
Abstract
Since their first discovery by Louis Dunoyer and Otto Stern, molecular beams have conquered research and technology. However, it has remained an outstanding challenge to isolate and photoionize beams of massive neutral polypeptides. Here we show that femtosecond desorption from a matrix-free sample in high vacuum can produce biomolecular beams at least 25 times more efficiently than nanosecond techniques. While it has also been difficult to photoionize large biomolecules, we find that tailored structures with an abundant exposure of tryptophan residues at their surface can be ionized by vacuum ultraviolet light. The combination of these desorption and ionization techniques allows us to observe molecular beams of neutral polypeptides with a mass exceeding 20,000 amu. They are composed of 50 amino acids – 25 tryptophan and 25 lysine residues – and 26 fluorinated alkyl chains. The tools presented here offer a basis for the preparation, control and detection of polypeptide beams.
Similar content being viewed by others
Introduction
Molecular beam science was started at the beginning of the last century1 to explore the foundations of physics in early tests of the kinetic theory of gases and in the first matter–wave diffraction of atoms and diatomic molecules2. Since then, free molecular beams have found diverse applications ranging from atomic clocks3 and high-resolution molecular spectroscopy4, over cluster deposition5 to nanoparticle metrology6,7 and modern tests of fundamental quantum optics8,9,10,11,12. Even functionalized silicon nanocrystals8 and silver-sulfur nanoparticles9 have been thermally transferred as neutral particles into high vacuum.
In recent years, unsolvated biomolecules have attracted interest13,14,15 since beam experiments allow one to extract intrinsic electronic or structural information of molecules free from any perturbing interactions with a local environment. While charged molecules have been successfully trapped in vacuum for spectroscopy16, photodissociation17,18, or photodetachment studies19, many electronic, magnetic, and optical properties are particularly interesting for neutral particle beams20. Beams of isolated molecules21 were also essential for the analysis of optical16 and electronic properties of oligopeptides22 and vitamins23.
Extending such experiments to large biopolymers and biomimetic particles requires novel methods for transferring them into the gas phase and for detecting them efficiently as well as mass-selectively. While some amino acids24, nucleotides25, and functionalized tripeptides26 can be sublimated or evaporated, large peptides and proteins typically decompose in thermal sources. Nanosecond laser desorption can reduce the heat load and an adiabatically expanding noble gas27 can cool and carry the molecules toward their subsequent analysis28. However, for more than 20 years, it has remained a challenge to desorb and photoionize biopolymers with masses exceeding 2,000 amu29,30.
Several arguments have been put forward to explain this observation: For instance, internal state relaxation competes with multi-photon ionization29,31. This can be reduced using femtosecond ionization32 but dissipation to non-ionizing states still often dominates in large biopolymers. Single-photon ionization (SPI) can avoid the intermediate levels and close such relaxation channels but it requires typically a photon energy >10 eV, which is beyond the reach of sufficiently intense table-top laser sources33,34. Tryptophan (Trp) is the only natural amino acid that can be ionized by a single photon of 7.89 eV as emitted by vacuum ultraviolet (VUV) F2 lasers.
Gramicidin D and a number of other antibiotic peptides22 owe their ionization properties to their Trp residues35. The role of Trp as an ionizable moiety has been shown for amino acid clusters up to Trp30, which can be abundantly detected using 157.6 nm light36. In contrast, photoionization of large covalently bound polypeptides has remained a challenge. A model for the reduced photoionization yield, based on the assumption of rapid electron recapture within the molecule, predicted an exponential decrease with particle mass31 and negligible efficiency for peptides beyond 2,000 amu, as observed in many experiments so far. We therefore hypothesized that ionization might still be possible if a high number of chromophores could be exposed to vacuum.
Here we show that polypeptides with alternating Trp residues and lysine residues that are amidated with fluoroalkyl chains can be efficiently laser desorbed and postionized. Femtosecond laser desorption enables >25 times higher material economy when compared to nanosecond methods. Polypeptides exceeding 20,000 amu were successfully desorbed and postionized.
Results
Peptide design
Based on these reasonings, we have designed and synthesized five model compounds (see Fig. 1) to build on the antibiotic polypeptide gramicidin D (1), which was among the most massive neutral biomolecules in previous beam experiments. Commercial gramicidin D is composed of 15 amino acids, and the most abundant A1 form (molecular weight 1882 amu) contains four Trp residues. Our first tailored model system, decatryptophan Trp10 (2), contains more than double the number of chromophores at a comparable mass. Since peptide solubility decreases with the number of Trp residues, Trp10 represents a practical upper bound to the chain length for a quantitative synthesis of such molecules. The second model compound combines good solubility with high Trp content by alternating Trp with lysine to Ac-(Lys-Trp)5-OH (3). This sequence of alternating amino acids is scalable to long peptides. Compound (4) displays the same (Trp-Lys)n motif as (3) for n = 10 and each NH2 has been amidated with a fluorinated alkyl chain. The total mass therefore exceeds that of gramicidin already by a factor of 4.5. The high fluorine content is intended to reduce intermolecular interactions and to enhance volatility.

Iterative design of high mass oligopeptides for molecular beam experiments. Gramicidin A1, as shown here (1) is the principal component of commercial gramicidin D. Decatryptophan (2) has the highest tryptophan-to-mass ratio of all peptides in our study. Alternating Trp with Lys as in (3) provides better solubility and synthetic scalability. The molecular mass and the volatility can be enhanced by capping the NH2 groups with CO(CH2)2(CF2)7CF3 groups as in (4)–(6). (5) extends (4) to n = 14, with a total of 29 amino acids. (6) extends (5) to n = 25, i.e., it contains 50 amino acids. It is functionalized to increase its molecular weight to 20,201 amu and to expose copious amounts of tryptophan to the vacuum
In order to maximize the molecular weight at constant Trp ratio, we synthesized the model polymers (5) and (6) with n = 14 and n = 25 blocks of (Lys-Trp)n. They were functionalized in the same way as (3). Peptide (6) holds 1,960 atoms in 50 amino acid building blocks with a total mass of 20,201 amu. It resembles a protein in macromolecular architecture and mass. The molecules were prepared by standard solid-phase peptide chemistry under Fmoc protection37. Fluorinated alkyl groups were introduced by amidation with the respective N-hydroxysuccinimide ester after cleavage from the resin38.
From short-pulse to ultrafast laser desorption of heavy polypeptides
The molecular beam apparatus is sketched in Fig. 2. Analyte powder is picked up by a felt wheel, which transfers a thin layer of molecules onto a counter-rotating glassy carbon wheel. This layer is desorbed by a pulsed focused laser beam39. Different desorption lasers were used to answer different questions. Model compounds (1)–(3) were desorbed by short-pulse infrared laser light (7 ns, 1064 nm, 4–6 × 107 W cm−2) to explore the role of the Trp content. A quantitative assessment of the importance of the laser pulse duration was realized using gramicidin D (1). The high-mass peptides (4) and (5) show the influence of fluoroalkyl functionalization. They were volatilized using ultraviolet nanosecond pulses (7 ns, 266 nm, 5–8 × 107 W cm−2). The most massive polypeptide (6) was desorbed using ultrafast, ultraviolet laser desorption (292 fs, 343 nm) with peak intensities around 5 × 1011 W cm−2.

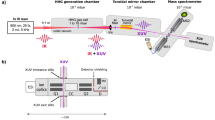
Molecular beam machine to launch and detect neutral polypeptides. Short or ultrashort laser pulses can be used to desorb the analyte molecules from a powder coated onto a rotating carbon wheel. The individualized polypeptides are entrained by an adiabatically expanding noble gas jet. The molecular beam enters the differentially pumped ionization chamber where a vacuum ultraviolet (VUV) laser pulse (ca. 1 mJ and 10 ns) ionizes it for subsequent analysis by a time-of-flight mass spectrometer (TOF-MS).
Once detached from the surface, the analyte molecules are immediately entrained by the atomic carrier gas (argon or neon) that is released by a valve with about 20 µs opening time. The neutral peptides fly 75 cm in high vacuum, across two differentially pumped vacuum chambers. They are then photoionized by a laser pulse at 157 nm to be extracted and detected by orthogonal time-of-flight mass spectrometry.
Influence of the Trp content and fluoroalkyl chains
Figure 3 compares a set of three tailored polypeptides with gramicidin D (1), which formed a molecular beam strong enough to saturate the detector after VUV postionization. Assuming that a high Trp content correlates with a high ionization cross-section, we have desorbed decatryptophan (2) but find additional fragmentation (For enlarged spectra and peak assignments, see Supplementary Figures 33–38). Substituting half of the Trp residues by lysine units enhances the solubility, facilitates the peptide synthesis and enables soft volatilization with reduced fragmentation, as shown for peptide (3). Compound (4) is built on this insight and raises the mass by more than a factor of five compared to (3), using fluoroalkyl functionalization. Even though peptide (4) exhibits more fragments than (3), it demonstrates that high-mass peptides can be tailored for neutral molecular beam experiments and be photoionized by VUV laser light.

Nanosecond desorption and VUV ionization of functional polypeptides (1)–(4). Gramicidin D (1) serves as a reference molecule for desorption/postionization experiments. Decatryptophan (2) has the highest tryptophan-to-mass ratio and lowest solubility in our series of unmodified peptides. Peptide (3) improves on that by substituting every second Trp by Lys. (4) is the first step on a new path toward neutral bio-inspired nanoparticle beams and toward a better understanding of the conditions for biopolymer ionization
We have verified the robustness of the mass spectra for various UV nanosecond desorption and ionization energies using compound (4) (8377 amu) and oligopeptide (P7, Supplementary Figures 41–43, Supplementary Table 2, Supplementary Notes 3–5) (12,320 amu). While we find a threshold behavior followed by saturation for the desorption process (Supplementary Figures 41 and 42 and Supplementary Table 2), the ion yield grows continuously with the ionization energy for the available range of laser pulse energies (Supplementary Figure 43).
On the importance of ultrafast desorption
At a pulse energy of up to 3 mJ, the Q-switched nanosecond laser beam was focused into a spot of 2 mm diameter and reached a peak intensity of about 107 W cm−2. This value is comparable to threshold values in matrix-assisted laser desorption ionization (MALDI) experiments40. However, several MALDI studies have shown that only a fraction of 10−4–10−7 of all desorbed material contributes to the final ion signal41. Similarly, in our experiments a substantial fraction of the material is released as nano- and microparticles, which visibly coat the vacuum chamber and are thus lost for the molecular beam.
This indicates an opportunity for substantial improvements and one is led to believe that ultrafast desorption might increase the desorption efficiency, since the absorbed heat will spread less before the pulse energy is transferred and molecules are ejected. This idea has caught on in recent mass spectrometric experiments that combined femtosecond laser desorption with electrospray postionization under atmospheric conditions42,43. Here we explore the benefits of ultrafast laser light on biomolecular layers in high vacuum, followed by rapid injection into an adiabatically expanding argon beam. Our ultrafast lasers cover pulse energies up to 80 µJ at 1030 nm and up to 40 µJ at 343 nm with a pulse duration down to 292 fs. We have characterized this idea using gramicidin D (1) because of its abundant commercial availability. The robustness of the qualitative form of the mass spectra was first tested for a wide range of desorption conditions, now including femtosecond light (Supplementary Table 2 and Supplementary Figure 40). Comparing then ultrafast desorption at 1030 and 343 nm for comparable pulse durations (292 fs) and energies (15 µJ, on the sample), we find the high UV photon energy (3.6 eV) to be essential for peptide desorption. The energy threshold at this wavelength amounts to about 10 µJ focused in a near-Gaussian waist of ca. 100 µm diameter, i.e., 100 mJ cm−2 or 3 × 1011 W cm−2 .
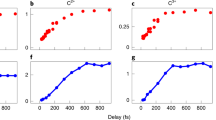
Most importantly, the femtosecond pulses improve the sample life time by a factor of >25 in comparison to nanosecond desorption. This is documented in Fig. 4a, where the molecular beam signal is traced as a function of time, while the laser is irradiating the same sample spot with a rate of 100 Hz. The hypothesis that ultrafast desorption favors individualized molecules over the formation of nanoparticles is also corroborated by the fact that the source chamber remains visibly clean.

Molecular beam strength and desorption efficiency as a function of laser pulse duration. a Desorption economy: Gramicidin D was desorbed from a thin film. We compare the longevity of a typical sample spot under ultrafast desorption (300 fs at 343 nm, 15 µJ red full circles and line) and short pulse desorption (7 ns at 355 nm, 3 mJ, blue full squares and line). For the same surface area of 100 × 100 µm2, femtosecond irradiation yields approximately the same initial molecular beam signal but at substantially longer sample life time. The solid lines are exponential fits to the data, with a decay constant of τns = 27 and τfs = 676 min. b Desorption laser pulse duration: When varying the pulse duration of infrared light (1030 nm, 40 µJ on 100 × 100 µm2) irradiating gramicidin D (see inset), we find a smooth increase in the number of detected molecules when the pulse duration is ramped through a factor of 25. The solid line is a square root fit to guide the eye
Figure 4b might suggest that, for the chosen parameters, a laser pulse duration of 10–20 ps already achieves this goal. Reducing the duration by another factor of 33, from 10 ps to 300 fs, the signal decreases by only about a factor of 2. This is consistent with the assumption that most particles depart already as isolated molecules. Reducing the laser pulse length at constant energy and thus increasing the peak intensity may foster multiphoton processes that reduce the molecular beam signal.
Improving the material economy by up to two orders of magnitude is a precious advantage in experiments with rare materials, but how would the tremendous laser intensity increase of ultrafast desorption affect the fragmentation pattern? Figure 5 illustrates that ultraviolet desorption can generate a neutral and photoionizable beam of the high-mass peptide (5) in both the nanosecond and the femtosecond mode. Comparable integrated signals and fragmentation patterns were obtained; however, at 100 times lower pulse energy, 200 times higher peak intensity and 2 orders of magnitude reduced material consumption in the femtosecond case.

Mass spectra of the polypeptide (5) in comparison between nanosecond and femtosecond desorption. Desorption conditions: short-pulse: λ = 355 nm, E = 3 mJ, τ = 7 ns (red line, foreground). Ultrashort pulse: λ = 343 nm, E = 15 µJ, τ = 292 fs (blue line, background). The different desorption conditions change the desorption efficiency but hardly influence the fragmentation pattern. For peak assignments, see Supplementary Figure 37
Further exploring the scalability of our new synthesis, desorption, and detection methods, Fig. 6 shows that even the most massive polypeptide in this series can be transferred into a beam of neutral molecules that can be postionized using 157 nm VUV light. This is noteworthy since compound (6) is about an order of magnitude more massive than other biopolymers whose photoionization has been reported in the literature so far.

A neutral molecular beam of massive functionalized polypeptides. A mixture of model peptides (5) and (6) was prepared in a ratio of 15 mg:140 mg and exposed to ultraviolet femtosecond desorption (λ = 343 nm, τ = 292, 15 µJ in 100 × 100 µm2). Even compound (6) stands out with good signal-to-noise ratio, proving the successful neutral launch and VUV postionization of this massive polypeptide. The fragments are assigned to the cleavage of Trp-Lys(fluoroalkyl) groups (as in Fig. 5) and are also found in the MALDI spectra of the same compounds (Supplementary Figures 24 and 28, see also Supplementary Figures 38 and 39)
To explore the role of the molecular structure and folding on the exposure of the Trp chromophores to vacuum, canonical-ensemble molecular dynamics simulations were performed for different temperatures. While even at low temperatures a plethora of stretched and coiled structures can stably coexist, we explore the extreme case, starting from the stretched state. While molecule (6) can initially extend over >7 nm, our numerical simulations show that it collapses within <100 ps into an energetically more favorable coiled-up tertiary structure upon heating, regardless of the initial state, which is kinetically trapped at low temperature. As a measure of compactness, the average radius of gyration 〈RG〉 was calculated as a function of temperature. Figure 7a traces the collapse toward an equilibrium end-to-end distance of about 2 nm beyond T > 350 K and 〈RG〉 ≃ 1.5 nm. One might worry that the close conformation might bury most chromophores and render photoionization difficult. To obtain an estimate for the exposure of all moieties to vacuum, we have calculated the solvent accessible surface area for the entire molecule as well as for its relevant components, i.e., the Trp and Lys residues as well as the fluoroalkyl chains. Figure 7b shows that the exposed surface area of Trp remains essentially constant over the 0-400 K temperature range and Trp residues are still abundantly exposed to vacuum even in the collapsed state. This can facilitate the escape of electrons after photoionization and distinguishes these Trp-rich polypeptides from many natural proteins (Computational details can be found in the Supplementary Note 2).

Structural parameters obtained with canonical-ensemble molecular dynamics simulations. a Temperature dependence of the radius of gyration 〈RG〉 of polypeptide (6). Even when starting from a stretched state, the molecule rapidly coils into a closed tertiary structure with increasing temperature. b The average solvent accessible surface area 〈SASA〉 quantifies the exposure of different parts of the molecule, here to vacuum. The total area (blue squares) decreases with increasing temperature, as expected but slowly increases beyond 300 K because of the increased exposure of the fluoroalkyl chains (green circles). The tryptophan residues (magenta diamonds) remain almost fully exposed at all temperatures, while the lysine surface (orange triangles) shrinks slightly. Error bars correspond to the standard deviation of the mean.
Polymer length and desorption/detection efficiency
While earlier papers had speculated that there might be an exponential decrease in the ionization efficiency with increasing mass of the biopolymer, the compounds of our present study were explicitly tailored to avoid this: the number of ionizable Trp chromophores increases in proportion with the polymer length and the simulations show that for all tested circumstances they are arranged abundantly on the molecular surface. While the absorption and ionization probability should thus remain constant with increasing polymer length, the dependence of the volatilization on chain length is less obvious. On the one hand, the equally increasing number of perfluoroalkyl chains reduces the potential stacking of aromatic rings and increase the mass/polarizability or mass/dipole moment ratios. This was already the idea and success of earlier high-mass molecular quantum optics experiments12. However, desorbing a given mass and volume of biopolymers must result in 10 times fewer clicks for compound (6) at about 20,000 amu compared to gramicidin at not even 2,000 amu. Finally, the secondary electron multiplication efficiency inside the time-of-flight mass spectrometer (TOF-MS) strongly depends on the velocity rather than the kinetic energy of the incident ions. While all molecules are accelerated by the same 16 kV voltage, they reach different velocities, whose influence is critical but not exactly determined for this particular molecular species. This makes it impossible to provide a clear functional dependence. However, we do not observe any substantial decay in signal/noise across the biopolymer mass range of 3,832–12,320 amu.
Discussion
Our study shows that a combined approach of synthetic design and ultrafast desorption enables the soft launch of neutral high-mass polypeptides up to the complexity of small proteins into neutral molecular beams and their successful soft photoionization. This could be achieved for bio-inspired nanoparticles composed of 50 amino acids and exceeding 20,000 amu, i.e., an order of magnitude higher than seen on native peptides. The functionalized polypeptide (6) is 350% more massive and contains 250% more atoms than insulin (51 residues, 5803 amu, 788 atoms).
Exploiting bio-mimetic growth principles, the tailored compounds can be synthesized with atomically defined composition and structure. It seems possible to ligate two or more of these chains to design bio-mimetic molecules with defined mass and atomic sequence to much higher masses, in future.
The peptide design focused on the abundant inclusion of Trp, which is the only natural amino acid compatible with single-photon VUV postionization using table top laser sources at 157 nm. The high Trp abundances also ensures that these chromophores will be exposed to vacuum, even in coiled-up peptides. This has been crucial for the successful in situ mass analysis of the initially neutral molecular beam. For the largest polypeptide in our series, ultrafast ultraviolet desorption was essential. And even for peptides up to 2,000 amu, fs-desorption was >25 times more material efficient than nanosecond methods. Ultrafast desorption therefore promises to also improve on mass spectrometry in combination with novel ionization methods. Intriguing progress on native proteins had been reported using electrospray postionization before43,44.
The high susceptibility of Trp-rich peptides to VUV SPI will also be of importance for advanced macromolecular matter–wave experiments where photo-depletion gratings45,46 provide novel beam splitters for complex biomolecular matter–waves. Polypeptides exceeding 20,000 amu are therefore interesting candidates for the next generation of matter–wave interference experiments, which can cope with de Broglie wavelengths as small as 50 fm.
Methods
Sample preparation and desorption
A homogeneous distribution of the molecules is established by first dissolving them and loading a felt wheel, which rubs against the counter-rotating glassy carbon wheel39. For the nanosecond desorption of compound (4), the sample was premixed with Trp in a ratio of 1:20. This procedure was not necessary for the femtosecond experiments. The Vienna TOF-MS was calibrated to the Swiss MALDI TOF results via a linear transformation using all sizeable peaks of the mass distribution of (5) and (6) up to 20,000 amu.
Four different laser types were compared in the desorption experiments (See Supplementary Table 2 for detailed conditions of every single experiment):
Short-pulse ultraviolet light, emitted by the fourth harmonic of the flash-lamp pumped Innolas Spitlight 400: wavelength λ = 266 nm, duration τd = 7±2 ns.
Short-pulse ultraviolet light, emitted by an EKSPLA “NT200 OPO” laser: λ = 355 nm, τd = 7±2 ns, E = 1.5 mJ.
Ultrafast infrared light, emitted by a TOPAG “Carbide” fs-fiber laser: λ = 1030 nm, τd = 292 fs–10 ps, Ed < 40 µJ on the sample
Ultrafast ultraviolet light, 343 nm, ultrafast pulse, emitted by a TOPAG “Carbide” fs-fiber laser: λ = 343 nm, E = 40 µJ.
Supersonic expansion and molecular beam velocity
A noble gas jet is released by an Even–Lavie valve47 with a backing pressure between 2 and 50 bar and a pulse duration of 20–22 µs. The experiments were performed using argon or neon with a purity of 99.999%. The gas and the polypeptides travel through a differentially pumped vacuum chamber, separated by a 3-mm skimmer, 55 cm behind the source and 20 cm from the ionization region. During operation at 100 Hz, the residual pressure is ca. 5 × 10−5 mbar in the source chamber and 5 × 10−7 mbar or 2 × 10−8 mbar in the detector chamber, for the nanosecond tests on (1)–(4) and the fs-tests on (5) and (6), respectively. For gramicidin D (1), we observe a mean velocity of 590 m s−1 with a full width at half maximum of 80 m s−1. The absence of any major velocity slip and the width of the distribution suggest that the number of collisions suffices to thermalize the translational energy spread of gramicidin to a temperature of 18 K. This is a lower limit to the expected rotational and vibrational temperature of this peptide.
Ion detection
The peptides were ionized by light from a fluorine laser (Coherent Excistar) with λ = 157 nm, τi = 10 ns, Ei = 1–2 mJ, A = 3× 3 mm2 and analyzed using an orthogonal TOF-MS (Kaesdorf München). Model compounds (1)–(4) were detected using an orthogonal TOF-MS Δm:m = 1:500 and baseline corrected. For (5)–(6), we used an orthogonal reflectron TOF-MS (Δm:m = 1:500). Both spectrometers accelerate the ions by 16 kV and count them by electron multiplication using a triple-stack of multi-channel plates. Figure 4: Every point was integrated 5 times over 1000 mass spectra. Figure 6: Each spectrum contained 106 points and was smoothed using a running average of 100 points (SI).
Synthetic procedures and characterization of compounds
See Supplementary Methods and Supplementary Note 1, Supplementary Table 1, Supplementary Figures 1–32.
Computational details
See Supplementary Note 2.
Laser desorption/photoionization mass spectra and conditions
See Supplementary Figures 33–39, 40, 42, 43 and Supplementary Notes 3–5, Supplementary Table 2.
Influence of the desorption laser wavelength and pulse length on the character of the mass spectrum
See Supplementary Note 3 and Supplementary Figure 40.
Influence of the desorption energy/fluence
See Supplementary Note 4 and Supplementary Figure 41.
Influence of the ionization energy/fluence on the normalized mass peak
See Supplementary Note 5 and Supplementary Figures 42 and 43.
Data availability
All relevant data are available from the authors upon reasonable request.
References
Stern, O. Zur Methode der Molekularstrahlen. I. Z. Phys. 39, 751–763 (1926).
Estermann, I. & Stern, O. Beugung von Molekularstrahlen. Z. Phys. 61, 95–125 (1930).
Rabi, I. I., Millman, S., Kusch, P. & Zacharias, J. R. The molecular beam resonance method for measuring nuclear magnetic moments. The magnetic moments of 3Li6, 3Li7and 9F19. Phys. Rev. 55, 526–535 (1939).
Bordé, C. J. et al. Observation of optical Ramsey fringes in the 10 µm spectral region using a supersonic beam of SF6. J. Phys. Coll. 42, C8-15–C8-18 (1981).
Barth, J. V., Costantini, G. & Kern, K. Engineering atomic and molecular nanostructures at surfaces. Nature 437, 671–679 (2005).
de Heer, W. A. The physics of simple metal clusters: experimental aspects and simple models. Rev. Mod. Phys. 65, 611–676 (1993).
Schmidt, M., Kusche, R., Issendorff, B. V. & Haberland, H. Irregular variations in the melting point of size-selected atomic clusters. Nature 393, 238–240 (1998).
Schöllkopf, W. & Toennies, J. P. Nondestructive mass selection of small Van der Waals clusters. Science 266, 1345–1348 (1994).
Chapman, M. S. et al. Optics and interferometry with Na2 molecules. Phys. Rev. Lett. 74, 4783–4786 (1995).
Arndt, M. et al. Wave-particle duality of C60 molecules. Nature 401, 680–682 (1999).
Akoury, D. et al. The simplest double slit: interference and entanglement in double photoionization of H2. Science 318, 949–952 (2007).
Hornberger, K., Gerlich, S., Haslinger, P., Nimmrichter, S. & Arndt, M. Colloquium: quantum interference of clusters and molecules. Rev. Mod. Phys. 84, 157–173 (2012).
Scherman, J.-P. Spectroscopy and Modelling of Biomolecular Building Blocks (Elsevier, Amsterdam, 2008).
Jarrold, M. F. Peptides and proteins in the vapor phase. Annu. Rev. Phys. Chem. 51, 179–207 (2000).
Meyer, T., Gabelica, V., Grubmüller, H. & Orozco, M. Proteins in the gas phase. Wires Comp. Mol. Sci. 3, 408–425 (2013).
Laure, J. et al. Ultraviolet spectroscopy of peptide and protein polyanions in vacuo: signature of the ionization state of tyrosine. J. Am. Chem. Soc. 129, 8428–8429 (2007).
Viglino, E., Shaffer, C. J. & Turecek, F. UV/Vis action spectroscopy and structures of tyrosine peptide cation radicals in the gas phase. Angew. Chem. Int. Ed. 55, 7469–7473 (2016).
Brodbelt, J. S. Photodissociation mass spectrometry: new tools for characterization of biological molecules. Chem. Soc. Rev. 43, 2757–2783 (2014).
Gabelica, V. et al. Electron photodetachment dissociation of DNA polyanions in a quadrupole ion trap mass spectrometer. Anal. Chem. 78, 6564–6572 (2006).
Antoine, R. et al. Electric susceptibility of unsolvated glycine-based peptides. J. Am. Chem. Soc. 124, 6737–6741 (2002).
Grotemeyer, J., Bösel, U., Walter, K. & Schlag, E. W. Biomolecules in the gas phase. 1. Multiphoton-ionization mass spectrometry of native chlorophylls. J. Am. Chem. Soc. 108, 4233–4234 (1986).
Antoine, R. et al. Application of molecular beam deflection time-of-flight mass spectrometry to peptide analysis. Anal. Chem. 75, 5512–5516 (2003).
Mairhofer, L. et al. Quantum-assisted metrology of neutral vitamins in the gas phase. Angew. Chem. Int. Ed. 56, 10947–10951 (2017).
Weinkauf, R., Schermann, J. P., de Vries, M. S. & Kleinermanns, K. Molecular physics of building blocks of life under isolated or defined conditions. Eur. Phys. J. D 20, 309–316 (2002).
Gabelica, V. Nucleic Acids in the Gas Phase (Springer, Heidelberg, New York, Dordrecht, London, 2014).
Schätti, J. et al. Tailoring the volatility and stability of oligopeptides. J. Mass Spectrom. 52, 550–556 (2017).
Meijer, G., de Vries, M. S., Hunziker, H. E. & Wendt, H. R. Laser desorption jet-cooling of organic molecules cooling characteristics and detection sensitivity. Appl. Phys. B 51, 395–403 (1990).
Grotemeyer, J., Boesl, U., Walter, K. & Schlag, E. W. Biomolecules in the gas phase. II. Multiphoton ionization mass spectrometry of angiotensin I. OMS Lett. 21, 595–597 (1986).
Schlag, E., Grotemeyer, J. & Levine, R. Do large molecules ionize? Chem. Phys. Lett. 190, 521–527 (1992).
Becker, C. H. & Wu, K. J. On the photoionization of large molecules. J. Am. Soc. Mass Spectrom. 6, 883–888 (1995).
Schlag, E. W. & Levine, R. D. Ionization, charge separation, charge recombination, and electron transfer in large systems. J. Phys. Chem. 96, 10608–10616 (1992).
Weinkauf, R., Aicher, P., Wesley, G., Grotemeyer, J. & Schlag, E. W. Femtosecond versus nanosecond multiphoton ionization and dissociation of large molecules. J. Phys. Chem. 98, 8381–8391 (1994).
Akhmetov, A., Moore, J. F., Gasper, G. L., Koin, P. J. & Hanley, L. Laser desorption postionization for imaging MS of biological material. J. Mass Spectrom. 45, 137–145 (2010).
Hanley, L. & Zimmermann, R. Light and molecular ions: the emergence of vacuum UV single-photon ionization in MS. Anal. Chem. 81, 4174–4182 (2009).
Hanley, L. et al. 7.87eV postionization of peptides containing tryptophan or derivatized with fluorescein. Appl. Surf. Sci. 252, 6723–6726 (2006).
Marksteiner, M. et al. Gas-phase formation of large neutral alkaline-earth metal tryptophan complexes. J. Am. Soc. Mass Spectrom. 19, 1021–1026 (2008).
Chan, W. C. & White, P. D. Fmoc Solid Phase Peptide Synthesis (Oxford University Press, Oxford, 2000).
Malik, L. et al. Perfluoroalkyl chains direct novel self-assembly of insulin. Langmuir 28, 593–603 (2011).
Gahlmann, A. P., Sang, T. & Zewail, A. H. Structure of isolated biomolecules by electron diffraction-laser desorption: uracil and guanine. J. Am. Chem. Soc. 131, 2806–2808 (2009).
Hillenkamp, F., Karas, M., Beavis, R. C. & Chait, B. T. Matrix-assisted laser desorption/ionization mass spectrometry of biopolymers. Anal. Chem. 63, 1193A–1203A (1991).
Knochenmuss, R. & Zenobi, R. MALDI ionization: the role of in-plume processes. Chem. Rev. 103, 441–452 (2003).
Brady, J. J., Judge, E. J. & Levis, R. J. Mass spectrometry of intact neutral macromolecules using intense non-resonant femtosecond laser vaporization with electrospray post-ionization. Rapid Commun. Mass Spectrom. 23, 3151–3157 (2009).
Brady, J. J., Judge, E. J. & Levis, R. J. Nonresonant femtosecond laser vaporization of aqueous protein preserves folded structure. Proc. Natl Acad. Sci. USA 108, 12217–12222 (2011).
Cui, Y. et al. Molecular imaging and depth profiling of biomaterials interfaces by femtosecond laser desorption postionization mass spectrometry. ACS Appl. Mater. Interfaces 5, 9269–9275 (2013).
Haslinger, P. et al. A universal matter-wave interferometer with optical ionization gratings in the time domain. Nat. Phys. 9, 144–148 (2013).
Dörre, N. et al. Photofragmentation beam splitters for matter-wave interferometry. Phys. Rev. Lett. 113, 233001 (2014).
Bahat, D., Cheshnovsky, O., Even, U., Lavie, N. & Magen, Y. Generation and detection of intense cluster beams. J. Phys. Chem. 91, 2460–2462 (1987).
Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant Nr. 320694), the Austrian Science Fund (FWF) within program W1210-N25, the Swiss National Science Foundation 200020-178808), and the Swiss Nanoscience Institute (P1403). The computational results were obtained using the Vienna Scientific Cluster (VSC); PROBIOTIQUS 70918. A.S. acknowledges funding by the (FWF) within the Lise-Meitner fellowship M 2364. G.G.R. is thankful for funding from the Brazilian National Council for Scientific and Technological Development (206729/2014-6). We gratefully acknowledge the loan of a Carbide femtosecond laser by TOPAG Germany and a Monaco fs-laser from Coherent Inc. Germany.
Author information
Authors and Affiliations
Contributions
The experiments were conceived by M.A., V.K., M.M., U.S. and A.S. The molecular design and synthesis was realized by J.S., V.K. and M.M. The nanosecond experiments were realized by U.S., P.R. and P.G. The femtosecond experiments were realized by P.R., G.R., and A.S. Data analysis was performed by J.S., U.S., G.R., P.R., A.S. and M.A. NMR experiments were carried out and analyzed by J.S. and D.H. Molecular dynamics simulations were designed and analyzed by A.S., G.G.R. and M.A. and realized by G.G.R. The manuscript was written by M.A., V.K. and J.S. with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schätti, J., Rieser, P., Sezer, U. et al. Pushing the mass limit for intact launch and photoionization of large neutral biopolymers. Commun Chem 1, 93 (2018). https://doi.org/10.1038/s42004-018-0095-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-018-0095-y
This article is cited by
-
Matter-wave interference of a native polypeptide
Nature Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
