
Abstract
Formamides represent an abundant class of compounds in organic synthesis. They can be made efficiently by the direct catalytic coupling of methanol with amines in the presence of metal-based catalysts. However, these catalysts require complicated organic ligands, susceptible to oxidative self-degradation, restricting their practical applications. Here, we describe an inorganic ligand-supported chromium (III) catalyst, (NH4)3[CrMo6O18(OH)6], which consists of a central chromium (III) single-atomic core supported by a cycle-shaped inorganic ligand consisting of six MoVIO6 octahedra, shows excellent activity and selectivity. Various primary amines and secondary amines are successfully transformed into the corresponding formamides under mild conditions, and the formylation of primary diamines is also achieved. The chromium catalyst can be reused several times with little loss of the activity. Mechanistic insight is provided based on the observation of an intermediate and control experiments.
Similar content being viewed by others
Introduction
Formamides are key chemical feedstock for the manufacture of valuable heterocycles, bioactive molecules, and pharmaceuticals1,2,3,4. They are also widely used as solvents (e.g., N-methylformamide or N, N-dimethylformamide) and softeners in industrial synthesis and processing. In addition, formamides are used as versatile intermediates for Leuckart and Vilsmeier–Haack reactions, and especially in the synthesis of formamidines and isocyanates5,6,7,8. More importantly, formamides are closely related with prebiotic chemistry and useful reagents in biochemistry and molecular biology, particularly in nucleic acids research9,10,11,12,13,14,15,16,17,18.
Traditionally, N-formamide synthesis involves the use of excessive formylating reagents such as formic acid9,10, chloral11, formate10,11,12, methanol13,14, formaldehyde14, and carbon dioxide15,16,17,18,19,20, thus generating copious waste and resulting in poor atom-economy. In the context of sustainability and green chemistry, direct coupling of methanol with amines to produce formamides is an atom-efficient and green alternative, with either H2 or H2O as sole by-products (Fig. 1a). Indeed, methanol is a versatile source of C1 feedstock that can be converted into a variety of key building blocks21. However, the activation of methanol appears to be more difficult than most of common organic chemicals because of its low reactivity and uphill energy barrier, therefore it generally requires harsher conditions to overcome the high-thermaldynamic demands. Recent progress has showed that the direct catalytic coupling of methanol with amines can be achieved in high effiency in the presence of a series of noble metal-based catalysts under mild conditions (Fig. 1b). In 2013, a NHC ligand supported ruthenium complex was developed by Glorius and co-workers for dehydrogenative coupling of methanol and amines to produce formamides with excess styrene as the hydrogen acceptor22. Soon after Glorius’s pioneering work, Asao and co-workers reported the oxidative coupling route promoted by Au nanoparticle catalyst using O2 as an oxidant23. In 2015, Hong and Kang developed another N-formylation catalytic system for dehydrogenative coupling of methanol and amines using an N-heterocyclic carbene coordinated ruthenium dihydride complex24. Very recently, Milstein demonstrated that N-formamides can be generated by dehydrogenative couplings of methanol and amines catalyzed by manganese–PNP pincer complex, representing the first example of base-metal catalysts25.

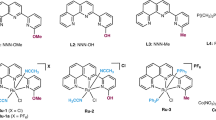
Catalytic systems for synthesis of N-formamides. a Synthesis of N-formamides. b Previous work: organic-ligand supported metal (ruthenium or manganese) catalysts. c This work: inorganic ligand-supported chromium catalyst
Despite remarkable advantages of homogeneous catalysis system in direct N-formylation with methanol26,27,28,29,30,31,32,33,34,35,36,37,38, most of them are based on precious noble-metal catalysts. Other shortcomings such as complicated/commercially unavailable organic ligands, poor stability of the organometallic complex, and the difficulty to recover and reuse catalysts, which restrict their practical applications. Therefore, the development of novel catalytic system that is facilely built from inexpensive and earth-abundant transition metals with good stability and recyclability is high desired but remains great challenges.
Polyoxometalates (POMs)39,40,41,42,43,44,45,46, as a class of metal–oxide clusters with unmatched structural diversity and functionality, offer great potentials as stable inorganic ligands to support transition-metal catalytic sites. Different from classical organic ligands in transition-metal complexes, POMs exhibit reversible redox and strong acidity, thus rendering themselves high resistance toward hydrolysis and oxidative degradation. In this regard, POMs have been widely used as robust inorganic alternatives in transition-metal catalysis47. Very recently, our group has reported for the first time that the Anderson-type POMs can be used as the inorganic ligand to support earth-abundant Fe and Cu as the so-called single-atomic-site catalyst for highly efficient aerobic oxidation of aldehydes to carboxylic acids in water48, and the oxidation of amines to imines under mild conditions49. Inspired by this, the versatile tunability of Anderson-type POMs prompts us to further extend the scope of this type of catalysts for catalytic oxidation transformations by replacing the central core with other base metals.
Herein, we report an inorganic ligand-supported chromium (III) catalyst 1a, (NH4)3[CrMo6O18(OH)6]50,51,52,53 (Fig. 1c, Supplementary Figs. 1–3), which can efficiently catalyze the oxidation coupling of various primary amines and diamine as well as secondary amines with methanol to afford the corresponding formamides using H2O2 as the sole oxidant. Mechanistic insight based on the observation of key intermediate and control reactions is presented. To our best knowledge, 1a represents the first example of chromium-based catalyst that could be used for this transformation, and the N-formylation of primary diamines is also achieved for the first time. Compared with previous homogeneous catalyst systems, the pure inorganic 1a can avoid the use of complicated/sensitive organic ligands. Moreover, this inorganic ligand-supported trivalent chromium catalyst is potentially nontoxic and may be safer than the high-valence chromium catalysts widely used in organic synthesis. More importantly, 1a can be recycled and reused for at least six times with negligible loss of activity due to high stability.
Results
Reaction optimization
Initially, our studies began with the oxidation coupling of methanol and benzylamine (Fig. 2) with 30% H2O2 as the sole oxidant to assess the catalysts 1 (Table 1, entries 1–5). The reaction of benzylamine (1.0 mmol) with CH3OH (1.5 mL) in the presence of 1a (0.1 mol%) at 80 °C resulted in the formation of N-benzylformamide in 60% yield after 40 min (Table 1, entry 1). Analysis of the crude reaction mixture by GC revealed selective formation of the corresponding formamide as major product. In contrast, replacing the centric chromium atom with other metals such as cuprum, nickel, cobalt, and iron (1b–1e), none of benzylformamide was detected under the same conditions (Table 1, entries 2–5), being indicative of the key role of chromium in catalysis.

Model system for screening of catalysts. Screening conditions and results are detailed in Table 1
Control experiments showed that no oxidation product was observed when the starting materials used for synthesis of 1a were employed as catalysts, indicating the synergesticly catalytic effect from both chromium and the polyoxomolybdate ligand (Table 1, entries 6–7). When the reaction was performed under nitrogen atmosphere, the desired product was obtained in <5% yield (Table 1, entry 8), showing that H2O2 is an essential oxidant for the reaction. Under one atm of O2, N-benzylformamide can also be generated but in much lower yield (57%) even after 24 h, which might be ascribed to the lower activity of molecular O2 (Table 1, entry 9). To improve the yield of N-benzylformamide, a series of additives were tested. Na2SO3 (0.05 equiv.) was proved to be very efficient, and the yield can be dramatically enhanced up to 95% (Table 1, entry 10). This is most likely due to the electron-transfer mediation of SO32− which greatly improves the electron-transfer efficiency.
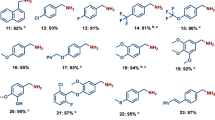
Using the optimized reaction conditions (Supplementary Tables 1–3), some readily available amines were transformed into the corresponding formamides in good to excellent yields (Fig. 3). The reaction of methanol with benzylamines bearing electron-rich groups afforded the corresponding formamides in good yields (compounds 2–7), while benzylamines bearing electron-deficient groups gave slightly diminished yields (compounds 9–13). Benzylamine with bulky substituent can also be converted into formamide smoothly (compound 8). Heteroaromatic benzylamines containing nitrogen, oxygen or sulfur atoms, which are well-known to poison organometallic catalysts due to their strong coordination to the metal center, could also be transformed into the corresponding formamides in good yields (compounds 14–16). The scope of primary amines is not limited to benzylamines. The reaction of methanol and cyclohexylamine afforded N-cyclohexylformamide in 73% yield (compound 18). Similarly, N-(naphthalen-1-ylmethyl)formamide and N-phenylformamide were obtained in 73 and 76% yield, respectively (compounds 17 and 19). Linear aliphatic amines can also undergo facile oxidation to produce the corresponding formamides in good yields (compounds 20–22). The reaction of secondary amines with methanol was also tested. Surprisingly, conversion of cyclic aliphatic amine to the corresponding formamides is achieved in almost quantitative yields (compounds 23–25). The secondary arylamine N-methyl-1-phenylmethanamine and 4-methoxy-N-methylaniline were converted into the corresponding formamides in good yields (compounds 26–27). The industrially important DMF was also obtained in 99% yield from dimethylamine and methanol (compound 28).

Oxidation coupling of amines with methanol catalyzed by 1a. Reaction conditions: Cat. 1a (0.1 mol%), methanol (1.5 mL), amines (1.0 mmol), and 30% H2O2 (4.0 equiv.) at 80 °C. Yields were determined by GC–MS analysis of the crude reaction mixture, values in parentheses are the isolated yields
Formylation of primary diamines with methanol is very challenging and has not been reported yet. This is due to the difficulty to manage the selectivity of product as both imines and ureas can compete with formamides. In particular, when linear diamines couple with methanol, polymeric urea can easily become the dominant species and crush out from the reaction, thus completely prohibiting the formation of formamides38,54,55. Encouraged by the high reactivity of primary and secondary amines using our methodology, a series of primary diamines were tested. As shown in Fig. 4, reaction of aromatic diamines o-phenylendiamine and m-phenylendiamine with methanol afforded the corresponding diformamides in 93% and 92% yields, respectively (compounds 29 and 30). The two important diformamide intermediates for the synthesis of isocyanate, methylene diphenyl diisocyanate and toluene diisocyanate, were also obtained in excellent yields (compounds 31 and 32). Sulfur-containing diamines that are widely used as monomers in polymer chemistry were converted into the corresponding formamides in excellent yields as well (compounds 33–35). These results indicate that the inorganic ligand-supported Cr catalyst has highly specific selectivity and activity towards oxidative formylation of primary diamines. To further demonstrate the practicality of this catalytic system, a 50 mmol reaction of 4-methylbenzene-1,3-diamine was conducted to give 7.298 g of N,N′-(4-methyl-1,3-phenylene)diformamide in 82% isolated yield.

Oxidation coupling of diamines with methanol catalyzed by 1a. Reaction conditions: Cat. 1a (0.1 mol%), methanol (1.5 mL), diamines (1.0 mmol) and 30% H2O2 (4.0 equiv.) at 80 °C. Yields were determined by 1H-NMR, values in parentheses are the isolated yields
After the reaction finished, the solid Cr catalyst 1a was easily isolated by filtration and used directly for the subsequent coupling without further purification. 1a could be recovered and reused at least six times with little loss of the activity (Supplementary Fig. 4). To confirm the high stability of the catalyst and its associated performance, the structure and morphology of the catalyst were investigated. As confirmed by FT-IR and XRD, the recycled catalyst remains almost unchanged from its original state (Supplementary Figs. 5 and 6).
Mechanistic considerations
To gain insight into the mechanism, 1H nuclear magnetic resonance (NMR) study was performed to trace the progress of oxidative coupling of benzylamine with methanol every 10 min (Fig. 5a). To our delight, the production of formic acid was detected in the first 20 min, which disappeared soon in the next 20 mins. The observation of formic acid indicates that it is probably an intermediate serving as the formylation reagent. To further confirm the key role of formic acid, control experiments were conducted by use of various formylating reagents (Fig. 5b). Reaction of benzylamine with paraformaldehyde gave no N-benzylformamide under the same conditions as methanol (Fig. 5b, (1)). On the contrary, formylation with formic acid yielded N-benzylformamide in high yield (Fig. 5b, (2)). In the absence of catalyst 1a, no reaction occurred with formic acid (Fig. 5b, (3)).

Mechanism study of the Cr-catalyzed formylation of amine with methanol. a 1H-NMR study of the reaction mixture at different time points. b Control experiments using different formylating reagents: formaldehyde (1), formic acid (2), and formic acid without catalyst (3). c Plausible reaction mechanism
Based on the observation of the key intermediate and control experiments, a tentative mechanism for the oxidation coupling of methanol and amines has been proposed (Fig. 5c). Methanol is firstly oxidized by hydrogen peroxide in the presence of Cr catalyst to give formaldehyde. Then, two pathways would be possible for the conversion of formaldehyde to formamide: one is through the formation of formic acid and the other is through the production of hemiaminal. Since formic acid is clearly observed by 1H-NMR, the pathway via formic acid would be the dominated one, but the other route can not be ruled out.
Discussion
In conclusion, we have demonstrated highly efficient N-formylation of amines with methanol catalyzed by an inorganic ligand-supported chromium (III) catalyst, which is potentially nontoxic, green and may be safer than the widely used high-valence chromium catalysts including CrO3 and K2Cr2O7. A variety of primary and secondary amines have been transformed to N-formamides in good to excellent yields and high selectivity under mild conditions. In particular, the formylation of primary diamines has been achieved for the first time, to our knowledge. Furthermore, this chromium catalyst can be used for large scale preparation of diformamide and recycled for at least six times with little loss of the activity. The generality of this methodology gives it the potential to be used on an industrial scale.
Methods
General procedure A
Catalyst 1a (0.1 mol%), an amine (1.0 mmol), 30% H2O2 (4 equiv.) and Na2SO3 (0.05 equiv.) were added into 1.5 mL CH3OH with stirring at 80 °C for 40 min in pressure tube. Afterward, a small amount of ethyl acetate was added into the reaction mixture and the solution was quickly filtered. The filtered solid was washed, dried and then recycled. Reaction mixture was analyzed by gas chromatography–mass spectrometry (GC–MS) analysis. Finally, the solvent was removed in vacuo, and the corresponding formamideds was purified by washing through base-washed silica gel column (petroleum ether:ethyl acetate = 2:1).
General procedure B
Catalyst 1a (0.1 mol%), secondary amine (1.0 mmol), 30% H2O2 (4 equiv.) and Na2SO3 (0.05 equiv.) were added into 1.5 mL CH3OH with stirring at 80 °C for 120 min in pressure tube. Afterwards, a small amount of ethyl acetate was added into the reaction mixture and the solution was quickly filtered. The filtered solid was washed, dried and then recycled. Reaction mixture was analyzed by GC–MS analysis. Finally, the solvent was removed in vacuo, and the corresponding formamideds was purified by washing through base-washed silica gel column. (Petroleum ether: Ethyl acetate = 2: 1)
General procedure C
Catalyst 1a (0.1 mol%), diamine (1.0 mmol), 30% H2O2 (4 equiv.), and Na2SO3 (0.05 equiv.) were added into 1.5 mL CH3OH with stirring at 80 °C for 12 h in pressure tube. Afterward, a small amount of ethyl acetate was added into the reaction mixture and the solution was quickly filtered. The filtered solid was washed, dried and then recycled. Reaction mixture was analyzed by 1H-NMR analysis. Finally, the solvent was removed in vacuo, and the corresponding formamideds was purified by washing through base-washed silica gel column (petroleum ether:ethyl acetate = 2:1)
Synthesis and characterization
Fully synthetic procedures are available in the Supplementary Methods. The structure of 1a is available in Supplementary Figures 1–3. Comparision of the fresh and sixth cycling catalyst is available in Supplementary Figures 4–6. NMR spectra of purified organic compounds are available in Supplementary Figures 7–41.
Screening conditions
Futher details of reaction conditions screened are available in Supplementary Tables 1–3.
Data availability
The authors declare that all data supporting the findings of this study are available within in the article and Supplementary Information files,and are also available from the corresponding author upon reasonable request.
References
Bipp, H. & Kieczka, H. Formamides. Ullmann’s Encyclopedia of Industrial Chemistry. (Wiley-VCH, Weinheim, Germany, 2003).
Valeur, E. & Bradley, M. Amide bond formation: beyond the myth of coupling reagents. Chem. Soc. Rev. 38, (606–631 (2009).
Weissermel, K. & Arpe, H.-J. Industrial Organic Chemistry. (Wiley-VCH: Weinheim, Germany, 1997) 13–58.
Wuts, P. G. M. & Greene, T. W. Protective Groups in Organic Synthesis. (Wiley, Hoboken, 2014).
Downie, I. M., Earle, M. J., Heaney, H. & Shuhaibar, K. F. Vilsmeier formylation and glyoxylation reactions of nucleophilic aromatic compounds using pyrophosphoryl chloride. Tetrahedron 49, 4015–4034 (1993).
Jones, S. & Warner, C. J. A. Trichlorosilane mediated asymmetric reductions of the C = N bond. Org. Biomol. Chem. 10, 2189–2200 (2012).
Jagtap, S. B. & Tsogoeva, S. B. First enantioselective organocatalytic allylation of simple aldimines with allyltrichlorosilane. Chem. Commun. 38, 4747–4749 (2006).
Han, Y. & Cai, L. An efficient and convenient synthesis of formamidines. Tetrahedron Lett. 31, 5423–5426 (1997).
Allen, C. L. & Williams, J. M. J. Metal-catalysed approaches to amide bond formation. Chem. Soc. Rev. 40, 3405–3415 (2011).
Gerack, C. J. & McElwee-White, L. Formylation of amines. Molecules 19, 7689–7713 (2014).
Olah, G. A., Ohannesian, L. & Arvanaghi, M. Formylating agents. Chem. Rev. 87, 671–686 (1987).
Mhoy, E. D. T., Evans, D., Rouden, J. & Blanchet, J. Formamide synthesis through borinic acid catalysed transamidation under mild conditions. Chem. Eur. J. 22, 5894–5898 (2016).
Ishida, T. & Haruta, M. N-formylation of amines via the aerobic oxidation of methanol over supported gold nanoparticles. ChemSusChem 2, 538–541 (2009).
Preedasuriyachai, P., Kitahara, H., Chavasiri, W. & Sakurai, H. N-formylation of amines catalyzed by nanogold under aerobic oxidation conditions with MeOH or formalin. Chem. Lett. 42, 1174–1176 (2010).
Phill, G., Jessop., Hisao, Y., Ikariya, T. & Noyori, R. Homogeneous catalysis in supercritical fluids: hydrogenation of supercritical carbon dioxide to formic acid, alkyl formates, and formamides. J. Am. Chem. Soc. 118, 344–355 (1996).
Kröcher, O., René, A., Köppel. & Baiker, A. Highly active ruthenium complexes with bidentate phosphine ligands for the solvent-free catalytic synthesis of N,N-dimethylformamide and methyl formate. Chem. Commun. 5, 453–454 (1997).
Daw, P. et al. Selective N-formylation of amines with H2 and CO2 catalyzed by cobalt pincer complexes. ACS Catal. 7, 453–454 (2017).
Zhang, L., Han, Z., Zhao, X., Wang, Z. & Ding, K. Highly efficient Ruthenium‐catalyzed N‐formylation of amines with H2 and CO2. Angew. Chem. Int. Ed. 54, 6186–6189 (2015).
DasNeves Gomes, C. et al. A diagonal approach to chemical recycling of carbon dioxide: organocatalytic transformation for the reductive functionalization of CO2. Angew. Chem. Int. Ed. 51, 187–190 (2012).
Federsel, C. et al. A well-defined iron catalyst for the reduction of bicarbonates and carbon dioxide to formates, alkyl formates, and formamides. Angew. Chem. Int. Ed. 49, 9777–9780 (2010).
Cifre, P. G. & Badr, O. Renewable hydrogen utilisation for the production of methanol. Energy Convers. Manag. 48, 519–527 (2007).
Ortega, N., Richter, C. & Glorius, F. N-formylation of amines by methanol activation. Org. Lett. 15, 1776–1779 (2013).
Tanaka, S. et al. Selective aerobic oxidation of methanol in the coexistence of amines by nanoporous gold catalysts: highly efficient synthesis of formamides. Chem. Eur. J. 19, 11832–11836 (2013).
Kang, B., Fu, Z. & Hong, S. H. Ruthenium-catalyzed redox-neutral and single-step amide synthesis from alcohol and nitrile with complete atom economy. J. Am. Chem. Soc. 32, 11704–11707 (2013).
Chakraborty, S. et al. Manganese-catalyzed N-formylation of amines by methanol liberating H2: a catalytic and mechanistic study. Angew. Chem. Int. Ed. 56, 4229–4233 (2017).
Moran, J., Preetz, A., Mesch, R. A. & Krische, M. J. Iridium-catalysed direct C–C coupling of methanol and allenes. Nat. Chem. 3, 287–290 (2011).
Chan, L. K. M., Poole, D. L., Shen, D., Healy, M. P. & Donohoe, T. J. Rhodium-catalyzed ketone methylation using methanol under mild conditions: formation of α-Branched products. Angew. Chem. Int. Ed. 53, 761–765 (2014).
Ogawa, S. & Obora, Y. Iridium-catalyzed selective α-methylation of ketones with methanol. Chem. Commun. 50, 2491–2493 (2014).
Morton, D. & Cole-Hamilton, D. J. Molecular hydrogen complexes in catalysis: highly efficient hydrogen production from alcoholic substrates catalysed by ruthenium complexes. J. Chem. Soc. Chem. Commun. 50, 1154–1156 (1988).
Shinoda, S. & Yamakawa, T. One-step formation of methyl acetate with methanol used as the sole source and catalysis by RuII–SnII cluster complexes. J. Chem. Soc. Chem. Commun. 21, 1511–1512 (1990).
Li, Y., Li, H., Junge, H. & Beller, M. Selective ruthenium-catalyzed methylation of 2-arylethanols using methanol as C1 feedstock. Chem. Commun. 50, 14991–14994 (2014).
Nielsen, M. et al. Low-temperature aqueous-phase methanol dehydrogenation to hydrogen and carbon dioxide. Nature 495, 85–89 (2013).
Monney, A. et al. Base-free hydrogen generation from methanol using a bi-catalytic system. Chem. Commun. 50, 707–709 (2014).
Rodriguez-Lugo, R. E. et al. A homogeneous transition metal complex for clean hydrogen production from methanol–water mixtures. Nat. Chem. 5, 342–347 (2013).
Hu, P., Diskin-Posner, Y., Ben-David, Y. & Milstein, D. Reusable homogeneous catalytic system for hydrogen production from methanol and water. ACS Catal. 4, 2649–2652 (2014).
Fujita, K., Kawahara, R., Aikawa, T. & Yamaguchi, R. Hydrogen production from a methanol–water solution catalyzed by an anionic iridium complex bearing a functional bipyridonate ligand under weakly basic conditions. Angew. Chem. Int. Ed. 54, 9057–9060 (2015).
Li, F., Lu, L. & Liu, P. Acceptorless dehydrogenative coupling of o-aminobenzamides with the activation of methanol as a C1 source for the construction of quinazolinones. Org. Lett. 18, 2580–2583 (2016).
Kothandaraman, J. et al. Efficient reversible hydrogen carrier system based on amine reforming of methanol. J. Am. Chem. Soc. 139, 2549–2552 (2017).
Pope, M. T. & Müller, A. Polyoxometalate chemistry: an old field with new dimensions in several disciplines. Angew. Chem. Int. Ed. 30, 34–48 (1991).
Rhule, J. T., Hill, C. L. & Judd, D. A. Polyoxometalates in medicine. Chem. Rev. 98, 327–358 (1998).
Müller, Kögerler,A. P. & Dress, A. W. M. Giant metal–oxide-based spheres and their topology: from pentagonal building blocks to keplerates and unusual spin systems. Coord. Chem. Rev. 222, 193–218 (2001).
Long, D. L., Tsunashima, R. & Cronin, L. Polyoxometalates: building blocks for functional nanoscale systems. Angew. Chem. Int. Ed. 49, 1736–1758 (2010).
Mizuno, N. & Kamata, K. Catalytic oxidation of hydrocarbons with hydrogen peroxide by vanadium-based polyoxometalates. Coord. Chem. Rev. 255, 2358–2370 (2011).
Kikukawa, Y., Yamaguchi, K. & Mizuno, N. Zinc(II) containing gamma-Keggin sandwich-type silicotungstate: synthesis in organic media and oxidation catalysis. Angew. Chem. Int. Ed. 49, 6096–6100 (2010).
Ben-Daniel, R., Alsters, P. & Neumann, R. Selective aerobic oxidation of alcohols with a combination of a polyoxometalate and nitroxyl radical as catalysts. J. Org. Chem. 66, 8650–8653 (2001).
Weinstock, I. A. et al. Equilibrating metal–oxide cluster ensembles for oxidation reactions using oxygen in water. Nature 414, 191–195 (2001).
Wang, S. S. & Yang, G. Y. Recent advances in polyoxometalate-catalyzed reactions. Chem. Rev. 115, 4893–4962 (2015).
Yu, H. et al. An efficient Iron (III)-catalyzed aerobic oxidation of aldehydes in water for the green preparation of carboxylic acids. Angew. Chem. Int. Ed. 56, 3867–3871 (2017).
Yu, H. et al. Transition-metal-controlled inorganic ligand-supported non-precious metal catalysts for the aerobic oxidation of amines to imines. Chem. Eur. J. 56, 13883–13887 (2017).
Anderson, J. S. Constitution of the poly-acids. Nature 3550, 850 (1937).
Evans, H. T. Jr. The crystal structures of ammonium and potassium molybdotellurates. J. Am. Chem. Soc. 70, 1291–1292 (1948).
Hasenknopf, B., Delmont, R., Herson, P. & Gouzerh, P. Anderson-type heteropolymolybdates containing tris (alkoxo) ligands: synthesis and structural characterization. Eur. J. Inorg. Chem. 2002, 1081–1087 (2002).
Wu, P. F. et al. Single-side organically functionalized Anderson-type polyoxometalates. Chem. Eur. J. 17, 12002–12005 (2011).
Kim, S. H. & Hong, S. H. Ruthenium-catalyzed urea synthesis using methanol as the C1 source. Org. Lett. 47, 212–215 (2016).
Zeng, H. & Guan, Z. Direct synthesis of polyamides via catalytic dehydrogenation of diols and diamines. J. Am. Chem. Soc. 133, 1159–1161 (2011).
Acknowledgments
We are grateful for the National Natural Science Foundation of China (Nos. 21871183, 21471087, 21631007, 21225103, and 21221062), Doctoral Fund of Ministry of Education of China No. 20130002110042, Tsinghua University Initiative Foundation Research Program No. 20131089204, Shanghai Natural Science Foundation of China (No. 18ZR1437900) and the State Key Laboratory of Natural and Biomimetic Drugs K20160202. The start-up fund of Shanghai Institute of Technology is also gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
H.Y. conceived the project idea; H.Y., S.H., and Y.W. designed the study; Z.K.W. and Z.Y.W. performed the experiments; H.Y., Z.K.W., and Z.Y.W developed methods, Y.Z., S.R., Q.Z., and J.W. analyzed and interpreted the data; H.Y. and Z.K.W. wrote the manuscript. Funding acquisition: H.Y., S.H., and Y.W.; supervision: H.Y., S.H., and Y.W. All the authors discussed the results and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, H., Wu, Z., Wei, Z. et al. N-formylation of amines using methanol as a potential formyl carrier by a reusable chromium catalyst. Commun Chem 2, 15 (2019). https://doi.org/10.1038/s42004-019-0109-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-019-0109-4
This article is cited by
-
Dimensional regulation in gigantic molybdenum blue wheels featuring {(W)Mo5} motifs for enhanced proton conductivity
Nano Research (2024)
-
Mechanistic insights into aerobic oxidative cleavage of glycol catalyzed by an Anderson-type polyoxometalate [IMo6O24]5−
Journal of Molecular Modeling (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
