acting independently of each other.
To investigate the importance of the biorthogonal link we constructed a classic disulfide-based link by mutating H148 to cysteine. The sfGFPH148C variants dimerised but only in the presence Cu2+ (Supplementary Fig. 4). The spectral properties suggested that the dimer was less fluorescent compared with sfGFP148x2 and sfGFPWT (Supplementary Fig. 4). The sfGFPH148C monomer displayed the expected switch from CRO B to CRO A. While a switch from CRO A to CRO B was observed on dimerization of sfGFPH148C, the dimer had a lower per CRO molar absorbance than sfGFPWT and significantly less than sfGFP148x2; a significant population of the A state was still observed. Fluorescence emission on excitation at 490 nm for dimeric sfGFPH148C was circa half that of sfGFPWT. Thus, the biorthogonal approach generated a better performing dimeric species than classical disulfide bond linkage.
Molecular basis for functional switching in sfGFP148x2
The crystal structure of sfGFP148x2 (see Supplementary Table 2 for statistics) reveals that the monomers forms an extensive dimer interface with long-range interactions linking the two CRO centres. The monomer units of sfGFP148x2 arrange in a quasi-symmetrical head-to-tail arrangement offset by ~45° in relation to each another (Fig. 3a). The anti-parallel side-by-side monomer arrangement is closest to that of the highest ranked model (Supplementary Fig. 1 and Supplementary Table 1). The electron density of the new triazole crosslink is clearly defined (Fig. 3b) and forms the elongated anti-1,4-triazole link that is partially buried and intimately associated with both monomer units so forming an integral part of the dimer interface (Fig. 3c). The CROs are 15 Å apart pointing toward each other (Fig. 3a).

Structure of sfGFP148x2. The azF bearing protein is coloured green and SCO bearing protein is coloured cyan. a Overall monomer arrangement, including a schematic outline of the relationship of the two monomers. CROs are shown as spheres and the residues 148 as sticks. b The electron density map (2Fo-Fc, 1.0 sigma) for the crosslink is shown confirming the formation of the anti-regioisomer. c The hydrophobic packing around the dimer interface with the SPAAC crosslink shown as transparent spheres. d H-bond network contributing to the dimer interface. PDB submission code 5nhn
The interface has similar characteristics to natural dimers48. Interface buried area is ~1300 Å2, with generally the same residues from each monomer contributing (Fig. 3c, d). H-bonding plays an important role with residues E142, N146, S147, N149 and N170 from both monomers contributing to eight inter-subunit H-bonds (Fig. 3b). The structure shows that natural dimer interfaces can be mimicked and stabilised through the use of Click-linked monomers, which the original modelling suggested were feasible but where probably too weak or transient to persist without the embedded link. Thus, it may be that our approach could be used to stabilise more broadly transient weak protein–protein interactions so forming defined interfaces.
Dimerisation induces a series of conformational changes to form a long-range interaction network that underlies the mechanism by which sfGFP is switched ON and brightness enhanced. The sfGFP148azF structure (PDB 5BT0)34 shows that 148azF occupies a similar position to H148 in sfGFPWT but cannot from the critical H-bond to the CRO phenol OH group that promotes formation of the CRO B state. On dimerisation, modification of 148azF through formation of the triazole link with 148SCO in the cognate monomer results a change in both its backbone and side chain position causing a hole that can now be occupied by a water molecule in the dimer (W1azF in Fig. 4a). The water can H-bond to CROazF and the backbone carbonyl of 148azF (Fig. 4). An equivalent water is present in the sfGFP148SCO monomer unit, (W1SCO) which forms similar interactions. These structured water molecules have the potential to replace the H-bond interaction lost on removal of H148 so activating the dimer through promoting formation of CRO B in the ground state. The water molecules are also buried at the dimer interface so dynamic exchange with the bulk solvent will be much reduced. Furthermore, the two CROs are now linked by an extended predominantly water network that spans the dimer interface (Fig. 4b, c). Analysis of the tunnel composition revealed that three water molecules in each unit (W1azF/SCO, W2azF/SCO and W3azF/SCO) are symmetrical; W4 combined with the backbone of F145SCO provide the bridge across the dimer interface to link the two water networks together. Thus, dimerisation generates an extended, inter-monomer water-rich H-bond network so promoting a switch from the A state CRO to the B form.

Activation via conformational changes and inter-subunit communication networks on formation of sfGFP148x2. The azF bearing protein is coloured green and SCO bearing protein is coloured cyan. a Conformational change to azF148 on dimerisation. The sfGFP148azF (PDB 5bt034) is coloured in magenta. b CAVER69 analysis of a proposed channel linking the two CRO of sfGFP148x2. c Water dominated long-range H-bond network linking the CRO from the azF (CROazF) and SCO (CROSCO) monomers
Single molecule fluorescence analysis of sfGFP148x2
Total internal reflection fluorescence (TIRF) microscopy was used to investigate the fluorescent behaviour of sfGFP148x2 dimers at the single molecule level. The fluorescence intensity time course of single sfGFP148x2 dimers demonstrated a range of intensity states, with fluorescence at ~80–100 counts predominating and displaying greater longevity than the sub-population of higher intensity states, with these characterised by brief forays to a range of intensities from ~100 to 300 counts (Fig. 2c). The fluorescence traces also demonstrate prolonged photostability with long periods to photobleaching (Fig. 2c and Supplementary Fig. 5). In comparison, sfGFPWT photobleaches more rapidly, with fluorescence traces showing a single intensity state in which the on states generally last for shorter periods (Supplementary Fig. 6). Furthermore, monomeric sfGFPWT was sometimes found to exist in an initial dark, non-fluorescent state, prior to initiation of fluorescence and subsequent photobleaching (Supplementary Fig. 6). Extraction of the average consecutive fluorescence ‘on time’ prior to photobleaching and occupancy of transient non-fluorescent states (blinking) finds that sfGFP148x2 displays longer periods of continuous fluorescence (mean 0.9 s), compared with sfGFPWT (0.65 s). Given the similarity in measured single molecule fluorescence intensity, the increased ON times and photobleaching lifetime likely contribute toward the increased fluorescence observed in steady state ensemble measurements of sfGFP148x2 (Fig. 2a).
In an attempt to rationalise the range of fluorescence states observed in the dimer traces, a histogram of all measured intensities was generated (Fig. 2c). Unlike sfGFPWT (Supplementary Fig. 6) that shows a single log-normal distribution49, sfGFP148x2 favoured a two-component fit50 (Fig. 2c). The measured intensity distribution shows a predominant lower intensity peak (~90 counts) and a partially overlapping higher intensity peak, as a consequence of the brief forays to higher intensity states observed in the single molecule fluorescence traces. Whilst a bimodal intensity distribution might ordinarily be expected in a dimer comprised of two co-located independently active fluorophores, with each fluorophore sequentially photobleaching, the single molecule intensity time-course traces are not consistent with this model and show a lack two well defined states. The simple on/off state behaviour of sfGFPWT is infrequently observed in the dimer traces which themselves do not present as the anticipated adduct of two monomeric traces, instead showing more complex behaviour.
Enhanced function on forming sfGFP204x2 dimer
To explore how different linkage sites can elicit functional affects, we investigated the alternative dimer sfGFP204x2 (Fig. 5a) constructed above (Fig. 1d). We found dimerisation enhanced the spectral properties above that of simple addition of the monomeric or sfGFPWT proteins highlighting again the synergistic benefits of dimerisation. Incorporation of either azF or SCO at residue 204 had little effect on spectral properties compared with sfGFPWT 46 (Fig. 5b and Table 1). The B CRO form predominated in the monomeric forms; both molar absorbance and emission intensities where similar to each other and sfGFPWT. The fluorescence emission of sfGFP204SCO was slightly reduced (80% of sfGFPWT; Table 1). On forming the sfGFP204x2 dimer (see Fig. 1d and Supplementary Fig. 2 for evidence) spectral analysis showed functional enhancement in terms of the core spectral parameters: molar absorbance coefficient (ε) and fluorescence emission (Fig. 5b and Table 1). On dimerization, ε increased up to 400% compared with the starting monomers to 160,000 M−1 cm−1. This equates to an average per CRO molar absorbance of 80,000 M−1 cm−1, almost doubling the brightness compared with the starting monomers, and 31,000 M−1 cm−1 higher compared with sfGFPWT. In line with increased capacity to absorb light, fluorescence emission was also enhanced; the normalised per CRO emission was 180% higher than the sfGFP204azF monomer. Using the Strickler-Berg51 calculation (website huygens.science.uva.nl/Strickler_Berg/) fluorescence lifetimes drop from 3.2 ns for sfGFPWT to 0.92 ns for sfGFP204x2. Thus, as with sfGFP148x2 the dimeric structure of sfGFP204x2 has an increased probability of electronic excitation and fluorescence output compared with monomeric forms (see Supplementary Fig. 8 for spectral comparison of dimers). This is all the more impressive for both dimeric forms as sfGFPWT is a benchmark for green fluorescent protein performance.

Spectral properties of sfGFP204 variants before and after dimerisation. a schematic of dimerisation of sfGFP204azF and sfGFP204SCO to form sfGFP204x2, b Absorbance and c fluorescence (on excitation at 487 nm) of sfGFP204x2 (blue), sfGFP204SCO (black dashed), sfGFP204azF (black) and sfGFPWT (green). Fluorescence emission was normalised to wt GFP. The red dashed line represent the molar absorbance value for a simple addition of two individual sfGFPWT at λmax
The importance of symmetry to synergy
Natural protein homodimers are generally symmetrical1,52 and such symmetry was mimicked in our artificial dimer through a common crosslink residue. We investigated the importance of a common crosslink residue (as a mimic of structural symmetry) to functional synergy. The advantage of biorthogonal chemistry is that the mutually compatible reaction handles allows for construction of defined pairs (i.e., 148 + 204 = 148–204 and not 148–148 or 204–204) so preventing undesirable products from forming which will be difficult to separate.
Dimers were generated that linked residue 148 and 204 in the two available combinations (148SCO+204azF and 148azF+204SOC). Steady state fluorescence revealed that in both dimeric forms, the protonated and deprotonated forms of CRO were clearly present (Fig. 6a, b). The 148azF-204SCO linked dimer exhibited a change in the relative populations of A and B forms, with a significant increase in the molar absorbance coefficient at 490 nm (Fig. 6a); it was almost double the original sfGFP204SCO and ~30% higher than that predicted by simple addition of the monomer spectra. The relative height of the 400 nm peak remains similar in the both sfGFP148azF and the 148azF-204SCO linked dimer suggesting the population of the deprotonated form is similar in the dimer as the original monomer. Dimerisation via the 148SCO-204azF combination changed the relative populations of the protonated and deprotonated forms but the reduction in the 400 nm absorbance peak was not matched by a concomitant increase in the 490 nm peak (Fig. 6b). In fact, dimerisation was largely detrimental as both major absorbance peaks had a lower molar absorbance coefficient than the simple addition spectra of the monomers (Fig. 6b). It is clear that the asymmetrically linked dimers are less fluorescent and contain a significant mixed population of the two CRO states compared with the symmetrically linked dimers so in this case, symmetry has important functional implications.

Non-symmetrical dimers sfGFP148azF-204SCO and sfGFP148SCO-204azF. a Absorbance spectra of sfGFP148azF-204SCO (red line) compared with sfGFP148azF (black line) and sfGFP204SCO (blue line). b Absorbance spectra of sfGFP148SCO-204azF (red line) compared with sfGFP148SCO (black line) and sfGFP204azF (blue line). c Model of the structural consequence of linking different residues to generate a non-symmetrically linked sfGFP dimer
Heterodimers and functional integration
Heterodimers, in which a dimer is composed of two different proteins, is a commonly observed alternative dimerisation state1,2. It also allows us to design new complexes in which functionally distinct proteins can be linked. The advantage of bioorthogonal coupling is the ability to generate defined, single species (hetero)dimers comprised of two different protein units (i.e., A + B = A–B not A–A, B–B, A–B mixture which can be difficult to separate). The yellow fluorescent protein Venus29 was chosen as the partner protein to sfGFP, given the spectral overlap between the two (Supplementary Fig. 9). Sequence differences are shown in Supplementary Fig. 10.
SfGFP148SCO was combined with the equivalent azF containing Venus (Venus148azF) to generate GFVen148 (see Supplementary Fig. 11 for evidence). New spectral characteristics emerge suggesting an integrated system has been generated. Formation of GFVen148 generates a dimer that shows improved brightness compared with either the sfGFP148SCO or Venus148azF (Fig. 7a and Supplementary Table 3). Interestingly, the dimer has spectral properties intermediate of individual monomers without any significant peak broadening (Fig. 7a, b and Supplementary Fig. 12a) suggesting that the two CRO centres have become functionally integrated in terms of fluorescence emission. The major λmax is 505 nm, intermediate between sfGFP (492 nm) and Venus (517 nm). The ε equivalent to the B CRO form (490–510 nm region) increases significantly (~4–5-fold), higher than the simple additive spectra of the monomers, while the A CRO population decreases but is still observed (Fig. 7a). This is matched by a ~4-fold increase in emission intensity on excitation at 505 nm (Fig. 7b). A single emission peak is observed that is also intermediate between the two monomers, irrespective of the excitation wavelength (λEM at 517 nm; Fig. 7b, c); a single rather than a double or broadened peak was observed on excitation at 490 nm (capable of exciting both CROs) suggesting that a single species is emitting. An additive spectrum of individual monomer spectra that simulates two independently acting proteins supports the idea of a new integrated function as it is broader and red-shifted compared with the measured GFVen148x2 emission profile (Supplementary Fig. 12b). Emission on excitation at 400 nm was also measured as Venus148azF has little absorbance at this wavelength compared with GFVen148. Emission intensity was 30-fold higher for GFVen148 compared with monomeric Venus148azF with emission peaking at 517 nm (Fig. 7c and Supplementary Fig. 12c). Rather than displaying classical FRET, as might be expected (vide supra), GFVen148 appears to act as a single entity in terms of fluorescence output. This could suggest that two CROs are now acting predominantly as one species with the structural aspects observed for sfGFP148x2 (such as the water network) playing a role. The presence of a significant neutral A state of the CRO does suggest the two monomeric units are not fully synchronised. This does not however, negate the clear impact dimerisation can have in the generation of novel spectral properties such as those seen in GFVen148.

Communication between heterodimers. a Absorbance spectra of GFVen148. Red, gold, green, black dashed lines represent GFVen148, Venus148azF, sfGFP148SCO and monomer addition spectrum, respectively. b Emission intensity of 0.5 μM GFVen148 (red) and Venus148azF (gold) on excitation at 505 nm. c Normalised emission of GFVen148 (red) and Venus148azF (gold) on excitation at 400 nm. d Spatial arrangement of GFP and Venus CRO based on the GFP148x2 structure. e Absorbance spectra of GFVen204. Red, gold, green, black dashed lines represent GFVen204, Venus204azF, sfGFP204SCO and monomer addition spectrum, respectively. f Emission spectra for GFVen204 (blue) and Venus204azF (gold). Unbroken, dashed and dotted lines represent excitation at 510 nm and 450 nm, respectively. Inset is emission spectra on excitation at 400 nm. g Spatial arrangement of sfGFP and Venus CRO based on sfGFP204x2 structure (PDB 5ni3)
As with sfGFP, incorporation of azF in place of Q204 (termed Venus204azF) had little effect on Venus’ spectral properties (Supplementary Table 3). Covalent linkage via 204 SPAAC (making GFVen204) successfully generated a dimer (Supplementary Fig. 11). GFVen204 combined the spectral features of both monomers generating a species with λMax at both 490 and 514 nm (ratio of 1:1.2) (Fig. 7e, f and Supplementary Table 3). Venus204 also absorbs at 490 nm but at a ratio of 1:2.7 to 514 nm. Formation of dimers again enhanced molar absorbance above that of the individual monomers; notably ε increased by ~26,000 M−1 cm−1 (~27%) for the Venus associated λmax (514 nm) where there is little contribution for sfGFP. To investigate communication between the monomers, fluorescence emission on excitation at four separate wavelengths was monitored: 400 nm (sfGFP only); 450 nm (sfGFP, minor Venus); 490 nm (sfGFP λmax, Venus shoulder); 510 (Venus, minor sfGFP). At all excitation wavelengths, the only clear emission peak was at 528 nm (Fig. 7b), corresponding to Venus indicating communication by Förster resonance energy transfer (FRET). An emission peak correlating to sfGFP204SCO (~510 nm) was not observed even on excitation at the lower wavelengths specific for sfGFP. Nor was the intermediate emission peak characteristic of GFVen148 observed, highlighting the novel characteristics of the 148 heterodimer. The most significant difference was observed on excitation at 400 nm where there is a 14-fold increase in emission intensity at 528 nm for the GFVen204 compared with Venus204azF (Fig. 7f, inset). The calculated relative FRET efficiency after spectral decomposition was circa 90%. Thus, the two functional centres are communicating through energy transfer (Fig. 7g) in a highly efficient manner.
Discussion
While historically the drive in protein engineering has been to make oligomers monomeric (including fluorescent proteins53), converting monomeric proteins to oligomers allows new architectures to be sampled and thus altered functional properties. Our use of in silico interface identification and genetically encoded Click chemistry provides an additional avenue by which to construct such complexes. Implementation of this approach led to the generation of two GFP dimers with improved brightness, and in the case of sfGFP148x2 functional switching. The use of the azF-SCO crosslink proved important as classical disulfide crosslinking did not improve protein function. Furthermore, the bioorthogonal nature of the reaction allowed the generation of bespoke heterodimers, including the observation of novel emission characteristics for GFVen148.
The structure of sfGFP148x2 highlights the structural role of internal water molecules and provides a rationale for the observed synergy: the formation of a buried long-range symmetrical interaction networks that link the two functional centres (Fig. 3). Formation of the triazole link is critical as it results in a local conformational change allowing entry of a water molecule to replace the role of the original histidine, which is important for promoting deprotonation of the chromophore and so the CRO B form (Fig. 4a). Long-range polar networks and charge transfer processes play an important role in chemistry and biology, including autofluorescent proteins54,55. We34,56 and others39,54,57,58 have proposed that water networks and dynamics are key to GFP fluorescence. In sfGFP and other monomeric A. victoria FPs, water molecules beyond the directly bonded CRO water molecule (termed W2 and W3; see Fig. 4) that contribute to an extended network are largely exposed to the solvent (Supplementary Fig. 7) and thus subject to dynamic exchange with the bulk solvent. In the dimer, these water molecules now lie at the dimer interface forming a buried putative H-bond network (Fig. 4). The simple effect of surface burial and reduced exchange with bulk solvent [dynamics] may be an important contributor to the improved functional effects we see here in terms increased brightness and can potentially be applied in other fluorescent proteins. In addition, water burial is likely to be important to permitting the formation of a more permanent, organised and concerted H-bond network, as observed for sfGFP148x2 resulting in changes to the inherent fluorescence properties. Symmetry generated by covalently linking identical residues in each monomer proved important at both a structural and functional level. Non-symmetrical crosslinks between residues 148 and 204 did not generate significant synergistic effects (Fig. 6) suggesting newly formed interaction pathways rather than simple close proximity of the two CROs is important. Given that the CROs lie close to the dimer interface, a second dynamical effect of dimerisation that may contribute to fluorescence enhancement is rigidification of the chromophore environment, similar to that observed for naturally occurring oligomeric DsRed fluorescent proteins53,59.
A potential explanation for sfGFP148x2 single molecule fluorescence behaviour may be found by considering the effect of protonation states on the sfGFP148 monomers and the inter-CRO bond network of the dimer as revealed by the crystal structure (Fig. 4). Both sfGFP148 monomers predominantly occupy the protonated CRO A form in the ground state with absorbance maxima at ~400 nm. This would be expected to give rise to no or extremely limited fluorescence under the 473 nm TIRF illumination. However, the buried water network connecting the CROs in the dimer interior provides a putative proton wire to shuttle a proton from the CRO in the ground state60. For such a mechanism to occur the carbonyl oxygen of F145SCO will have to be involved in the proton transfer pathway, in a similar manner carbonyl groups from adjacent residues N146 and H148 are proposed to contribute to proton shuttling within monomeric GFP54,58. Indeed, carbonyl oxygens have been proposed to facilitate proton shuttling in other proteins, such as cytochrome c oxidase61,62. This could be achieved in sfGFP148x2 through classical keto-enol tautomerization, promoted by the local organised water molecules surrounding the F145-N146 peptide bond. It is interesting to speculate that rapid proton shuttling between the two CROs in a synchronised “ping-pong” mechanism of action may manifest63 in a situation where the one CRO in the dimer transiently exists in its minimally fluorescent protonated (A) state, while the other exists in the fluorescent deprotonated (B) state that is observable in our TIRF measurements. This is supported by the steady state ensemble absorbance data in which a shoulder around 400 nm is indicative of a minor CRO A population in the dimer, a feature absent from sfGFPWT absorbance data (Fig. 2b). Such a scenario, with a single CRO of the dimer active at any one time, would give rise to the dominant intensity peak as measured in the sfGFP148x2 single molecule intensity histogram arising from the lower persistent intensity state seen in the single molecule traces (Fig. 2c and Supplementary Fig. 5a, g, i, m, o), and be consistent with the absence of classical two-step sequential photobleaching. It is notable that comparable single molecule intensities (~100 counts) are observed in the sfGFPWT, where a single CRO is present and predominantly exists in its CRO B form. In the dimer, the less frequented higher intensity state which is transiently visited during the fluorescent time course may be indicative of a more rarely encountered simultaneous activity of both CROs.
In conclusion, dimers can be constructed from normally monomeric units using genetically encoded bioorthogonal chemistry through predicting protein interface regions. Importantly, we go beyond simple passive linking of individual proteins by constructing truly structurally integrated complexes that enhance inherent function. The generation of new protein oligomerisation systems is currently a hot topic in protein engineering7,16 as it allows us to understand a commonly observed molecular feature in biology, explore new functional and structural space, and expand uses in the nanosciences. Here we have addressed a key challenge in engineered protein oligomerisation systems: interunit communication and networking beyond the direct interface region. In this work, homodimers displayed enhanced function, including switching ON through assembly. The latter aspect could have applications for the generation of proximity-based biosensors that do not rely on the inherent complexities of FRET64. Our structure reveals an extensive dimer interface is formed through mutually compatible interactions, mimicking natural dimer interfaces without having to extensively engineer such weak interactions; the new Click link enables otherwise weak and/or transient interactions to persist, likely through an entropic mechanism. Indeed, the ability to predict potential interaction interfaces and stabilise them through covalent linkage may provide a general strategy of constructing defined and structurally interacting protein dimers and higher order oligomers, so moving beyond simple passive linkage and the use of classical disulfide crosslinking. The approach is not restricted to the ncAA used here, with different strained alkyne regioisomers and linking chemistries available18 allowing a broader sampling of dimer conformations. Nor are the sites or targets of coupling restricted, with alternative non-symmetrical architectures and linkage of disparate proteins not easily accessible by disulfide linking becoming feasible. This could include artificial enzyme cascades and integrated light harvesting/energy transfer systems. With developments in codon reprogramming for incorporation of multiple bioorthogonal chemistries into a single protein at defined positions65 and codon replacement cell lines66,67, coupled with integration of more classical protein engineering techniques, our approach can aid the design of higher order functional oligomeric species.
Methods
Protein engineering and production
Detailed methods are provided in the Supplementary Methods.
In silico modelling of sfGFP dimer interfaces
ClusPro is a global docking rigid-body approach that requires no prior information on interface regions, and has been shown to be a good predictor of dimer interfaces40. ClusPro was used in multimer mode (set to dimers) using the structure of wild type sfGFP (sfGFPWT, 2B3P) as a starting model. ClusPro generates ~100,000 structures and scores them using balanced energy coefficients as described by Kozakov et al.40 (Eq. (1)). E is the energy score of the complex; Erep is the energy of the repulsive contribution of van der Walls interactions and Eatt is the attractive interaction equivalent. Eelec is a term generated by electrostatic energy and EDARS is a term that mainly accounts for free energy change due to exclusion of water from the interface.
E = 0.40Erep + −0.40Eatt + 600Eelec + 1.00EDARS (1)
The server then takes the 1000 models with the lowest scores and clusters them using pairwise to generate I-RMSD (interface root mean squared deviation). Doing so creates clusters centred on the structure with the most neighbours within a 9 Å radius. Of the remaining models that do not fall within the first cluster the one with the most neighbours becomes the centre of the next cluster and so on until all models are part of a cluster. The centre models of each cluster are energy minimised using the CHARMM force-field for 300 steps with fixed backbone to minimise steric clashes.
A model for each cluster was downloaded and run through ROSETTA’s high resolution docking protocol. This added extra rotamers and subsequent minimisation of side chains. The docking protocol also rescores the models and adds an interface score41,42. The interface score is the total complex score minus the sum of the separate monomer energies and is used as a metric of how good a model is. Total score and interface score were plotted against I-RMSD to highlight any outliers and remove them. The top five models were then used as a basis for determining which residues would be most suitable for crosslinking to form dimers.
Protein dimerisation by SPAAC
Concentrations of monomer variants were determined using the Bio-RAD DC Protein Assay using sfGFPWT as a standard and correlated to the 280 nm absorbance. Dimers were generated by mixing azF and SCO monomers (100 µM, 50 mM Tris-HCl) overnight at room temperature. Dimers were purified by size exclusion chromatography and concentrations determined, as described above. The spectral properties of the dimers were characterised as described below. Fluorescence spectra were taken at a concentration of 0.25 µM (equivalent chromophore number to 0.5 μM of monomers). Protein dimerisation was also monitored by SDS PAGE gel mobility assays. Dimer yields were between 35 and 80%. Mass spectrometry to verify formation of dimer is outlined in the Supplementary Methods.
Protein spectral analysis
Proteins were diluted to 5 µM and 0.5 µM in 50 mM Tris-HCl pH 8.0 for absorbance and fluorescence spectra, respectively. Absorbance spectra were taken on a Cary Win UV, using a 300 nm/min scan rate at 1 nm intervals. Absorbance at λmax for each variant, was used to determine the molar extinction coefficients (ε) for each variant, using the Beer-Lambert equation. Emission spectra were collected on a Cary Varian fluorimeter at a scan rate of 150 nm/min and either 0.5 or 1 nm intervals. Emission and excitation slit widths were set to 5 nm and a detector voltage of 600 mV. Samples were excited at various wavelengths as stated in the main text and emission was scanned from the excitation wavelength to 650 nm. Quantum yields were calculated as previously described using a fluorescein standard dissolved in 0.1 M NaOH33,34. In brief, samples were diluted to an optical density of 0.05 at the chosen excitation wavelength, in 50 mM Tris-HCl pH 8.0. An emission spectrum was then taken as above with a reduced slit width of 2.5 nm and increased PMT voltage of 800 mV. The integral of the emission spectrum from 5 nm after the excitation wavelength to 650 nm.
Protein structure determination
The procedures for determining sfGFP148x2 are provided in the Supplementary Methods.
Single molecule imaging and data processing
Single molecule imaging was performed using a custom built total internal reflection fluorescence (TIRF) microscope based on a Nikon Ti-U inverted microscope, with illumination provided by a Venus 473 nm DPSS laser with a power output of 100 mW and detection via a Andor iXon ultra 897 EMCCD camera as outlined in detail in the Supplementary Methods. Single-molecule imaging data were processed and analysed using ImageJ68 and Matlab (R2017a) (MathWorks USA) as outlined in detail in the Supplementary Methods.
Data availability
Crystal structure data that supported the findings of this study have been deposited in the PDB with the accession code 5nhn. The characteristic of the fluorescent proteins used in the study will be added to FPbase. All other experimental data that support the findings of this study are available from the corresponding author upon reasonable request. Alternatively, data sets related to this publication are available via the https://doi.org/10.17035/d.2019.0078708057.
Change history
20 August 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Ali, M. H. & Imperiali, B. Protein oligomerization: how and why. Bioorg. Med. Chem. 13, 5013–5020 (2005).
Goodsell, D. S. & Olson, A. J. Structural symmetry and protein function. Annu Rev. Biophys. Biomol. Struct. 29, 105–153 (2000).
Marianayagam, N. J., Sunde, M. & Matthews, J. M. The power of two: protein dimerization in biology. Trends Biochem. Sci. 29, 618–625 (2004).
Mei, G., Di Venere, A., Rosato, N. & Finazzi-Agro, A. The importance of being dimeric. FEBS J. 272, 16–27 (2005).
Norn, C. H. & Andre, I. Computational design of protein self-assembly. Curr. Opin. Struct. Biol. 39, 39–45 (2016).
Kobayashi, N. & Arai, R. Design and construction of self-assembling supramolecular protein complexes using artificial and fusion proteins as nanoscale building blocks. Curr. Opin. Biotechnol. 46, 57–65 (2017).
Yeates, T. O., Liu, Y. & Laniado, J. The design of symmetric protein nanomaterials comes of age in theory and practice. Curr. Opin. Struct. Biol. 39, 134–143 (2016).
Miller, S., Lesk, A. M., Janin, J. & Chothia, C. The accessible surface area and stability of oligomeric proteins. Nature 328, 834–836 (1987).
Thomson, A. R. et al. Computational design of water-soluble alpha-helical barrels. Science 346, 485–488 (2014).
Mou, Y., Huang, P. S., Hsu, F. C., Huang, S. J. & Mayo, S. L. Computational design and experimental verification of a symmetric protein homodimer. Proc. Natl Acad. Sci. USA 112, 10714–10719 (2015).
Der, B. S. et al. Metal-mediated affinity and orientation specificity in a computationally designed protein homodimer. J. Am. Chem. Soc. 134, 375–385 (2012).
Brodin, J. D. et al. Metal-directed, chemically tunable assembly of one-, two- and three-dimensional crystalline protein arrays. Nat. Chem. 4, 375–382 (2012).
Song, W. J. & Tezcan, F. A. A designed supramolecular protein assembly with in vivo enzymatic activity. Science 346, 1525–1528 (2014).
Leibly, D. J. et al. A suite of engineered GFP molecules for oligomeric scaffolding. Structure 23, 1754–1768 (2015).
Sahasrabuddhe, A. et al. Confirmation of intersubunit connectivity and topology of designed protein complexes by native MS. Proc. Natl Acad. Sci. USA 115, 1268–1273 (2018).
Fallas, J. A. et al. Computational design of self-assembling cyclic protein homo-oligomers. Nat. Chem. 9, 353–360 (2017).
Chevalier, A. et al. Massively parallel de novo protein design for targeted therapeutics. Nature 550, 74–79 (2017).
Patterson, D. M. & Prescher, J. A. Orthogonal bioorthogonal chemistries. Curr. Opin. Chem. Biol. 28, 141–149 (2015).
Kim, C. H. et al. Synthesis of bispecific antibodies using genetically encoded unnatural amino acids. J. Am. Chem. Soc. 134, 9918–9921 (2012).
White, C. J. & Bode, J. W. PEGylation and dimerization of expressed proteins under near equimolar conditions with potassium 2-pyridyl acyltrifluoroborates. ACS Cent. Sci. 4, 197–206 (2018).
Schoffelen, S., Beekwilder, J., Debets, M. F., Bosch, D. & van Hest, J. C. Construction of a multifunctional enzyme complex via the strain-promoted azide-alkyne cycloaddition. Bioconjug Chem. 24, 987–996 (2013).
Torres-Kolbus, J., Chou, C., Liu, J. & Deiters, A. Synthesis of non-linear protein dimers through a genetically encoded Thiol-ene reaction. PLoS One 9, e105467 (2014).
Eger, S., Scheffner, M., Marx, A. & Rubini, M. Synthesis of defined ubiquitin dimers. J. Am. Chem. Soc. 132, 16337–16339 (2010).
Hatzakis, N. S. et al. Synthesis and single enzyme activity of a clicked lipase-BSA hetero-dimer. Chem. Commun. 21, 2012–2014 (2006).
Hudak, J. E. et al. Synthesis of heterobifunctional protein fusions using copper-free click chemistry and the aldehyde tag. Angew. Chem. Int. Ed. Engl. 51, 4161–4165 (2012).
Peng, X., Chen, J., Misewich, J. A. & Wong, S. S. Carbon nanotube-nanocrystal heterostructures. Chem. Soc. Rev. 38, 1076–1098 (2009).
Sletten, E. M. & Bertozzi, C. R. From mechanism to mouse: a tale of two bioorthogonal reactions. Acc. Chem. Res 44, 666–676 (2011).
Pedelacq, J. D., Cabantous, S., Tran, T., Terwilliger, T. C. & Waldo, G. S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24, 79–88 (2006).
Rekas, A., Alattia, J. R., Nagai, T., Miyawaki, A. & Ikura, M. Crystal structure of venus, a yellow fluorescent protein with improved maturation and reduced environmental sensitivity. J. Biol. Chem. 277, 50573–50578 (2002).
Niu, W. & Guo, J. Expanding the chemistry of fluorescent protein biosensors through genetic incorporation of unnatural amino acids. Mol. Biosyst. 9, 2961–2970 (2013).
Bae, J. H. et al. Expansion of the genetic code enables design of a novel "gold" class of green fluorescent proteins. J. Mol. Biol. 328, 1071–1081 (2003).
Wang, F., Niu, W., Guo, J. & Schultz, P. G. Unnatural amino acid mutagenesis of fluorescent proteins. Angew. Chem. Int. Ed. Engl. 51, 10132–10135 (2012).
Reddington, S. C. et al. Different photochemical events of a genetically encoded phenyl azide define and modulate GFP fluorescence. Angew. Chem. Int. Ed. Engl. 52, 5974–5977 (2013).
Hartley, A. M., Worthy, H. L., Reddington, S. C., Rizkallah, P. J. & Jones, D. D. Molecular basis for functional switching of GFP by two disparate non-native post-translational modifications of a phenyl azide reaction handle. Chem. Sci. 7, 6484–6491 (2016).
Freeley, M. et al. Site-specific one-to-one click coupling of single proteins to individual carbon nanotubes: a single-molecule approach. J. Am. Chem. Soc. 139, 17834–17840 (2017).
Marth, G. et al. Precision templated bottom-up multiprotein nanoassembly through defined click chemistry linkage to DNA. ACS Nano 11, 5003–5010 (2017).
Zaki, A. J. et al. Defined covalent assembly of protein molecules on graphene using a genetically encoded photochemical reaction handle. RSC Adv. 8, 5768–5775 (2018).
Remington, S. J. Fluorescent proteins: maturation, photochemistry and photophysics. Curr. Opin. Struct. Biol. 16, 714–721 (2006).
Salna, B., Benabbas, A., Sage, J. T., van Thor, J. & Champion, P. M. Wide-dynamic-range kinetic investigations of deep proton tunnelling in proteins. Nat. Chem. 8, 874–880 (2016).
Kozakov, D. et al. The ClusPro web server for protein-protein docking. Nat. Protoc. 12, 255–278 (2017).
Leaver-Fay, A. et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzym. 487, 545–574 (2011).
Alford, R. F. et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 13, 3031–3048 (2017).
Plass, T., Milles, S., Koehler, C., Schultz, C. & Lemke, E. A. Genetically encoded copper-free click chemistry. Angew. Chem. Int. Ed. Engl. 50, 3878–3881 (2011).
Chin, J. et al. Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc. 124, 9026–9027 (2002).
Reddington, S., Watson, P., Rizkallah, P., Tippmann, E. & Jones, D. D. Genetically encoding phenyl azide chemistry: new uses and ideas for classical biochemistry. Biochem Soc. Trans. 41, 1177–1182 (2013).
Reddington, S. C., Tippmann, E. M. & Jones, D. D. Residue choice defines efficiency and influence of bioorthogonal protein modification via genetically encoded strain promoted Click chemistry. Chem. Commun. 48, 8419–8421 (2012).
Seifert, M. H. et al. Backbone dynamics of green fluorescent protein and the effect of histidine 148 substitution. Biochemistry 42, 2500–2512 (2003).
Larsen, T. A., Olson, A. J. & Goodsell, D. S. Morphology of protein-protein interfaces. Structure 6, 421–427 (1998).
Mutch, S. A. et al. Deconvolving single-molecule intensity distributions for quantitative microscopy measurements. Biophys. J. 92, 2926–2943 (2007).
Dijkman, P. M. et al. Dynamic tuneable G protein-coupled receptor monomer-dimer populations. Nat. Commun. 9, 1710 (2018).
Strickler, S. J. & Berg, R. A. Relationship between absorption intensity and fluorescence lifetime of molecules. J. Chem. Phys. 37, 184–822 (1962).
Ahnert, S. E., Marsh, J. A., Hernandez, H., Robinson, C. V. & Teichmann, S. A. Principles of assembly reveal a periodic table of protein complexes. Science 350, aaa2245 (2015).
Campbell, R. E. et al. A monomeric red fluorescent protein. Proc. Natl Acad. Sci. USA 99, 7877–7882 (2002).
Shinobu, A., Palm, G. J., Schierbeek, A. J. & Agmon, N. Visualizing proton antenna in a high-resolution green fluorescent protein structure. J. Am. Chem. Soc. 132, 11093–11102 (2010).
van Thor, J. J. Photoreactions and dynamics of the green fluorescent protein. Chem. Soc. Rev. 38, 2935–2950 (2009).
Reddington, S. C. et al. Genetically encoded phenyl azide photochemistry drives positive and negative functional modulation of a red fluorescent protein. RSC Adv. 5, 77734–77738 (2015).
Agmon, N. Proton pathways in green fluorescence protein. Biophys. J. 88, 2452–2461 (2005).
Shinobu, A. & Agmon, N. Proton wire dynamics in the green fluorescent protein. J. Chem. Theory Comput. 13, 353–369 (2017).
Wall, M. A., Socolich, M. & Ranganathan, R. The structural basis for red fluorescence in the tetrameric GFP homolog DsRed. Nat. Struct. Biol. 7, 1133–1138 (2000).
Acharya, A. et al. Photoinduced chemistry in fluorescent proteins: curse or blessing? Chem. Rev. 117, 758–795 (2017).
Kamiya, K., Boero, M., Tateno, M., Shiraishi, K. & Oshiyama, A. Possible mechanism of proton transfer through peptide groups in the H-pathway of the bovine cytochrome c oxidase. J. Am. Chem. Soc. 129, 9663–9673 (2007).
Tsukihara, T. et al. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc. Natl Acad. Sci. USA 100, 15304–15309 (2003).
Frank, R. A., Titman, C. M., Pratap, J. V., Luisi, B. F. & Perham, R. N. A molecular switch and proton wire synchronize the active sites in thiamine enzymes. Science 306, 872–876 (2004).
Ding, Y. et al. Ratiometric biosensors based on dimerization-dependent fluorescent protein exchange. Nat. Methods 12, 195–198 (2015).
Wan, W. et al. A facile system for genetic incorporation of two different noncanonical amino acids into one protein in Escherichia coli. Angew. Chem. Int. Ed. Engl. 49, 3211–3214 (2010).
Rovner, A. J. et al. Recoded organisms engineered to depend on synthetic amino acids. Nature 518, 89–93 (2015).
Lajoie, M. J. et al. Genomically recoded organisms expand biological functions. Science 342, 357–360 (2013).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Chovancova, E. et al. CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 8, e1002708 (2012).
Acknowledgements
We would like to thank the Edward Lemke and his group at EMBL Heidelberg for donating the pEVOL-SCO plasmid. We would like to thank the staff at the Diamond Light Source (Harwell, UK) for the supply of facilities and beam time, especially Beamline I03 and I04 staff. We thank BBSRC (BB/H003746/1 and BB/M000249/1), EPSRC (EP/J015318/1) and Cardiff SynBio Initiative/SynBioCite for supporting this work. H.L.W. was supported by a BBSRC-facing Cardiff University PhD studentship, H.S.A. by the Higher Committee for Education Development in Iraq, and R.L.J. by KESS studentship. We would like to thank Mr Thomas Williams of the Mass Spectrometry Suite in the School of Chemistry, Cardiff University, and the Protein Technology Hub, School of Biosciences, Cardiff University for use of facilities.
Author information
Authors and Affiliations
Contributions
All authors contributed to the writing of the paper and analysing data. H.L.W. undertook the modelling work, structural biology and functional characterisation of the homodimers; H.S.A. undertook the heterodimer work; J.R.P. and A.W. performed the initial homodimer synthesis and gel mobility assays; R.B. performed the disulfide-based dimer construction and analysis; R.L.J. and D.W.W. helped prepare mutant proteins. W.J.D. and O.K.C. undertook the TIRF measurements and analysis. P.R. collected structural data and helped with structure determination. D.D.J. conceived and directed the project, and analysed data.
Corresponding author
Ethics declarations
Competing interest:
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Worthy, H.L., Auhim, H.S., Jamieson, W.D. et al. Positive functional synergy of structurally integrated artificial protein dimers assembled by Click chemistry. Commun Chem 2, 83 (2019). https://doi.org/10.1038/s42004-019-0185-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-019-0185-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
