
Abstract
Amides are prevalent in pharmaceuticals, agrochemicals, and materials. Hydroaminocarbonylation of alkenes has emerged as one of the most direct and rapid approaches to amides. Nowadays, these reactions remain largely confined to using alkylamines as nitrogen sources, leading to sluggish reactions due to their strong binding ability and basicity. Here we show the application of abundantly available hydroxylamine hydrochloride (NH2OH·HCl) as a surrogate of ammonia for the relay hydroaminocarbonylation of simple alkenes in the presence of a convenient palladium catalyst system. The in situ formation of alkylamines from hydroxylamine hydrochloride by hydroaminocarbonylation reaction and Lossen rearrangement allows us to establish an efficient relay hydroaminocarbonylation. Notably, this transformation allows the catalytic formation of two C–N bonds with hydroxylamine hydrochloride as an amine source by incorporation of two molecules of alkene and avoids the use of alkylamines.
Similar content being viewed by others
Introduction
Nitrogen atoms are ubiquitous in natural products, pharmaceuticals, polymers, and synthetic functional materials. Therefore, the development of novel catalytic reactions for the efficient construction of C–N bonds that take place with unactivated feedstocks under mild conditions while generating few byproducts has been a theme of modern synthesis1,2. Arguably, the direct synthesis of nitrogen-containing molecules with the inexpensive commodity chemical ammonia as a nitrogen source and simple alkenes as carbon sources represents a highly attractive approach. Unfortunately, many common reactions with simple alkenes as starting materials catalyzed by transition-metal complexes do not occur with ammonia. This observation is probably attributed to its easy formation of unreactive Lewis acid–base adducts with transition metals owing to its basicity, small size, and strong N–H bond3,4. An attractive and complementary approach would use ammonium salts as surrogates of ammonia gas for establishing transition-metal-catalyzed reactions. Some ammonium salts have been utilized as ammonia surrogates for transition metal-catalyzed C–N bond formation reactions5,6, yet catalytic reactions between simple alkenes and ammonium salts remain largely unexplored7.
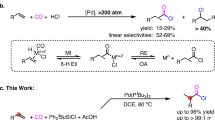
Amides are an important class of nitrogen-containing compounds that constitute the major body of bioactive natural products, pharmaceuticals, and the backbones of all natural peptides and proteins8. As such, the construction of amidic C–N bond is of utmost importance in synthetic organic chemistry, and has been the subject of tremendous research activity for many years9,10,11. Among the numerous strategies for the preparation of amides, transition metal-catalyzed hydroaminocarbonylation of alkenes has emerged as one of the most promising methods, enabling a rapid access to various amides from abundant and cost-effective feedstock materials12,13,14,15,16,17,18,19,20,21,22. At present, these catalytic hydrocarbonylative C–N bond formation reactions remain largely confined to the utilization of primary or secondary amines as amine sources (Fig. 1a). Unfortunately, the higher binding ability of these secondary and primary amines to transition metals coupled with their stronger basicity can inhibit the desired reaction to some extent12. One potential method to circumvent this problem is using ammonia or its surrogates as amine sources to in situ generate alkylamines (Fig. 1b).

Design relay hydroaminocarbonylation with hydroxylamine chloride. a Classical hydroaminocarbonylation with alkylamines. b Hydroaminocarbonylation via in situ generation of alkylamine. c This work: relay hydroaminocarbonylation with hydroxylamine hydrochloride
In this context, we have previously established a palladium-catalyzed hydroaminocarbonylation of alkenes to primary amides with simple NH4Cl as an ammonia surrogate16. In this process, the reaction was initiated via palladium hydride species to form alkylpalladium i, which underwent CO insertion to form acylpalladium species ii. Direct aminolysis of the resultant intermediate ii with NH4Cl in the presence of CO and NMP afforded the primary amides in excellent yields under very lower catalyst loading (Fig. 1c). Encouraged by this result, we sought to establish a catalytic sequential hydroaminocarbonylation and hydroamination reaction manifold to access secondary amides with NH4Cl as a nitrogen source, in which the alkylpalladium species i was thought to be intercepted by the newly formed primary amide to construct the second C–N bond. Unfortunately, many attempts to realize the aforementioned sequential reaction were failed, although the palladium-catalyzed hydroamination of alkenes with amides has long been well-established23,24,25,26,27,28,29,30. The underlying reason might be largely stemmed from the inherently lower nucleophilicity of the primary amide, as well as the lower concentration of the alkylpalladium species i that exists in the presence of CO. Thus, it is highly desirable to identify an alternative strategy that is capable of producing alkylamines from the simple alkenes and further able to transfer the alkylamines into secondary amides. However, to the best of our knowledge, there is currently no reported strategy that is capable of generation of primary amines via hydroamination of unactivated alkenes with simple ammonia in the presence of CO27.
To this end, we envisioned that a relay hydroaminocarbonylation reaction, in which alkylamine would originate from a simple alkene and hydroxylamine hydrochloride salt (NH2OH·HCl) via consequence hydroaminocarbonylation and Lossen rearrangement31,32,33,34, instead of from hydroamination of alkene with ammonia (Fig. 1c). Hydroxylamine hydrochloride salt is an attractive choice of abundant, cost-effective, and stable nitrogen source, which has been widely utilized as an important nucleophilic reagent in organic synthesis. On the basis of our programmatic focus on the development of Pd-catalyzed hydroaminocarbonylation reactions, we hypothesized that the hydroxylamine hydrochloride could be utilized as a nucleophilic amine source for hydroaminocarbonylation reaction and the tandem hydroaminocarbonylation/Lossen rearrangement sequence would yield the desired alkylamine intermediate. Specifically, aminolysis of acylpalladium intermediate ii with NH2OH·HCl would lead to the formation of hydroxamic acid iii, which underwent Lossen rearrangement to give the primary alkylamine iv by extrusion of CO2. The resultant primary alkylamine iv is nucleophilic in nature, which should be intercepted by the electrophilic acylpalladium intermediate ii to give the desired secondary amides. The key to this strategy is the discovery that the combination of hydroaminocarbonylation and Lossen rearrangement as key strategic steps is uniquely effective for transformation of the hydroxylamine hydrochloride salt to primary alkylamines. Such strategy would be considerably more practical and versatile, as it would not require alkylamines as amine sources, which can overcome the basicity barrier imparted by alkylamines.
Herein, we describe the design and development of Pd-catalyzed relay hydroaminocarbonylation of unactivated alkenes, hydroxylamine hydrochloride salt, and CO coupled with Lossen rearrangement. This study demonstrates that an alkylamine is not necessary for the success of the secondary amide synthesis via hydroaminocarbonylation.
Results
Catalytic system and solvent optimization
At the outset, we anticipated several key challenges associated with this transformation (Fig. 1c). First, it was unclear whether such a protocol could be implemented, as the Lossen arrangement has rarely been accomplished under the carbonylation reaction conditions. Second, HCl, a Brønsted acid will be released over the course of this transformation; thus, a suitable acid-binding reagent must be identified and must not be inhibited in the reaction to drive the reaction forward. Third, there were four potential regioisomers to be generated, thus, the catalyst must be highly selective. With these challenges in mind, we started our proposed catalytic protocol by evaluating the relay hydroaminocarbonylation of styrene 1a with NH2OH·HCl in the presence of CO.
In the presence of 2.5 mol% Pd(t-Bu3P)2, conducting the reaction in NMP under 20 atm of CO for 12 h failed to give any product (Table 1, entry 1). To our delight, when anisole was employed as the solvent, the desired transformation occurred, thus providing the branched–branched secondary amide 2a in 40% yield with a trace amount of linear-branched amide 4a (Table 1, entry 2). Other two potential regioisomers were not detected. These results suggested that the anisole played a key role, which may act as a weak base to capture the released HCl by formation of oxonium salts to drive the reaction forward35,36. Inspired by this promising result, the effect of palladium-catalyst was systematically investigated (Table 1, entries 2–9). Several commonly used palladium-catalysts, such as Pd(PPh3)4, Pd(Xantphos)Cl2, and Pd(PPh3)2Cl2, were firstly evaluated, however no good results were observed (Supplementary Table 1). Further screening of phosphine ligands with Pd(CH3CN)2Cl2 as a catalyst precursor disclosed that sterically congested monodentate phosphine ligands could deliver the desired amide with good regioselectivity, and among the monophosphines that have been evaluated (Supplementary Table 2), the RuPhos (L4) showed highest efficiency (71% yield) and was thus kept for the rest of the study. These results revealed that subtle modifications on the monophosphine ligand backbone led to profound changes on the reaction outcome (entries 6–9), suggesting that a reasonable steric bulk was critical for stabilizing the transient reaction species. Furthermore, subsequently experiments demonstrated that variation of the ratio of alkene/NH2OH·HCl from 1/2.4 to 2.4/1 resulted in higher reactivity and selectivity (71% vs. 89% yield). In line with our expectations, the nature of the solvent used for this reaction proved to be a crucial parameter (Supplementary Table 3). Reactions conducted in anisole gave the best yield and selectivity. Notably, excellent yield (96% isolated yield) and regioselectivity were achieved when the catalyst loading was increased to 5 mol% (entry 17, Supplementary Table 4). Control experiment revealed that a catalyst is necessary for the reaction to take place (entry 18).
Scope of the reaction
After identifying the optimal reaction conditions, we next shifted our focus to investigating the scope of this transformation (Fig. 2). Styrenes bearing different substituents underwent smooth carbonylation to furnish the corresponding regioisomerically pure amides. A variety of electronically disparate ortho-, meta-, and para-substituted styrenes were found to be suitable for this transformation, affording the corresponding branched–branched amide products in 32–98% yields (2a–2r). Notably, no other regioisomers were observed for all of alkenes tested here except 2-methylstyrene (the branched-linear product was isolated in 13% yield, see Supplementary Information (SI)). A good range of functional groups, such as methyl, alkyl, ether, halogen (F, Cl, Br), CF3, OCF3, CN and even thioether, were well tolerated, which provided a potential synthetic handle for further coupling reactions. It was noted that the electronic nature on the phenyl ring of the styrene had a strong influence on the reactivities. Styrenes bearing electron-donating groups proceeded smoothly to give the corresponding amides in excellent yields with complete regioselectivities. Compared with the results of the substrates with an electron-donating group, the presence of a strong electron-withdrawing group, such as CN (2j) and CF3 (2k) led to moderate yields (32–38% yields). The reactions of 1-vinylnaphthalene and 2-vinylnaphthalene delivered 2s and 2t in 54% and 85% yields, respectively. In addition, heteroaromatic 2-vinylthiophene also provided product 2u in 26% yield with good regioselectivity. To further test the applicability of this method for late-stage functionalization, we examined a family of estrone and testosterone substrates. Alkenes derived from these two compounds underwent the present reaction smoothly, leading to the corresponding amides 2w and 2y in good yields with excellent regioselectivities. These transformations indicated that the reaction system was amenable to functionalization and modification of complex alkenes bearing the skeleton of natural products, and showed high potential applications in biological evaluation. In addition, the reaction was applicable to internal alkenes, as exemplified by the reactions of 1H-indene (1v) and prop-1-en-1-ylbenzene (1x), which provided the corresponding products (2v and 2x) in moderate yields with excellent regioselectivities. Notably, although the diastereoisomer ratio in all cases is about 1:1, nearly all of the two diastereoisomers could be isolated by flash column chromatography (see Supplementary Methods). The structures of 2l and 4a were confirmed by X-ray single-crystal diffraction analysis (Supplementary Figs. 3 and 4, Supplementary Data 1 and 2).

Substrate scope of palladium-catalyzed relay hydroaminocarbonylation with aromatic alkenes. Reaction conditions: 1 (1.2 mmol), NH2OH·HCl (0.5 mmol), Pd(CH3CN)2Cl2 (5.0 mol%), L4 (11 mol%), CO (20 atm), anisole (1.0 mL), 120 °C, 12 h, isolated yield. Diastereoisomer ratio in all cases is about 1:1, which was determined by GC or 1H NMR analysis of crude reaction mixture. a140 °C, 24 h. b1 (2.0 mmol), 140 °C, 24 h
Encouraged by our success with aromatic alkenes, the reactions of unactivated aliphatic cyclic alkenes were then explored (Fig. 3). After extensively optimizing the reaction conditions (Supplementary Tables 5–10), we found that the catalytic system composed with [Pd(allyl)Cl]2 and Xantphos could catalyze the desired reaction with anisole as solvent, allowing for significant expansion of the substrate scope. Under the optimized reaction conditions, a broad range of cyclic alkenes, including cyclopentene (6a), cyclohexene (6b), cycloheptene (6c), and cis-cyclooctene (6d) were amenable to the desired reaction to give the corresponding amides (7a–7d) with moderate to good yields (entries 1–4, 42–74% yields). Other cyclic alkenes, such as norbornene and benzonorbornadiene, as well as its derivatives (6e, 6g and 6h) were also applicable to give secondary amides in 64–85% yields with good diastereoselectivity. Interestingly, the simple linear aliphatic alkenes and functionalized alkenes proceeded smoothly with slightly modified reaction conditions to afford a series of secondary amides in good yields as well (entries 6, and 9–11). It is worth mentioning that the steric hindrance of the alkenes strongly affects the regioselectivity, and the linear–linear amides were exclusively formed for the steric congested vinylcyclohexane and t-butyl ethylene. Finally, the bulk-scale ethylene was applicable to give the corresponding amides 8 in 54% yield with three molecules of alkene incorporated.

Substrate scope of palladium-catalyzed relay hydroaminocarbonylation with aliphatic alkenes. Reaction conditions: 6 (2.0 mmol), [Pd(allyl)Cl]2 (2.5 mol%), Xantphos (6.0 mol%), NH2OH·HCl (0.5 mmol), anisole (2.0 mL), CO (20 atm) at 140 °C for 24 h, isolated yield. a120 °C, b48 h. cEthylene (5 atm), NH2OH·HCl (5 mmol), [Pd(allyl)Cl]2 (0.5 mol%), Xantphos (1.2 mol%), anisole (3.0 mL), CO (20 atm) at 140 °C for 24 h. dThe dr ratio was determined by GC or 1H NMR analysis of crude reaction mixture. eThe isomer ratio of (linear–linear)/(linear–branched)/(branched–linear) was determined by GC or 1H NMR analysis of crude reaction mixture
Mechanism study
To gain insight into the reaction mechanism, we performed several experiments (Supplementary Figs. 6–18). To test whether the key palladium hydride species could be formed by the reaction of Pd(0) with acidic hydroxylamine hydrochloride. The reaction of Pd(t-Bu3P)2 with NH2OH·HCl was initially investigated and monitored by in situ 1H NMR and 31P NMR. After heating the mixture for 5 min, a new signal at −16.35 ppm appeared in the 1H NMR spectra (Fig. 4c, Supplementary Figure 14), which corresponded to the Pd–H resonance of HPd(t-Bu3P)2Cl species (Fig. 4a, Supplementary Figure 14)13. As prolonging the reaction time, the amount of Pd–H species increased accordingly (Fig. 4d, Supplementary Figure 14). At the same time, the 31P NMR analysis of the reaction mixture showed that a signal at 81.84 ppm grew up simultaneously (Fig. 4g–i, Supplementary Figure 15), which was assigned to the HPd(t-Bu3P)2Cl species (Fig. 4f, Supplementary Figure 15)13. In addition, the signal of Pd(t-Bu3P)2 in 31P NMR spectra is at 98.04 ppm (Fig. 4e, Supplementary Figure 15). Moreover, the standard reaction could be catalyzed by HPd(t-Bu3P)2Cl to afford the desired product with good yield (see Supplementary Methods). These results supported that the Pd–H species was indeed formed by the reaction of Pd(t-Bu3P)2 and NH2OH·HCl.

Monitoring the formation of Pd–H species. In situ NMR spectra (a-i) of Pd(t-Bu3P)2 with NH2OH·HCl in benzene-d6 and anisole supports the formation of HPd(t-Bu3P)2Cl
Analysis of gas phrase of reaction mixture of the standard reaction by using mass spectrometer demonstrated that carbon dioxide (CO2) was produced (Supplementary Figure 7), which revealed that the oxygen contained in NH2OH converted into CO2 and indicated that the Lossen rearrangement was most likely involved in the present reaction. Moreover, monitoring the standard reaction by ESI-MS disclosed that the hydroxamic acid 9 and α-methylbenzylamine 10 were produced when the reaction was stopped in 2 h (Supplementary Figure 9). However, the N-(1-phenylethyl)hydroxylamine, which could be potentially generated from the alkene and hydroxylamine via Cope-type hydroamination reaction37, could not be detected. These results supported the intermediates of hydroxamic acid and primary amine in the catalytic cycle.
It was reported that hydroxamic acids can be converted into secondary amides via Lossen rearrangement and transamidation along with extrusion of NH2OH38,39. Thus, we considered the possibility that the desired amide 2a might be produced from the above self-transformation of hydroxamic acids. However, only <10% conversion to 2a was observed when the hydroxamic acid 9 was heated at 120 °C in the presence of palladium catalyst or not (Fig. 5, Eqs. (1) and (2)), which indicated that self-propagative Lossen rearrangement of hydroxamic acids might be invoked in the transformation of hydroxamic acids into amines. In contrast, treatment of hydroxamic acid 9 with styrene 1a in the presence of CO and stoichiometric amount of Et2O·HCl under other identical reaction conditions affords the desired 2a in 85% yield (Eq. (3). These results support that the above self-transformation of hydroxamic acid 9 to 2a is not the major reaction pathway, but the aminolysis of acylpalladium species with primary amine 10 generated via Lossen rearrangement is likely the major pathway. Moreover, a crossover reaction between hydroxamic acid 9 and 4-methylstyrene 1b was carried out under the standard carbonylation conditions, and the products 2a and 2b were obtained together with the crossover products 11 and 12. This result together with the fact that relatively higher yields obtained for the 4-methylstyrene-derived amides (2b, 11 and 12) further supports our original mechanistic hypothesis.

Control experiments. Transformation of the proposed reaction intermediate
Although the detailed mechanism of the present reaction is not clear, we suggest the following catalytic cycle (Fig. 6) based on our preliminary observations and previous work on hydroaminocarbonylation of alkenes with NH4Cl15. Initially, the palladium-hydride species would be generated in situ from the oxidative addition of Pd(0) and NH2OH·HCl. Reversible coordination and insertion of alkene into the palladium-hydride yields a Pd-alkyl intermediate A, which undergoes CO insertion to form intermediate B. Aminolysis of B with NH2OH·HCl under the assistance of anisole leads to hydroxamic acid C along with palladium hydride species, in which the released HCl might be trapped by anisole. Finally, Lossen rearrangement of hydroxamic acid C takes place to give the primary amine D, which was then intercepted by acylpalladium complex B, leading to the desired amide 2 and regeneration of the palladium hydride species. Alternatively, the minor reaction pathway by transamidation of hydroxamic acid C with amine D to afford 2 might be also involved in the catalytic reaction.

Plausible reaction mechanism. The proposed mechanism involves a palladium-hydride species and a Lossen rearrangement sequence
Discussion
In summary, we have disclosed an approach to secondary amides via Pd-catalyzed relay hydroaminocarbonylation of simple alkenes with the simple and abundant NH2OH·HCl as an ammonia surrogate. In this process, the requisite alkylamine coupling partner for secondary amide preparation is produced in situ via hydroaminocarbonylation/Lossen rearrangement sequence, which enables two molecules of alkene to be incorporated into the functionalized amides. This approach allows for the facile and highly regioselective formation of C–N bonds from various aromatic alkenes, cyclic aliphatic alkenes, and NH2OH·HCl, where an hydroxylamine hydrochloride salt can now be thought of as a surrogate of ammonia in alkene-functionalization reactions. This study lays a foundation for the use of hydroxylamine chloride as versatile ammonia equivalent in transition metal-catalyzed reactions.
Methods
General procedure for the palladium-catalyzed relay hydroaminocarbonylation
In a glove box, NH2OH·HCl (35.0 mg, 0.5 mmol), Pd(CH3CN)2Cl2 (6.6 mg, 0.025 mmol), RuPhos (25.8 mg, 0.055 mmol), styrene (124.8 mg, 1.2 mmol), and anisole (1.0 mL) were added into a glass tube which was placed in an autoclave. The autoclave was taken out from the glove box and then the autoclave was purged and charged with CO (20 atm). The resulting reaction mixture was then stirred at 120 °C for 12 h. After the reaction finished, the autoclave was cooled to room temperature and the gas was carefully released in the fume hood. The diastereoisomer ratio of the secondary amide was determined by GC or 1H NMR analysis of the crude reaction mixture. Then the corresponding reaction mixture was purified by flash column chromatography on a silica gel column (petroleum ether/ethyl acetate = 100/1 – 1/1) to give the desired product 2a.
Synthesis and characterization
Full synthetic procedures and characterization for products are available in the Supplementary Methods. Full synthetic procedures for 2a and 7j are available in Supplementary Figs. 1 and 2. The ORTEP drawing of products 4a and 2l′ are available in Supplementary Figs. 3 and 4. Full procedures for the synthesis of 8 are available in Supplementary Figure 5.
Mechanistic study
Full procedures for the mechanism study are available in the Supplementary Methods and Supplementary Figs. 6–18.
Synthetic transformations
Full procedures for synthetic transformations of compounds 2a and 2n are available in the Supplementary Methods and Supplementary Figs. 19 and 20.
NMR spectra
1H, 13C, and 19F NMR spectra of purified compounds are available in Supplementary Figs. 21–87.
Crystallography
X-ray crystallographic CIF files for compounds 4a and 2l′ are available in Supplementary Data 1 and 2. X-ray-derived ORTEP representations of 4a and 2l′ are available in Supplementary Figs. 3 and 4.
Data availability
The X-ray crystallographic data for this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) with the accession code CCDC 1863093 (2l) and 1863095 (4a). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK; fax: (+44)1223-336-033; or deposit@ccdc.cam.ac.uk). The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files, and also are available from the corresponding author upon reasonable request.
References
Henkel, T., Brunne, R. M., Müller, H. & Reichel, F. Statistical investigation into the structural complementarity of natural products and synthetic compounds. Angew. Chem. Int. Ed. 38, 643–647 (1999).
Hili, R. & Yudin, A. K. Making carbon-nitrogen bonds in biological and chemical synthesis. Nat. Chem. Biol. 2, 284–287 (2006).
Roundhil., D. M. Transition metal and enzyme catalyzed reactions involving reactions with ammonia and amines. Chem. Rev. 92, 1–27 (1992).
Zhao, J., Goldman, A. S. & Hartwig, J. F. Oxidative addition of ammonia to form a stable monomeric amido hydride complex. Science 307, 1080–1082 (2005).
Kim, J. & Chang, S. Ammonium salts as an inexpensive and convenient nitrogen source in the Cu-catalyzed amination of aryl halides at room temperature. Chem. Commun. 0, 3052–3054 (2008).
Green, R. A. & Hartwig, J. F. Palladium-catalyzed amination of aryl chlorides and bromides with ammonium salts. Org. Lett. 16, 4388–4391 (2014).
Klinkenberg, J. L. & Hartwig, J. F. Catalytic organometallic reactions of ammonia. Angew. Chem. Int. Ed. 50, 86–95 (2011).
Crespo, L. et al. Peptide and amide bond-containing dendrimers. Chem. Rev. 105, 1663–1682 (2005).
Valeur, E. & Bradley, M. Amide bond formation: beyond the myth of coupling reagents. Chem. Soc. Rev. 38, 606–631 (2009).
Allen, C. L. & Williams, J. M. J. Metal-catalysed approaches to amide bond formation. Chem. Soc. Rev. 40, 3405–3415 (2011).
Figueiredo, R. M., Suppo, J. S. & Campagne, J. M. Nonclassical routes for amide bond formation. Chem. Rev. 116, 12029–12122 (2016).
Zhang, G., Gao, B. & Huang, H. Palladium-catalyzed hydroaminocarbonylation of alkenes with amines: a strategy to overcome the basicity barrier imparted by aliphatic amines. Angew. Chem. Int. Ed. 54, 7657–7661 (2015).
Hu, Y., Shen, Z. & Huang, H. Palladium-catalyzed intramolecular hydroaminocarbonylation to lactams: additive-free protocol initiated by palladium hydride. ACS Catal. 6, 6785–6789 (2016).
Hu, Y. & Huang, H. Highly selective construction of medium-sized lactams by palladium-catalyzed intramolecular hydroaminocarbonylation of aminoalkynes. Org. Lett. 19, 5070–5073 (2017).
Gao, B. & Huang, H. Palladium-catalyzed hydroaminocarbonylation of alkynes with tertiary amines via C–N bond cleavage. Org. Lett. 19, 6260–6263 (2017).
Gao, B., Zhang, G., Zhou, X. & Huang, H. Palladium-catalyzed regiodivergent hydroaminocarbonylation of alkenes to primary amides with ammonium chloride. Chem. Sci. 9, 380–386 (2018).
Fang, X., Li, H., Jackstell, R. & Beller, M. Selective palladium-catalyzed aminocarbonylation of 1,3-dienes: atom-efficient synthesis of β,γ-unsaturated amides. J. Am. Chem. Soc. 136, 16039–16043 (2014).
Fang, X., Jackstell, R. & Beller, M. Selective palladium-catalyzed aminocarbonylation of olefins with aromatic amines and nitroarenes. Angew. Chem. Int. Ed. 52, 14089–14093 (2013).
Li, H., Dong, K., Neumann, H. & Beller, M. Palladium-catalyzed hydroamidocarbonylation of olefins to imides. Angew. Chem. Int. Ed. 54, 10239–10243 (2015).
Liu, J. et al. Selective palladium-catalyzed aminocarbonylation of olefins to branched amides. Angew. Chem. Int. Ed. 55, 13544–13548 (2016).
Liu, H., Yan, N. & Dyson, P. J. Acid-free regioselective aminocarbonylation of alkenes. Chem. Commun. 50, 7848–7851 (2014).
Xu, T., Sha, F. & Alper, H. Highly ligand-controlled regioselective Pd-catalyzed aminocarbonylation of styrenes with aminophenols. J. Am. Chem. Soc. 138, 6629–6635 (2016).
Hartwig, J. F. Development of catalysts for the hydroamination of olefins. Pure Appl. Chem. 76, 507–516 (2004).
Beller, M., Seayad, J., Tillack, A. & Jiao, H. Catalytic Markovnikov and anti-Markovnikov functionalization of alkenes and alkynes: recent developments and trends. Angew. Chem. Int. Ed. 43, 3368–3398 (2004).
Hultzsch, K. C. Transition metal-catalyzed asymmetric hydroamination of alkenes (AHA). Adv. Synth. Catal. 347, 367–391 (2005).
Müller, T. E., Hultzsch, K. C., Yus, M., Foubelo, F. & Tada, M. Hydroamination: direct addition of amines to alkenes and alkynes. Chem. Rev. 108, 3795–3892 (2008).
Huang, L., Arndt, M., Gooßen, K., Heydt, H. & Gooßen, L. J. Late transition metal-catalyzed hydroamination and hydroamidation. Chem. Rev. 115, 2596–2697 (2015).
Michael, F. E. & Cochran, B. M. Room temperature palladium-catalyzed intramolecular hydroamination of unactivated alkenes. J. Am. Chem. Soc. 128, 4246–4247 (2006).
Cochran, B. M. & Michael, F. E. Mechanistic studies of a palladium-catalyzed intramolecular hydroamination of unactivated alkenes: protonolysis of a stable palladium alkyl complex is the turnover-limiting step. J. Am. Chem. Soc. 130, 2786–2792 (2008).
McGhee, A., Cochran, B. M., Stenmark, T. A. & Michael, F. E. Stereoselective synthesis of 2,5-disubstituted morpholines using a palladium-catalyzed hydroamination reaction. Chem. Commun. 49, 6800–6802 (2013).
Yale, H. L. The hydroxamic acid. Chem. Rev. 33, 209–256 (1943).
Bauer, L. & Exner, O. The chemistry of hydroxamic acids and N-hydroxyimides. Angew. Chem. Int. Ed. 13, 376–384 (1974).
Abdelhafez, E. M. N., Aly, O. M., Abuo-Rahma, G. E. A. A. & King, S. B. Lossen rearrangements under Heck reaction conditions. Adv. Synth. Catal. 356, 3456–3464 (2014).
Stafford, J. A., Gonzales, S. S., Barrett, D. G., Suh, E. M. & Feldman, P. L. Degradative rearrangements of N-(t-butyloxycarbonyl)-O-methanesulfonylhydroxamic acids: a novel, reagent-based alternative to the Lossen rearrangement. J. Org. Chem. 63, 10040–10044 (1998).
Maass, O. & McIntosh, D. The basic properties of oxygen compounds of the halogen acids with benzene derivatives containing oxygen. J. Am. Chem. Soc. 32, 70–71 (1910).
Burdham, R. A., Ludman, C. J., Lynch, R. J. & Waddington, T. C. The interaction of hydrogen chloride with ethers and other bases studied by 35C1 nuclear quadrupole resonance and 1H nuclear magnetic resonance. J. Magn. Reson. 34, 223–231 (1979).
Cooper, N. J. & Knight, D. W. The reverse Cope cyclisation: a classical reaction goes backwards. Tetrahedron 60, 243–269 (2004).
Allen, C. L., Atkinson, B. J. & Williams, J. M. J. Transamidation of primary amides with amines using hydroxylamine hydrochloride as an inorganic catalyst. Angew. Chem. Int. Ed. 51, 1383–1386 (2012).
Ohtsuka, N., Okuno, M., Hoshino, Y. & Honda, K. A base-mediated self-propagative Lossen rearrangement of hydroxamic acids for the efficient and facile synthesis of aromatic and aliphatic primary amines. Org. Biomol. Chem. 14, 9046–9054 (2016).
Acknowledgements
This research was supported by the National Natural Science Foundation of China (21790333, 21702197, and 21672199), the CAS Interdisciplinary Innovation Team, and the Anhui Provincial Natural Science Foundation (1708085MB28).
Author information
Authors and Affiliations
Contributions
H.H. designed the project and wrote the paper. J.L., S.W. and S.Z. carried out the experimental work. All authors analyzed the data and discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, J., Wang, S., Zou, S. et al. Palladium-catalyzed relay hydroaminocarbonylation of alkenes with hydroxylamine hydrochloride as an ammonia equivalent. Commun Chem 2, 14 (2019). https://doi.org/10.1038/s42004-019-0108-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-019-0108-5
This article is cited by
-
Palladium-catalyzed 3,4-hydroaminocarbonylation of conjugated dienes for formation of β,γ-unsaturated amides
Science China Chemistry (2023)
-
Palladium-catalyzed enantioselective carbonylation reactions
Science China Chemistry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
