
Abstract
Multiple methods with distinctive strengths and drawbacks have been devised so far to produce graphene. However, they all need post-synthesis transfer steps to characterize the product. Here we report the synthesis of pristine graphene inside the transmission electron microscope using gold as catalyst and self-removing substrate without employing a specialized specimen holder. The process occurs at room temperature and takes place within milliseconds. The method offers the possibility of precise spatial control for graphene production and immediate characterization. Briefly, the irradiating electrons generate secondary electrons leading to surface charging if the gold particles reside on a poorly conducting support. At a critical charge density, the particle ejects ions mixed with secondary electrons (plasma) causing the particle to shrink. Simultaneously, hydrocarbon contamination within the electron microscope is cracked, thus providing carbon for the growth of graphene on the particle’s surface. The Technique is potentially attractive for the manufacture of in situ graphene-based devices.
Similar content being viewed by others

Introduction
Graphene has rapidly emerged as a key material in less than two decades since its isolation by Novoselov and Geim1, such that early products incorporating graphene are already on the market (https://www.graphene-info.com/graphene-products). However, this is only the beginning in terms of potential applications using graphene. Its unique electrical, chemical, and mechanical properties have inspired numerous ideas such as flexible touch screens2, high-performance solar cells3, and even superconducting components4. However, if these and more applications are to materialize, the scientific community needs to develop new synthesis techniques that are tailored to streamline the incorporation of graphene into industrial assembly lines. The synthesis techniques developed to date have strong and weak points relative to the target application one has in mind5. Chemical vapor deposition (CVD) has made progress in terms of quantity and quality by producing large-area high-quality graphene sheets6,7,8, however, the sheets get easily contaminated (and damaged) during the transfer process required to mount them onto the target substrate. So-called “wet chemical methods” are cheap but the final products can suffer from structural inhomogeneity and toxic byproducts are common9. Exfoliation methods are versatile and can produce high-quality graphene, however, they tend to be low-yield and time consuming (this can be expedited significantly by increasing the synthesis temperature)10. The need for high temperatures plagues most synthesis approaches including arch discharge11 and epitaxial growth12. Furthermore, with the exception of a few variations of the CVD approaches13,14, fabrication methods in general suffer from two crucial weaknesses namely: spatial control and the need of a post-synthesis transfer step that can contaminate and/or damage the flakes. The use of metallic catalysts is widespread in many of the techniques mentioned above, however, to the best of our knowledge Au has only once been used as substrate for the thermal CVD production of graphene15. Gold, as well as copper and palladium nanoparticles have been used to graphitize carbon nanofibers in situ and inside a transmission electron microscope (TEM)16. However, the end products vary from resembling carbon fibers with small graphitized areas to short graphene nanoribbons interspaced by graphitic material rather than two-dimensional graphene flakes. Furthermore, all these studies used specialized biased holders with a controllable nanotip to drive the synthesis process which relied on Joule heating for the production of graphene16,17,18.
Here we show a synthetic approach that aside from yielding essentially instantaneous fabrication at room temperature, opens exciting possibilities for precise spatial control since it takes place in a TEM in which the spatial size and location of the electron beam can in principal be controlled. Our method is entirely carried out at room temperature; it employs Au particles that simultaneously serve as a substrate and catalyst that produce graphene flakes in a high vacuum environment, thus significantly mitigating contamination. The fact that the Au substrate is largely “consumed” during the process coupled with the high spatial control of the technique opens up the possibility of applying this method to rapidly manufacture devices in situ with spatial control avoiding the need for laborious transfer procedures. In addition, our method offers a more direct channel to study important aspects of the dynamics of the synthesis process and makes the characterization of the flakes more straightforward.
Results
Synthesis protocols
The typical specimen used for the production of graphene consists of dry Au powder mechanically smeared over a standard Cu/lacey C grid (see Methods section). The grids are loaded into a TEM which is usually operated at 300 kV (and at times at 80 kV). Once inside the TEM, the synthesis protocol proceeds as follows: initially, the Au particles are generally found aggregated in clusters over the lacey C threads of the grid although individual nanoparticles (radius 200–400 nm) can also be found scattered over the grid. Upon exposure to electron beam irradiation the clusters remain stable so long as the beam current density is below 0.1 A/cm2. Near this threshold value, which in this study is achieved by condensing the electron beam, the clusters change into single monocrystalline particles (Supplementary Figure 1) with radii that vary from 1 to around 5 µm. Graphene flakes are found to have formed at the surface of the Au particles. Some graphene flakes protrude freely from the particle’s surface while others from a bridge that anchors the particle to the lacey C. However, it is difficult to visualize the process as it occurs quasi-instantaneously. If one then further condenses the electron beam on the newly formed Au particle/graphene, the Au particle shrinks as the current density increases. As the particles shrink, more of the produced graphene flakes are exposed and new graphene also appears to form. The graphene synthesis process is summarized in Fig. 1 (and Supplementary Figure 2 which is an unedited micrograph of Fig. 1g), alternatively, the synthesis can be visualized in Supplementary Movie 1 which shows the process as viewed from the TEM flu-screen.

Overview of the synthesis process. a–c show a schematic representation of the synthesis process. a Schematic of the initial state of an Au multi-particle cluster supported on lacey C. b Drawing of a single larger Au particle formed as the multi-particle cluster fuses after the current density threshold is reached. Small graphene flakes bridge the particle to the lacey C. c The particle shrinks as the current density increases further. The exposed graphene is larger now. The inset shows a schematic balls-and-sticks model of the graphene sheet. d–f Screenshots from Supplementary Movie 1 showing synthesis process as seen on the microscope’s flu-screen and they respectively correspond to the stages depicted in a, b and c (scale bar: 1 μm). d Stable central cluster 1 while the current density is lower than 0.1 A/cm2. e Particle 1 which formed from cluster 1 after the current density is reached, ca. 0.1 A/cm2. f Particle 1 shrunken after being exposed to a current density of 3.8 × 103 A/cm2. g False color micrograph of the area in the white box in f. The lacey C, graphene flakes and the Au particle are respectively shaded in red, blue and yellow (an unedited version of this micrograph is shown in Supplementary Figure 2). h Same as image in g but showing only the outlines of the three materials materials (scale bars in g and h: 400 nm). i Area within the small red box in h. The atomic structure of the flake is now discernible. j Filtered TEM image of the red box in i. The uniform Moiré pattern typical of bilayer graphene is clear (scale bars in i and j: 1 nm)
Chemical and structural characterization
Electron energy loss spectroscopic (EELS) studies confirm that the flakes comprise sp2 carbon (Supplementary Figure 3) while X-ray dispersive spectroscopy (EDS) confirms that the Au particles have no trace contaminants (Supplementary Figure 4). High resolution low voltage spherical aberration (Cs) corrected TEM studies and complementary analysis of the as-produced graphene flakes are consistent with polycrystalline bilayer graphene as the primary product (Fig. 2). The graphene grain size varies greatly, from ca. 10 nm2 to more than 100 nm2.

Microstructure of the as-produced graphene. a Drawing of an Au particle anchored to the lacey C by the as-produced graphene. b Real micrograph of the area schematically represented in the red box in a (scale bar: 20 nm). c Higher magnification of the red box in b (scale bar: 1 nm). d Schematic balls-and-sticks model of a graphene bilayer this structure produces a range of Moiré patterns such as the one boxed in c. e, f FFT pattern from the boxed area in c. The pattern has 12 spots which stacked flakes rotated by and angle of around 18˚ with respect to each other forming a bilayer. g, h Filtered TEM images from the spots selected in c and d respectively. The filtered images show the honeycomb structures of the individual graphene sheets of the bilayer. i Filtered TEM image from all the diffraction spots. j Graphene flake (outlined in blue) extending from an Au particle to the lacey C (scale bar: 100 nm). k Higher magnification image of the region within the red box in j (scale bar: 1 nm). Two separate Moiré patterns are appreciated roughly at the left and right half of the image. l Filtered TEM image of k selecting 6 out of the 18 main diffraction spots of the FFT in the inset. The image shows a seamless honeycomb structure. m Filtered image from the remaining 12 diffraction spots (see in inset). A grain boundary runs vertically throughout the image. A way to interpret panels l and m is that we have a graphene bilayer where one sheet is a seamless graphene flake (l) and the second is a flake divided by a grain boundary (m)
Synthesis on various supports
The use of different supports on the TEM grids was also explored. The results show that it is possible to produce graphene flakes (albeit with much lower yields) from Au particles supported on insulating SiOX membranes (Fig. 3 and its unedited version in Supplementary Figure 5). The main difficulty is that the Au particles on top of the SiOX membranes get ejected almost instantaneously as the beam starts to converge upon them. This happens before the particles coalesce which seems to be an important stage of the graphene production process.

Graphene synthesis in C-based and C-free grids. a Illustration of a cluster of Au particles over a lacey C grid. b Real Au cluster corresponding to what is schematically represented in the red box of a. The cluster is shaded in yellow (as in the rest of the panels) while the lacey C threads are shaded in red. c Fused and shrunken particle resulting from irradiating the cluster in b with a current density of 4.6 A/cm2. A large graphene flake grows (shaded in blue). d Magnified image of the red box inc. The blue dashed line shows the outline of the flake. e shows a schematic drawing of a cluster on the edge of a hole on a SiOX membrane. f Real cluster as graphically represented in the red box in e. The membrane is shaded in green. g The cluster after being irradiated at a current density of 4.6 × 102 A/cm2. Some particles became partially fused. There is a region shaded in blue highlighting a small graphene flake on the particle in the red box. h Magnified image of the region in the red box. The outline of the flakes are indicated by blue dashed lines. i Schematic representation of an Au cluster on the edge of the metallic frame of a Mo grid. j The cluster in its initial state as it is represented in the red square of panel I. The cluster is unaffected when the current density is raised to around 4.2 × 103 A/cm2. A close up of the red box in h is shown in panel l. No graphene is visible. A version of Fig. 3 with unedited TEM micrographs is given in Supplementary Figure 5. (Scale bars in panels b, c, f and g: 500 nm, scale bars in panels d, h and l: 50 nm, scale bars in panels j and k: 200 nm)
Synthesis never occurred for Au particle clusters residing on conducting materials such as pure Molybdenum grids (Fig. 3i–l). Furthermore, when using pure metallic TEM grids the clusters remain unperturbed regardless of the current density used to irradiate them, suggesting a highly conducting support material hinders the fabrication process.
We also explored the use of holey C and C film grids to investigate the shrinking dynamics of the Au particles as a function of the current density for the different C-based supports as the contact area between the Au particles and the support varies considerably (Supplementary Figure 6). Particle clusters on lacey C grids (smaller contact area) shrink to form a single crystal single particle with a radius between 70–350 nm for current densities between 1.2–3 × 103 A/cm2. For particle clusters on holey C (medium contact area) radii between 400–450 nm at a current density of 4.5 × 103 A/cm2 were observed while for C films (large contact area) radii between 520–610 nm at current densities between 2.8–4.4 × 103 A/cm2 were found (Supplementary Figure 7). The data show a clear trend where a larger contact surface leads to less shrinkage. The same trend is reproduced when individual particles are irradiated instead of fused particle clusters (Supplementary Figure 8). Particles loosely attached to the lacey C shrink more than those that are fully supported on carbon.
Mechanical factors might also affect the shrinking process and the topology of the as-produced graphene. As the Au particles shrink they strain the carbon support by pulling on the graphene attached to a support (lacey carbon). The support can often be seen to deform as a consequence of the strain as the Au NPs shrink. Less robust lacey C threads bend easily towards a shrinking Au particle producing more wrinkled graphene. More robust supports such as holey C and C films deform less easily and apparently make shrinking more difficult, however, the produced flakes extend more due to the larger strain (Supplementary Figure 9 and further explained in Supplementary Note 1).
Rate of shrinkage and the role of charging by secondary electron emission
The rate of mass loss from the Au particles depends on the rate of increase of the current density irradiating the Au particles and it also varies at different stages of the shrinkage process. For instance, soon after coalescence the particles lose a significant fraction of their mass in an abrupt or “explosive” manner. A large number of nanoparticles form as a consequence of this explosion, they can be seen residing on various areas of the amorphous C support surrounding the irradiated particle (Supplementary Figure 10). Shrinkage continues after this initial explosion albeit in a less abrupt manner. If the current density increases as the current density increases smoothly, the shrinkage can be made approximately linear. Supplementary Movie 2 shows a short clip of an Au particle shrinking slowly due to the very gradual increase of the impinging current density. The shrinkage of this particle is confirmed to be close to linear as it can be observed in Supplementary Figure 11.
The collective data suggest two major physical phenomena are responsible for triggering and driving the synthesis process from its onset to its conclusion: superficial charging due to SE emission and hydrocarbon radiolysis induced cracking followed by carbon deposition. The former drives the shrinking of the Au particles (mass loss) while the latter provides the C feedstock necessary for the nucleation and growth of the as-produced graphene. We now reflect on these phenomena in greater detail.
All SEM and TEM specimens emit SEs (kinetic energy <50 eV) when irradiated by an e-beam19,20. Our Au specimens generally start as particle clusters that fuse into quasi-spherical Au particles when the current density of the 300 kV e-beam is of around 0.1 A/cm2 but it is not trivial to determine this threshold current accurately enough so as to perform a reasonably accurate calculation of its SE yield δSE and the emission current ISE. Thus, to estimate the SE yield as accurately as possible, we focused on the coalesced microparticle shown in Supplementary Figure 11 and partially recorded in Supplementary Movie 2 as it shrank under convergent irradiation. One can estimate ISE at various points through the shrinkage by measuring the particle’s radius through consecutive frames of Supplementary Movie 2 and applying the framework developed by Lin et al.20 which is developed in Supplementary Note 2. Briefly, δSE can be divided into two components, one fraction generated by the primary electrons (δPE) and one produced by the backscattered electrons (δBE). Thus we have:
where η is the backscattering coefficient and the ratio β = δPE/δBE is of around 2 for Au and energies larger than 10 keV21. Estimating δPE for the case of our fused Au particles we get δSE ≈ 0.05 which generates a current of SEs of around ISE = 1.52 × 10−8 A = 9.44 × 1010 electrons/s for a current density of 4.6 A/cm2 (the details of the calculation are shown in Supplementary Note 2). Most of the SE making up ISE are produced near the particle’s surface since their mean escape depth is λAu = 0.5 nm20. The surface depletion of electrons will lead to the accumulation of positive charge distributed in a surface “shell”, viz. Au ions. This is in agreement with the observations of Chen et al.22 who studied Ga microparticles (some coated with silica, some not) expelling their external layers as they were subjected to convergent e-beam irradiation. They explain that superficial charging of the Ga particles grows over time until a critical superficial charge density σ is reached. σ is the maximum amount of superficial charge that the particle can store before repulsive forces between the superficial ions (ionized atoms) overcome its surface energy, thus, causing the expulsion of the outermost atomic layer. σ is given by:
where αAu is the surface energy of gold (1.5 J/m2) and ε0 is the vacuum’s permittivity (8.85 × 10−12 s2A2/Jm). Thus, the analyzed particle has a critical surface charge value of σ = 6.04 × 10−3 C/m2. If the current of SE emitted from the particle is of around ISE = 1.52 × 10−8 A, then, the superficial charge density should be reached after only 2.64 µm. Now, Au has a FCC crystal structure with a lattice constant of 0.408 nm. The particle reduces its radius by one atomic layer every 2.64 µs. At first approximation we can imagine that this shrinking rate is constant, thus, the particle in question should reduce its radius by around 3 nm in a little longer than 18.48 µs. However, experimentally the data shows that the radius of the particle shrinks by about 3 nm in 0.2 s. The reason behind this lower shrinking rate is the electrical contact established between the particle and the substrate. A current of electrons IS supplied by the underlying substrate flows into the charging Au particle thus mitigating the surface charge build up rate. However, the contact resistances between the Au particle and the bilayer graphene and the bilayer graphene and the amorphous carbon of the TEM grid prevent the IS from the particle reaching electrostatic equilibrium fast enough. In particular, not fast enough to prevent the critical surface charge density to be reached eventually (in a timescale of around 10−2 s as bore by the data) thus resulting in the shrinkage of the Au particle. A more detailed explanation of this process and the influence of the substrate is provided in the Supplementary Note 3.
Summarizing, as the electron beam gradually converges, the increasing current density further enhances charging which in turn leads to the ejection of more superficial Au ions, thus shrinking the Au particles (see Fig. 4). We refer to the process as a localized electron-beam induced plasma (LEBIP). The expelled ions themselves, along with the emitted SE form a plasma-like cloud (ions and electrons) around the irradiated Au particles which subsequently condenses forming small Au nanoparticles at any nearby surface, for example, lacey C (Fig. 5a–d and Supplementary Figure 10). The SE emission is crucial for both, the synthesis of graphene and driving the LEBIP process. The expulsion of ions due to the LEBIP process leads to the mass reduction of the Au particle.

The graphene synthesis process revisited. a Schematic drawing of a multiparticle Au cluster irradiated with low current density. The particles in the cluster emit secondary electrons as long as they are irradiated, however, the SE yield is not high enough to build up a superficial charge sufficient to disrupt the structure of the cluster. b The yield of secondary electrons increases with the current density, as a result, electron-depleted superficial areas are generated. The net charge buildup is positive and comes from superficial Au ions left behind. Areas that are further away from the anchor points between the as-produced graphene and the lacey C (top surfaces) are prone to charge more since they cannot be efficiently resupplied with electrons from the C support. The bottom surface is more stable and a better substrate for the growth of graphene since it is closer to the entering channels of the electrons coming from the substrate. Eventually, the repulsive forces between Au ions on the top surface are large enough to cause their ejection. The structure is compromised and the individual Au particles fuse while a large fraction of their mass is expelled. c The fused particle sitting over the as-produced graphene flakes continues to shrink as the current density grows

Plasma generation–condensation and secondary electron-induced graphene deposition. a–d Show the plasma generation process which is responsible for the shrinking of the irradiated Au particles. The plasma is generated by the emission of secondary electrons and the subsequent expulsion of Au ions. a Shows a plasma cloud on top of an Au particle right after it has shrunk and produced graphene. b Shows a magnified schematic of the black rectangle in a. The ions in the plasma are free to interact. (Hydrocarbons are also present but are not central in these panels). c The lacey C provides nucleation sites for the neutral Au atoms produced in the plasma to condense into numerous Au nanoparticles. d TEM micrograph showing real Au nanoparticles condensed on the lacey C near an irradiated Au particle (scale bar: 500 nm). e–h Focus on the hydrocarbon radiolysis and graphene formation through the synthesis process. e Shows a drawing of an irradiated multipaticle cluster. f Magnification on the black box in e showing the emitted secondary electrons and the nearby volatile organic. g Several processes occur simultaneously. The cluster is disrupted and fuses while the secondary electrons and imaging electrons crack the hydrocarbon molecules through radiolysis reactions. Carbon is produced via successive radiolysis events and gets adsorbed on the lower surface of the Au particle. Graphene quickly starts to form connecting the Au particle with the lacey C. This creates an influx of electrons (IS) which helps to mitigate the superficial charging of the Au particle stabilizing its surface enough to provide a substrate for the formation of small graphene islands. h The graphene islands grow and diffuse superficially until they merge to create polycrystalline graphene flakes
The physical mechanisms in the process also explain why the process does not occur when irradiating Au particles residing on highly conductive supports, since no charge can build. The degree of charging is limited by the conductivity of the support and, more importantly, by the contact resistance between the particles and support. Thus, two factors are important: the electric contact resistances (between the particle and the graphene and the graphene with the amorphous carbon support) and the physical extent of the Au/substrate contact (both aspects are discussed in more depth in Supplementary Notes 2 and 3). The LEBIP process also explains why shrinking Au particles do not eventually vanish. This is because the SE current leaving the particles (ISE) scales down with particle size, therefore a smaller current from the support (IS) is required to restore electrostatic equilibrium. In other words, the substrate becomes more effective at mitigating the charge build-up in the particles as the particle’s size goes down, in this way, further shrinkage is prevented. A further implication of this is the fact that individual particles loosely attached to the carbon substrate shrink down to smaller sizes than individual particles fully supported on the substrate. The latter are in essence totally supported by a C film and thus better electrically contacted by the support. In such circumstances, IS can more effectively balance out the SE current ISE thus shrinking is more easily prevented.
Carbon feedstock supply and graphene growth
Our experiments, especially the successful synthesis of graphene using carbon-free grids (SiOX, membranes), suggest that the main source of carbon necessary for the growth of the as-produced graphene flakes comes from hydrocarbon contamination inside the TEM column or primarily from species adsorbed on the specimen/TEM grid23. Adsorbed molecules desorb under electron beam irradiation and form a gaseous cloud around the irradiated area24,25. These molecules can then be cracked by radiolysis26 providing a source of carbon and other volatile byproducts. In this case the radiolysis cracking of contaminant hydrocarbons can occur not only from the imaging electrons but also the relatively large number SE’s. We calculate ISE = 3.89 × 109 electrons/s when a particle with a radius of around 1 µm (typical dimensions at the moment of coalescence) irradiated with a current density of 0.1 A/cm2. This is equivalent to an outflow of 250 SE’s emitted per second per nm2 of Au. Considering that the surface density of C atoms in graphene is ca. 39 atoms/nm2 and that a high fraction of SE’s have sufficient kinetic energy (up to 50 eV) to trigger two consecutive radiolysis hydrocarbon cracking reactions, the outflow of SE’s is sufficient on their own to promote the hydrocarbon decomposition via radiolysis, nucleation, and growth of the first graphene islands. As the graphene islands continue to grow they also diffuse over the surface of the Au particles due to the very low miscibility of C on Au27,28. With time, the monocrystalline islands collide and merge producing the polycrystalline flakes that we observe. The shrinking of the particle via LEBIP helps in bringing the graphene islands together since their superficial concentration increases not only through continuous island nucleation but by the gradual shrinking of the particle’s surface. This whole process is illustrated in Fig. 5e–h.
There is another fact that highlights the importance of the LEBIP process for the synthesis of exposed graphene flakes. For instance, one cannot discard the formation of small flakes or islands on Au particles that do not undergo LEBIP, for example, those residing on conducting grids. These particles must also emit SE’s that should be able to crack volatile hydrocarbons, however, since the particles do not shrink, the surface concentration of graphene islands does not increase at the rate necessary to promote the efficient formation of larger graphene flakes. Furthermore, the graphene that possibly forms on them cannot protrude or become exposed since the frustration of the LEBIP process prevents the Au particles from acting as self-removing substrates.
The contribution of carbon feedstock coming from direct contact between the Au particles and the amorphous carbon substrate must be limited since, for instance, the particles are often entirely suspended by graphene “bridges” with no direct contact to the amorphous carbon support as they shrink and continue to produce graphene. Furthermore, small graphene flakes appear shortly after coalescence and they often protrude from areas of the particle that have no connection to the carbon support, thus indicating that the C feedstock is more likely to be from the vacuum column.
Some subtler but important considerations are also relevant. For instance, some graphene flakes nucleated early on may anchor the particles to a lacey C filament by the time the multi-particle clusters fuse into single Au particles. This also can explain the observed strain in the graphene in many instances. Moreover, these anchor points receive new graphene flakes from the upper and lower sides of the spherical particles as growth takes place. It is easy to envisage both the upper and lower supply of graphene (flakes) anchor and merge to two slightly different anchorage sites leading to bi-layer graphene as we observe.
The limited role of beam-induced heating
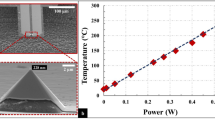
We also consider the role of heating due to electron beam irradiation. Our calculations based on the particle in Supplementary Figure 11, i.e., a 700 nm Au particle producing graphene as it shrinks, tends asymptotically towards a maximum value of 365.75 K when bombarded by a 300 KeV beam with a current density of 4.6 A/m2 (Supplementary Figure 12 shows a schematic model of the particle which is used for the calculations developed in Supplementary Note 4). In other words, beam-induced heating cannot be taken as the main mechanism driving the shrinkage of the Au particles since the melting point of Au is 1065 K.
The absorption of C into the Au particles should be very limited because, one the one hand, the limited heating effects do not enhance the absorption process and secondly the solubility of C in Au is minimal27.
Discussion
In conclusion, we have developed a nearly instantaneous synthesis route to obtain polycrystalline bilayer graphene flakes inside the TEM. The fabrication of the graphene mechanism relies on the emission of SEs from the irradiated Au particles leading to the ejection of the resultant surface ions being ejected forming a plasma around the Au particle and shrinking the particle in this manner. At the same time hydrocarbon contamination in the vicinity is cracked by these species through radiolysis providing a C feedstock. The C feedstock deposit combined with the shrinking Au particles allows for the nucleation and growth and merging of graphene flakes which ultimately yield free-standing bi-layer graphene. It is nevertheless important to stress that although the mechanism put forward to explain the synthesis of graphene and shrinkage of the irradiated particles is built upon reasonable assumptions and a number of reported findings that make it plausible, some of them need further experimental scrutiny in order to reduce the degree of speculation inherent to our proposed mechanism, most crucially, the precise I-V characteristics of the electrical contacts between the graphene and the aC support.
Since the technique takes place using an electron bean, it provides a high degree of special control. Moreover, with optimized synthesis conditions for the graphene, the Au (or other metal) nanoparticle can be fully removed, providing a self-removing catalyst in the process. The rapid growth (milliseconds), the spatial control and the self-removal of the catalyst makes the technique attractive as a viable means to manufacture graphene (and potentially other 2D based) devices in situ in TEM or in future dedicated instruments.
Methods
TEM samples preparation
The TEM specimens were prepared simply by gently smearing a TEM grid, typically a Cu/lacey carbon grid (Plano, 400 mesh), with gold powder (particle size <1 micron and 99.9% from Chempur. CAS-Nr.: 7440-57-5). A minute amount of the powder is sprinkled on a plastic petri container while the grid, held with metallic tweezers, is softly pressed and dragged over the sparse Au powder. This preparation procedure was identically replicated with every other kind of grid used for our experiments, namely: holey C grid/Cu grids (Plano, 300 mesh), Cu/carbon film grids (Plano, 200 mesh), molybdenum grids (Plano, 200 mesh) and silicon monoxide grids (Plano, 200 mesh).
In situ synthesis protocol
The grids were immediately introduced into the TEM to carry out the graphene synthesis process. The TEM in question is a FEI-Tecnai F30 Transmission Electron Microscope operated at 300 kV. EDS and EELS studies of the as-produced graphene and the Au particles were carried out in this device. After synthesis, the samples were taken to a FEI Titan 300-80 TEM with third-order spherical aberration
correction operated at 80 kV for high-resolution imaging without the risk of damaging the graphene due to knock-on interactions.
Data availability
The data that support the findings of this study are available on request from the corresponding author.
References
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Large, M. J. et al. Selective mechanical transfer deposition of langmuir graphene films for high-performance silver nanowire hybrid electrodes. Langmuir 33, 12038–12045 (2017).
Yan, K. et al. High-performance graphene-based hole conductor-free perovskite solar cells: Schottky junction enhanced hole extraction and electron blocking. Small 11, 2269–2274 (2015).
Gibney, E. Surprise graphene discovery could unlock secrets of superconductivity. Nature 555, 151–152 (2018).
Rümmeli, M. H. et al. Graphene: pieceing it together. Adv. Mater. 23, 4471–4490 (2011).
Li, X. et al. Large-area synthesis of high-quality and uniform graphene films on copper foils. Science 324, 1312–1314 (2009).
Kim, K. S. et al. Large-scale pattern growth of graphene films for stretchable transparent electrodes. Nature 457, 706–710 (2009).
Reina, A. et al. Large area, few-layer graphene films on arbitrary substrates by chemical vapor deposition. Nano Lett. 9, 30–35 (2009).
Zhong, Y. L., Tian, Z. M., Simon, G. P. & Li, D. Scalable production of graphene via wet chemistry: progress and challenges. Mater. Today 18, 73–78 (2015).
Cai, M. Z., Thorpe, D., Adamson, D. H. & Schniepp, H. C. Methods of graphite exfoliation. J. Mater. Chem. 22, 24992–25002 (2012).
Fang, X. Q., Shashurin, A. & Keidar, M. Role of substrate temperature at graphene synthesis in an arc discharge. J. Appl. Phys. 118, 103304 (2015).
Tetlow, H. et al. Growth of epitaxial graphene: theory and experiment. Phys. Rep. 542, 195–295 (2014).
Ding, D., Solis-Fernandez, P., Hibino, H. & Ago, H. Spatially controlled nucleation of single-crystal graphene on cu assisted by stacked Ni. ACS nano 10, 11196–11204 (2016).
van den Beld, W. T. E., van den Berg, A. & Eijkel, J. C. T. Spatial control of direct chemical vapor deposition of graphene on silicon dioxide by directional copper dewetting. RSC Adv. 6, 89380–89386 (2016).
Oznuluer, T. et al. Synthesis of graphene on gold. Appl. Phys. Lett. 98, 183101 (2011).
Takahashi, C., Yaakob, Y., Yusop, M. Z. M., Kalita, G. & Tanemura, M. Direct observation of structural change in Au-incorporated carbon nanofibers during field emission process. Carbon N. Y. 75, 277–280 (2014).
Rosmi, M. S. et al. Visualizing copper assisted graphene growth in nanoscale. Sci. Rep. 4, 7563 (2014).
Rosmi, M. S. et al. In situ TEM visualization of Pd assisted graphene growth in nanoscale. 2016 IEEE 16th International Conference on Nanotechnology (IEEE-NANO), Sendai: 622–623 https://doi.org/10.1109/NANO.2016.7751486 (2016).
Joy, D. C. A model for calculating secondary and backscattered electron yields. J. Microsc. 147, 51–64 (1987).
Lin, Y. & Joy, D. C. A new examination of secondary electron yield data. Surf. Interface Anal. 37, 895–900 (2005).
Reimer, L. & Drescher, H. Secondary electron emission of 10–100 KeV electrons from transparent films of Al and Au. J. Appl. Phys. D 10, 805–815 (1977).
Chen, Y. et al. Fabrication of nanoscale Ga balls via a Coulomb explosion of microscale silica-covered Ga balls by TEM electron-beam irradiation. Sci. Rep. 5, 11313 (2015).
Rummeli, M. H. et al. In Situ Room Temperature electron-beam driven graphene growth from hydrocarbon contamination in a transmission electron microscope. Materials 11, E896 (2018).
Kumao, A., Hashimoto, H. & Shiraishi, K. Studies on specimen contamination by Transmission Electron Microscopy. J. Electron Microsc. 30, 161–170 (1981).
Ennos, A. E. The origin of specimen contamination in the electron microscope. Br. J. Appl. Phys. 4, 101–106 (1953).
Leontowich, A. F. G. & Hitchcock, A. P. Secondary electron deposition mechanism of carbon contamination. J. Vac. Sci. Technol. B 30, 030601 (2012).
McLellan, R. B. The solubility of carbon in solid gold, copper and silver. Scr. Metall. 3, 389–392 (1969).
Rummeli, M. H. et al. New frontiers in electron-driven chemistry in and around graphene. Adv. Mater. 30, 1800715 (2018).
Acknowledgements
M.H.R. thanks the China National Science Foundation (NSFC, Project 51672181), the National Science Center (Poland) for the financial support within the frame of the Sonata Program (Grant agreement 2014/13/D/ST5/02853) and the Opus program (Grant agreement 2015/19/B/ST5/03399), and the Sino-German Research Institute for support (project: GZ 1400).
Author information
Authors and Affiliations
Contributions
M.H.R. and I.G.M. designed the in situ experiments. I.G.M. and T.G. (EELS) conducted the experiments. T.G., A.B., and B.T. helped with high-resolution imaging and image analysis. Z.L. helped with the development of the theoretical framework (charging and heating calculations). The project was led by M.H.R. at every stage. All authors contributed with the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gonzalez-Martinez, I.G., Bachmatiuk, A., Gemming, T. et al. Rapid synthesis of pristine graphene inside a transmission electron microscope using gold as catalyst. Commun Chem 2, 33 (2019). https://doi.org/10.1038/s42004-019-0134-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-019-0134-3
This article is cited by
-
Detection and remediation of bisphenol A (BPA) using graphene-based materials: mini-review
International Journal of Environmental Science and Technology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
