
Abstract
The atmospheric evolution of organic compounds encompasses many thousands of compounds with varying volatility, polarity, and water solubility. The molecular-level chemical composition of this mixture plays a major, yet uncertain, role in its transformations and impacts. Here we perform a non-targeted molecular-level intercomparison of functionalized organic aerosol from three diverse field sites and a chamber. Despite similar bulk composition, we report large molecular-level variability between multi-hour organic aerosol samples at each site, with 66 ± 13% of functionalized compounds differing between consecutive samples. Single precursor environmental laboratory chamber experiments and fully chemically-explicit modeling confirm this variability is due to changes in emitted precursors, chemical age, and/or oxidation conditions. These molecular-level results demonstrate greater compositional variability than is typically observed in less-speciated measurements, such as bulk elemental composition, which tend to show less daily variability. These observations should inform future field and laboratory studies, including assessments of the effects of variability on aerosol properties and ultimately the development of strategic organic aerosol parameterizations for air quality and climate models.
Similar content being viewed by others

Introduction
Organic aerosol (OA) is a major component of ambient fine particulate matter (20–90%)1, which has significant detrimental health effects and climate forcings2,3. Primary OA (POA) is emitted directly from anthropogenic and biogenic sources, while secondary OA (SOA) is formed through the oxidation of reactive gas-phase precursors in the atmosphere (including volatile organic compounds (VOCs)). The oxidation of both gas- and particle-phase organic compounds occurs via a diverse set of oxidation pathways, depending on molecular structure and atmospheric conditions. These reactions and their divergent branching ratios convert an often complicated mixture of emitted reactants into an increasingly diverse mixture of oxidation products, which is broadly characterized as SOA or oxygenated OA1,4,5. Products span a wide polarity and volatility space, with diverse oxygen-, nitrogen-, and sulfur-containing functionality6.
Molecular identity and structure are tied to the atmospheric transformations of organic compounds, and to the ultimate health and environmental impacts of OA7,8,9. Thus, detailed molecular-level speciation is necessary to advance fundamental understanding of atmospheric processes and chemical life cycles, and to provide effective parameterizations for air quality-climate models and detailed chemical mechanisms. However, this is challenging because the number of potential oxidation products and isomers to catalog increases with molecular size and elemental constituents4.
Knowledge of OA chemical composition has advanced considerably in recent years through online and offline measurement studies, but major uncertainties remain at the molecular level. Chemically detailed online measurement capabilities have improved through applications of mass spectrometry (MS) (e.g., aerosol MS (AMS), chemical ionization MS (CIMS), ion mobility spectrometry–MS) and gas chromatography (GC) with MS detectors1,10,11,12. Some recent studies have combined data from multiple mass spectrometers to measure across wider volatility and polarity ranges11,13, even achieving mass closure at one forested site (i.e., Σ(individual measured species) ≈ total observed carbon mass)11.
However, the current state of knowledge from both online and offline measurements is not chemically-comprehensive. Most online methods have limited molecular-level speciation due to limited MS resolution and capability to differentiate isomers14,15. Many online OA instruments only characterize the bulk OA mixture, which reduces chemical detail. However, these bulk measurements are central to most OA source apportionment studies and atmospheric chemistry models. For example, AMS data on bulk elemental ratios (oxygen-to-carbon and hydrogen-to-carbon, O/C and H/C) from chambers and ambient measurements are often used to parameterize the overall composition and photochemical age of OA in models1,16,17. Offline characterization of OA filter samples with MS is common and sometimes includes gas or liquid chromatographic separation (GC or LC)18,19,20, but is often limited to targeted analyses of a smaller subset of compounds. Very few studies have looked at compositional variability via a non-targeted approach with a sufficiently powerful MS, and those that have were limited by sample size or did not include LC6,21,22.
In this study, we perform a non-targeted multi-platform survey of functionalized OA compositional variability via LC and high-resolution MS, with more temporal resolution and a larger sample size than previous offline studies. With the objective of advancing the state of knowledge on molecular-level OA composition, variability, and dynamics, we: (a) carry out a non-targeted molecular-level speciation of complex OA mixtures from three diverse field sites and a set of environmental laboratory chamber experiments, resolving isomers using LC and identifying the molecular formula of each observed LC peak using high-resolution MS; (b) perform an intercomparison of functionalized OA composition in chamber experiments and at each field site to examine sample-to-sample variability in each data set and explore potential causes of this molecular-level variability; (c) model the multi-generational evolution of molecular-level variability in OA composition using chemically-explicit mechanisms, and compare this to our other results. Results from all platforms are supported by a sensitivity analysis.
Results
Limited variability in bulk speciation of functionalized OA
Summertime OA measurements were made at three field sites: a remote forest in Northern Michigan (PROPHET site), near downtown Atlanta, and New York City (Queens Co.). Particulate matter (PM10) samples were collected on polytetrafluoroethylene (PTFE) membrane filters using a passivated stainless steel sampler with a minimal inlet surface area to reduce sampling artifacts and losses. Multi-hour samples were collected during the day and night (site-dependent, 8 h on average), avoiding dusk and dawn. OA was extracted from the filters with methanol, and characterized via LC with electrospray ionization (ESI) in positive and negative mode, with high-resolution quadrupole time-of-flight MS (LC-ESI-Q-TOF).
Data collection was extensively quality-controlled through the use of field and laboratory blanks, deuterated internal standards, and consistent sample handling and analytical procedures. Similarly, data processing was thoroughly quality controlled. We set a conservative threshold for peak inclusion in all analyses (limit of detection (LOD) threshold), and assigned each analyte a molecular formula using high mass accuracy molecular ion m/z ratios and isotopic distributions. We scrutinized peaks and assigned formulas with a series of rules and standards, and catalogued them based on their molecular formula and LC retention time. To ensure an accurate interpretation of results, data collection and processing methods were validated with replicate runs of standards and samples, and run-to-run variability of identified compounds was reduced to less than 8% (see Methods, Supplementary Note 1, Supplementary Figure 1, Supplementary Table 1). Unless specified, all data presented here are based on number of compounds (see Supplementary Note 2 for details on abundance-weighted results).
The focus of this study was functionalized OA; many of these functionalized compounds are readily protonated/deprotonated, or can interact with other ions to form adducts, making them well-suited for electrospray ionization23. This includes SOA and functionalized POA, though the majority of the compounds sampled are expected to be SOA, as these samples were all collected during the summer. Henceforth, all references to the OA characterized by LC-ESI-Q-TOF are labeled OA, but represent functionalized POA and SOA. The majority of compounds observed at each field site contain at least one oxygen atom (92%-94%), with minor contributions of nitrogen-containing compounds without oxygen, which ionize well via ESI23. Sulfur-containing compounds without oxygen and non-functionalized OA do not ionize well via ESI, and are thus omitted here23,24. Overall, the average sum of speciated components represents ~2.6 ± 1.3 μg/m3, ~0.5 ± 0.3 μg/m3, and ~1.0 ± 0.3 μg/m3, in the forest, Atlanta, and New York City, respectively (see Supplementary Note 1), excluding sulfur-containing compounds without oxygen and non-functionalized OA.
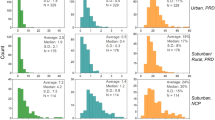
The bulk composition of OA filter samples at each site (i.e., mean ratios of oxygen-to-carbon (O/C), oxygen-to-nitrogen (O/N), and oxygen-to-sulfur (O/S)) shows limited variance across both daytime and nighttime samples (Fig. 1, Supplementary Figures 2, 3, Supplementary Tables 2–4), and is particularly consistent for the forested site. The compound classes observed are diverse and similar to previous high-resolution OA characterization6,18,21. Limited variance in average O/C was also observed via AMS in the forest and in Atlanta (Supplementary Figures 2–4). Such behavior is typical of many past AMS studies, where there is some variance in high-time resolution data, but diurnal profiles are consistent day-to-day, often falling within a narrow O/C range25,26. Approximately 50%-65% of observed compounds contain nitrogen or sulfur, including organonitrates and organosulfates, whose average molecular weights have been previously under constrained and are provided in Supplementary Figure 2. Compounds containing oxygen, nitrogen, and sulfur (CHONS), which have been detected previously in the ambient atmosphere27,28,29, were prevalent at the three sites (10% on average).

Bulk elemental composition shows limited variability. Functionalized OA composition averaged across all samples shows limited variability in overall elemental ratios and compound class distributions (a) at the forested site, (b) near downtown Atlanta, and (c) in New York City. In (a-c), elemental ratios (points on upper scales) are displayed as daytime (red) and nighttime (blue) values for LC-ESI-Q-TOF data, and shown along with AMS ratios (black). Error bars represent one standard deviation (LC-ESI-Q-TOF data) and 28% error (AMS data, as determined by Canagaratna et al. for improved-ambient analysis17). Values presented here are unweighted. O/N and O/S are computed for compounds with one or more N or S atoms. Bar charts show the fraction of compounds in each compound class, denoted by the elements present in each class (daily compound class distributions are shown in Supplementary Figures 10–11). Oxygen and nitrogen-containing organic compounds are split into two classes depending on O/N: CHON with O/N < 3, and CHON with O/N ≥ 3 (includes organic nitrates). Error bars represent one standard deviation from the mean. In Atlanta, the LC-ESI-Q-TOF O/C agrees with the AMS O/C. However, there is a larger discrepancy between these values in the forest, which decreases slightly in the abundance-weighted results (see Supplementary Note 2, Supplementary Figure 3)
Molecular-level speciation reveals extensive variability
Despite relatively constant overall composition, an isomer-specific molecular-level analysis shows significant sample-to-sample variability in chemical composition of OA at all three sites (Fig. 2, Supplementary Figures 5–9). With 1620–8100 compounds measured at each site, variability at the molecular-level was regularly 60–80%, meaning that 60–80% of compounds in a given sample were distinct and only 20–40% of compounds overlapped between any two compared samples. This reached a minimum of only 40% distinct compounds (60% overlapping) in two Atlanta comparisons. For each site, we compared subsets of similar samples (e.g., all daytime to all daytime) and all consecutive samples (e.g., day-to-day, day-to-night). Compounds were catalogued by both their molecular formula and LC retention time to differentiate isomers. Here, the definitions of “distinct” and “overlapping” are the percentages of observed compounds in a given sample that are either different or shared, respectively, in a comparison of two samples (see Methods, Equations 1 and 2). Distinct compounds are present above the LOD in one of the two compared samples, but absent or below the LOD in the other, and therefore contribute a negligible amount of mass to the second sample.

An omnipresent variability in molecular-level composition. The molecular-level composition of functionalized OA varies greatly between multi-hour samples at three diverse field sites, and this variability occurs across abundances, volatilities, and compound classes. a The average (±standard deviation, shown as error bars) percentage of distinct compounds for sample-to-sample comparisons is shown across various sample subsets at each field site, where overlapping compounds are determined by comparing molecular formulas and LC retention times. Markers above and below each average and standard deviation represent the minimum and maximum values of distinct compounds for each set of comparisons. All other sample-to-sample comparisons in each set fell in the range set by the minimum and maximum displayed here. “All consecutive samples” includes consecutive days, consecutive nights, consecutive day–night pairs, and consecutive night-next day pairs. Day- and night-only comparisons for all samples and consecutive samples are also shown. b–d Molecular-level variability in all sample comparisons across all three sites is shown as the distribution of compositional variability (i.e., distinct compounds) across (b) ion abundance ranges (divided into percentiles based on ion count, with corresponding individual analyte mass concentrations on the order of ng/m3), (c) volatility bins (i.e., intermediate/semi/low/extremely-low volatility organic compounds: IVOCs, SVOCs, LVOCs, ELVOCs, respectively), and d compound classes
On average, 73 ± 8%, 78 ± 13%, 70 ± 9% of compounds were distinct between any compared samples in the forest, Atlanta, and New York City, respectively. Large sample-to-sample variability was observed even with minimal changes in overall composition (Supplementary Note 3, Supplementary Figures 5–6, 10–11). Daytime and nighttime comparisons were equally variable. For all three sites, comparisons of consecutive samples had only slightly more overlap than comparisons of all samples, but were within standard deviations (Fig. 2), with consecutive sample-to-sample variability of 67 ± 9%, 65 ± 13%, 69 ± 7% in the forest, Atlanta, and New York City. Comparisons between daytime and nighttime samples are expected to vary somewhat, due to the distinctly different oxidation conditions in the presence or absence of sunlight. However, comparisons between consecutive daytime samples and consecutive nighttime samples also show significant variability, despite similar oxidation conditions from day-to-day and night-to-night. These highly variable results are notable in contrast to the consistent bulk OA composition across each field campaign (Fig. 1).
The sample-to-sample compositional variability we observe is due to differences in both detected molecular ions and isomers differentiated by LC. However, there are still prominent molecular formulas in our data sets spread across multiple isomers, including common markers of monoterpene, isoprene, and PAH oxidation (Supplementary Table 5). There are also compounds that appear relatively frequently at each site, but few compounds are above LOD thresholds in all samples (Supplementary Figure 12). Compounds were observed with up to eight isomers at the three field sites (Supplementary Figure 13). However, the majority of compounds at each site were present as only one isomer above LODs.
The observed compositional variability occurs across a distribution of abundances, volatilities, and compound classes (Fig. 2b–d, Supplementary Figures 15, 16). Though not displayed in Fig. 2, similar trends were observed for the chamber experiments. Distinct compounds from each sample-to-sample comparison are similarly spread across abundance quartiles, highlighting that this variability exists across a range of concentrations and is not attributed just to low abundance compounds. Furthermore, the distinct compounds contribute significantly to the total analyzed mass in the OA samples, representing 59 ± 17% of the abundance (i.e., mass) in the comparisons of all consecutive samples (e.g., Supplementary Figure 6b).
The volatility of each observed compound was computed according to a parameterization by Li et al.30, and compounds were then binned as intermediate-volatility (IVOC), semivolatile (SVOC), low-volatility (LVOC), and extremely low volatility (ELVOC) organic compounds. Distinct compounds span a wide volatility space, with similar contributions from IVOCs, SVOCs, ELVOCs, and fewer distinct LVOCs (Fig. 2c, Supplementary Figures 15–16). IVOCs represent 21% of observed variability on average (Supplementary Table 6); their presence in OA can be attributed to either: highly-polar water-soluble compounds often observed in OA (e.g., 2-methylglyceric acid (C4H8O5) from isoprene oxidation), misattribution without structural consideration in the above parameterization31, or fragments of larger species. Finally, distinct compounds occur across all compound classes (Fig. 2d, Supplementary Figures 15, 16), similar to the overall distributions in Fig. 1, and there is no statistically significant difference in the relative occurrence of compositional variability within each class (Supplementary Figure 17).
There are several considerations in the interpretation of these results. First, non-functionalized hydrocarbons (i.e., unoxidized precursor gases or unoxidized OA) are outside the scope of this study. However, the presence of fresh emissions could either homogenize OA if there are continuous sources, or exacerbate variability if there are intermittent diverse sources. The relative effect of any given intermittent source through non-functionalized hydrocarbons or their SOA depends on the complexity of emissions (e.g., motor vehicle exhaust)10.
Second, it is possible that longer duration samples (e.g., 24 h) with greater averaging times could reduce sample-to-sample variability by collapsing diurnal variations into a single aggregate sample. Here, sampling times vary depending on the site (4.5–10 h), but comparisons were always between samples of similar duration.
Third, the OA mass collected and instrument LOD affect the total number of compounds observed in a given sample. We observe an average of 553 ± 146 compounds per sample for the forest samples (mean ± standard deviation), 128 ± 41 compounds per sample for the Atlanta samples, 161 ± 66 compounds per sample for the New York City samples, and 240 ± 35 compounds per sample for the chamber samples. This is the result of strict quality control on all of the peaks detected and associated formulas produced (see Methods). We investigated the influence of LOD via a sensitivity analysis of ambient, chamber, and modeling results. Our sensitivity analysis (Fig. 3a and Supplementary Figure 18) demonstrates that the outcomes of this study are not prone to bias based on the LOD threshold, number of analytes detected, or sample mass collected. The number of compounds observed increases as the LOD threshold for inclusion is dropped. All LOD thresholds tested were above the instrument LOD (determined by a signal-to-noise ratio of three) in the sensitivity analysis. Improved instrument sensitivity (e.g., attogram-level) could enable the measurement of very low concentration compounds, which may reduce sample-to-sample variability in ambient data. However, this study demonstrates that variability occurs across a range of abundances (Fig. 2b), even in the most abundant OA components, which are most influential on OA properties and associated impacts.

Supplemental analyses to substantiate results. a A sensitivity analysis demonstrates minimal change in compositional variability results from the base case (100% of LOD threshold) with changes in LOD threshold for field, chamber, and modeling results. LOD thresholds were increased to 110%, 120%, 125%, and 190%, and decreased to 10%, 20%, 25% and 90%. The model threshold was set to a baseline of 0.5 ppq. The model threshold was varied as above, and was also reduced and increased by 1–3 orders of magnitude, resulting in less than 6.6% change in compositional variability. For each LOD threshold, all sample-to-sample comparisons were performed, and compositional variability values combined to yield an average change in compositional variability from the base case. The range denoted by the arrow on the right denotes the full extent of variability observed in the sensitivity analysis when varying all relevant threshold parameters together (a) and independently (Supplementary Figure 18). b The distributions of molecular mass differences between compounds modeled or measured at forested sites in this study, and in another published ultra-high-resolution (UHR) offline filter study, demonstrate small mass differences between many compounds that could be missed with typical online MS15. They are compared to published CIMS compound lists with demarcation of necessary mass differences for CIMS or other online MS to resolve peaks and assign formulas15. All measured or modeled data have been normalized to CIMS mass resolution (M/∆M = 4000) for comparison, but CIMS resolution limits do not apply to the others. Neither CIMS nor direct UHR-MS measurements employ LC separation prior to analysis. The MS resolution of each analyte from its nearest neighboring analyte is quantified as molecular mass difference/mass spectral peak half-width. The model data from this study shows the average of α- and β-pinene oxidation (individual precursor results were similar)
We acknowledge that the OA discussed here may not be representative of all particle-phase organic compounds at each of the sites we sampled, but rather represent a subset of compounds that are functionalized with oxygen, nitrogen, and/or sulfur. Filter extraction and analytical techniques were tailored based on best-practices in the literature with an emphasis on this functionalized fraction (see Methods). There is some uncertainty regarding the representativeness of the compositional variability we observe in this fraction of compounds compared to the total concentration of OA (especially when including POA), and results should be interpreted in this context. However, this highly functionalized fraction has been understudied, and warrants further characterization and assessment for its possible effects on the chemical/physical properties and impacts of SOA.
We only see a fraction of the 10,000’s of compounds estimated to be present in the atmosphere or the 1000’s anticipated based on GECKO-A results, but this is expected; many of these compounds are present at very small concentrations, below our conservative inclusion threshold. For example, though GECKO-A produces roughly 83,700 possible compounds for α- and β-pinene oxidation, most of these theoretical compounds exist at minimal concentrations (i.e., « ppq); for all model runs at the final simulation time, 2296 ± 2911 particle phase compounds are present above the 0.5 ppq LOD set for the model (discussed below), 88 ± 94 particle phase compounds are present above 1 ppt (which is on the order of our ambient measurement LOD), and none are present above 1 ppb. That said, there is no guarantee that all of these ~100 particle phase compounds theoretically above our ambient measurement LOD will form in a given ambient atmosphere due to variable chemical conditions, precursor/oxidant concentrations, or further particle-phase processing (see Methods).
The instrumentation used in this study is a key differentiator between this study and similar previous online work. A comparison of molecular ion mass differences between OA components in this study and other forested sites using both CIMS and offline ultra-high-resolution MS shows a large fraction of compounds that would not be resolved by online MS methods due to interferences from other analytes (Fig. 3b)15. This renders a significant fraction of the chemical diversity observed or modeled in this study inaccessible via current online MS measurements. In contrast, our offline LC-ESI-Q-TOF analysis has the advantage of isolating analyte peaks in LC space for accurate mass detection and formula assignment, without interference from other mass spectral peaks.
Exploring the potential driving factors of variability
To constrain the driving factors behind this observed functionalized OA variability, we examine the effects of diversity in precursor emissions, chemical oxidation conditions, and air parcel history, via a combination of environmental laboratory chamber experiments, chemically-explicit modeling, and backward trajectory analysis. Increasingly speciated measurements have advanced our understanding of molecular-level variability in primary emissions from both biogenic and anthropogenic sources. Gas- and particle-phase emissions are chemically-diverse across source types (e.g., trees versus motor vehicles), and differences also exist between source sub-types (e.g., orange versus pine trees). Composition of emissions can also vary temporally10,32,33.
To investigate molecular-level variability in oxidation products, a set of nighttime chamber experiments was performed using either α-pinene or β-pinene precursors. Precursor compounds were initially oxidized by NO3• radicals to form RO2• radicals, and then chemistry was varied between experiments, and either controlled by RO2• + NO3• or RO2• + HO2• pathways. We observe different product distributions between α-pinene and β-pinene experiments (84% distinct on average) in similar chemical conditions, while changing oxidation conditions with the same precursor produces more product overlap (60% distinct on average) (Table 1, Supplementary Figure 19)22. Isomers were observed in sets of up to four in the chamber experiments (Supplementary Figure 13).
Interestingly, the abundances of most overlapping compounds are similar between chamber experiments (Supplementary Note 4, Supplementary Figure 19). Compounds that are common to both experiments tend to fall near a 1:1 abundance line, suggesting the presence of shared prominent products across oxidation conditions for nighttime α- or β-pinene chemistry, and potentially other precursors or chemical conditions. This trend was not evident in the model chamber simulations, discussed below (Supplementary Figure 20). Several compounds observed in the chamber were also observed in the ambient forest and Atlanta samples (Supplementary Figure 23).
To supplement these chamber experiments, we modeled the gas-phase product distribution of α- and β-pinene, and their initial particle-phase composition after partitioning using GECKO-A (Table 1). Chemical structures were tracked between modeling experiments. With an average of 2296 ± 2911 particle phase compounds (range: 142–6934) and 2024 ± 2525 gas-phase compounds (range: 154–6012) above a 0.5 ppq LOD threshold at each final model simulation time, the results demonstrate similar molecular-level variability as in the chamber and field data. It is important to note that 0.5 ppq is below our instrument LOD threshold for ambient and chamber analyses, but was selected as a threshold for the GECKO-A simulated data because it provided an opportunity to further extend the LOD sensitivity analysis beyond the range of our ambient/chamber measurements (Fig. 3a).
We tested nighttime (NO3•) oxidation with either RO2• + NO3• or RO2• + HO2• reaction pathways, similar to the chamber experiments (chamber simulations), as well as daytime OH•-initiated oxidation under ambient conditions (forest simulations). Model runs with RO2• + NO3• controlled reactivity showed a significantly more diverse set of oxidation products than the product distribution from RO2• + HO2• chemistry. The chamber and model results are in agreement that variations in precursor structure are more influential contributors to compositional variability than the oxidation conditions tested. However, this chamber and modeling study was limited to α- and β-pinene, and should not be extrapolated to other precursors or oxidation conditions without testing. Other precursor comparisons with greater difference in structure or formula are likely to cause greater variability in OA composition. The inclusion of these modeling results acts as an independent verification of our ambient and chamber observations. The variability observed in the simulated data strongly supports the results by replicating the variability observed in ambient and chamber data.
Modeling results suggest that SOA compositional variability can be partially attributed to the preceding composition of oxidized organic gases, which, along with the initial particle-phase composition, provides a lower limit of variability just after partitioning but before additional particle- or aqueous-phase chemistry that could produce additional variability (such chemistry is excluded from GECKO-A). The LOD sensitivity analysis with modeling data also shows little sensitivity to changes in threshold (Fig. 3a, Supplementary Figure 18), down to LODs several orders of magnitude below the ambient or chamber LODs.
Time-resolved modeling results show that the evolution of compositional variability varies with chemistry and precursors (Fig. 4a–b), with variability increasing or decreasing over time for comparisons with like versus differing precursors, respectively. Additional intra-comparisons of individual model runs at fresh versus old “ages” (i.e., product generation) (Fig. 4c) demonstrates that air parcel age likely plays a role in accentuating compositional variability since reaction precursors are frequently not emitted and reacted at a uniform time in the ambient atmosphere. The precursor-dependent increase in potential compounds in chemically-explicit models has been shown previously to reach 105–106 over several generations, including diverse isomers5,13. The chemical diversity of a single precursor or simple mixture is expected to grow exponentially and peak with a diverse population of larger oxidized compounds, and ultimately converge on a smaller number of oxidized fragments at the end of the oxidation life cycle5,34. The extent to which this decrease in variability might occur in the ambient atmosphere is subject to the effects of aerosol deposition and emissions of fresh precursors.

Chemically-explicit model results show similar variability. GECKO-A modeling results with the dynamics of SOA compositional variability for α- or β-pinene daytime OH• oxidation (“OH”) or nighttime NO3• oxidation with RO2• reacting predominantly with HO2• or NO3• (“HO2”, “NO3”). a, b Model results show the evolution of compositional variability over reaction time, with variability increasing or decreasing over time for comparisons with like versus differing precursors, respectively. Oxidant exposure values for each modeling experiment are shown in Supplementary Figure 21, and the evolution of gas- and particle-phase mass and elemental ratios in the modeling experiments are shown in Supplementary Figure 22. c Intra-run comparisons of fresh (5 min simulation time) versus aged (last two simulation time points) compounds in the gas-phase GECKO-A output show compositional variability within single simulations. Bars represent maximum and minimum values for distinct compound fractions
To investigate the role of air parcel history, we modeled hourly backward trajectories covering all air parcels’ 24-hour history for each sample using HYSPLIT (Supplementary Note 5)35,36. For each site, we compared the statistical distribution of backward trajectories separately for all daytime and nighttime samples with 20 × 20 km2 resolution and observed a range of air parcel histories (Supplementary Figure 25). Despite some prevailing backward trajectories observed at each site, there is no observed correlation between dissimilar backward trajectories and chemical compositional variability (Supplementary Figure 26, and additional constraints on variability explored in Supplementary Figure 24). However, there is clustering around very dissimilar backward trajectories and very high OA compositional variability, meaning most sample-to-sample comparisons with significantly different backward trajectories do not share many compounds. This suggests that changes in air parcel backward trajectories may contribute to compositional variability, but do not control it.
Our observations are supported by evidence from other studies. Source apportionment via positive matrix factorization (PMF) with bulk OA measurements at the forested site shows major shifts in the relative contributions of three factors, representing more oxidized oxygenated OA, IEPOX-derived OA, and monoterpene-derived OA (Supplementary Note 3, Supplementary Figure 14). All three factors frequently approach negligible prevalence over long time periods, and at least two of the factors may also have significant variability in precursors or oxidation pathways. Similar shifts in the prevalence of PMF factors has been observed in other regions including the Southeastern U.S, though interpretation has been limited without chemical speciation37,38. In addition, our molecular-level compositional variability results (Fig. 2) are supported by two recent analogous studies using high-resolution mass spectrometry without LC separation. First, laboratory experiments on aqueous photochemistry of SOA from terpene precursors found that >50% of all observed oxidation products were unique to a single precursor22. Second, a comparison of four ambient OA samples from Los Angeles and Bakersfield, CA report 35–65% compositional variability between sites, and 35–91% when compared to chamber experiments with diesel fuel or isoprene21. These values are all lower limits of variability, since no LC separation was employed in either of these two studies to distinguish between isomers.
Discussion
In this study, we present a multiplatform analysis of compositional variability in the atmosphere, and show significant temporal variability in ambient, chamber, and modeled functionalized OA composition. Our observations highlight the complex interplay between precursor emissions, chemistry, and air parcel history in determining molecular-level functionalized OA composition, which is likely primarily composed of SOA. The atmosphere combines variability in precursor emissions with variability in the measured air parcels’ ages, and multiple levels of variability in multiphase chemical oxidation conditions (e.g., day versus night; concentrations of oxidants, NOX, and co-reactants (e.g., HNO3, H2SO4, NH3); concentration and composition of existing POA/SOA; particle acidity; relative humidity and aqueous chemistry;39 solar intensity; temperature). Additionally, differences in transport and meteorology can also affect dry and wet deposition timescales and OA lifetime. In all, the combination of these factors produces a large number of divergent chemical species, whose presence varies with changes in these underlying factors (Fig. 5). Shifts in one or more of these factors can significantly alter ambient molecular-level OA composition day-to-day, despite other factors, such as the presence of major precursors, remaining constant. The specific combination of factors driving SOA formation is constantly changing, and their combined effects on aerosol properties under constrained.

Potential causes and implications of variability. The compositional variability observed in this study is driven by a combination of several key factors, such as precursor identity, chemical oxidation conditions, and air parcel history. This compositional variability has potential implications for aerosol properties that are sensitive to chemical composition, and possible associated impacts on air quality, health, and climate
This and future molecular-level studies on aerosol composition should be used to strategically parameterize aerosol composition in models, or to inform strategic model simplifications to more efficiently yet accurately describe complex aerosol mixtures. Future research is required to expand on this study of aerosol variability and observe temporal trends in molecular-level aerosol composition at a variety of sites and throughout different seasons. Gas-phase composition should also be assessed to determine the link between variability in gas-phase precursors and resulting SOA. The variability of non-functionalized POA and its possible effect on compositional variability should be considered (see discussion above). Finally, the impacts on aerosol properties should be assessed, using molecular-level data to study mixture-wide aerosol viscosity, phase state, solubility, hygroscopicity, volatility, and other properties of interest (Fig. 5).
These results are important for fundamental understanding, monitoring and modeling practices, and air quality management policies for several main reasons. First, this molecular-level study reveals significant temporal variability in OA composition, in contrast with common bulk OA composition metrics which seemingly collapse variability in OA evolution within and between studies, and thus tend to oversimplify the underlying variability of complex OA mixtures. For example, tracking elemental ratios from our LC-ESI-Q-TOF analysis (e.g., O/C, O/N, O/S) shows minimal temporal variability (Fig. 1), while molecular-level data show major changes in chemical composition with time (Fig. 2). Molecular-level data should be used in combination with bulk metrics to evaluate, constrain, and improve the use of simplified metrics to represent aerosol composition, as well as age, evolution, and properties. SOA parameterizations in 3-D chemistry and climate models should be informed by these results and incorporate strategic simplifications that capture key changes in molecular-level composition. Existing bulk composition metrics include elemental ratios26, bulk carbon oxidation state34, mass spectra fragments (e.g., m/z 43/44)40, functional group fractions41, or often generic statistically-derived source apportionment factors (e.g., more oxidized oxygenated OA (MO-OOA), less oxidized oxygenated OA (LO-OOA))42. Non-targeted molecular-level speciation should be used to elucidate variations between seemingly similar bulk metric values within data sets, between studies, and in model evaluations. If such non-targeted data are unavailable, studies should avoid basing analyses on single representative days because they are uncertain to include typical OA composition for a given site.
Second, the molecular-level composition of OA (both elemental and structural) is a determining factor for its oxidation chemistry, its chemical/physical properties, the multiphase evolution of the whole organic mixture, and its interactions with aerosol inorganic species and water. The observed variability in aerosol composition may imply changes in oxidation chemistry and resulting aerosol properties, though this must be the subject of future research. Therefore, to efficiently develop comprehensive air quality and climate management strategies, it is essential that we understand the underlying processes that control this variability and thus influence the impacts of the coupled emissions-chemistry system. OA compositional variability could modify (and signify changes in) its impacts, such as urban air quality (i.e., SOA formation/evolution)1,43, human health (e.g., structurally dependent toxicity, carcinogenicity, and in vivo reactive oxygen species (ROS) generation)3,44, climate (e.g., changes in aerosol phase state, cloud condensation nuclei43), and feedbacks to chemical composition evolution through changes in aerosol phase state (e.g., glassy versus aqueous layer) that will affect chemistry in all phases.
For example, a recent study with α- and β-pinene showed that precursor structure caused greater variations in in vivo ROS generation than reaction conditions44. Similarly, molecular-level characterization is valuable because it can enable detailed estimates of physical/chemical properties like aerosol phase state, viscosity, solubility, hygroscopicity, and volatility, and allow for assessments of their ambient variability43,45. Future work should determine which chemical/physical properties are most prone to changes with compositional variability, and how observed compositional changes at different locations will affect SOA properties. For example, while there is some correlation between hygroscopicity and average O/C42,46, O/C distribution is also important in determining volatility42,47. Additionally, metrics like O/C, which do not reflect changes in the isomeric composition of a mixture, may be misleading in characterizing an aerosol mixture because overall O/C may remain fairly consistent with time, and therefore seemingly indicate consistent chemical composition. However, when chemical structure is taken into account, several structure-dependent properties may begin to show significant variability. For example, oxidation products in our GECKO-A analysis with the formula C10H16O3 (e.g., pinonic acid) span 1.8 orders of magnitude in saturation mass concentration, C* (i.e., volatility). Similarly, aerosol solubility will be sensitive to molecular structure45, and viscosity and phase state will be sensitive to molecular-level composition43. For these and other properties, understanding molecular-level variability will further advance understanding and improve our predictive capacity.
Third, these data-rich molecular-level characterizations of compositional variability represent an incredible statistical resource and opportunity to develop a fundamental understanding of the chemical evolution of complex OA mixtures and its driving factors. A broad survey and intercomparison of ambient sites and laboratory chamber experiments is needed to provide data to enable computationally-intense source apportionment and factor analysis studies with machine learning, neural networks, and other statistical methods. These offline methods and results are not intended to supplant, but rather augment, the valuable data produced by online methods and bulk characterization. Together with powerful, high-time resolution instruments (i.e., AMS, CIMS), future molecular-level compositional variability studies with greater temporal resolution and structural characterization of analytes with tandem MS can further deconvolve the factors that drive SOA formation and transformations.
Methods
Overview
The methods combined in this study each have strong foundations discussed in numerous previous studies, including filter handling and extraction, LC-ESI-Q-TOF analysis of OA extracts including QC/QA for formula assignment and formula evaluation, and non-targeted approaches to chemical characterization (all references herein).
Site descriptions
Ambient particle-phase samples were collected at three field sites during the summer to capture OA chemical complexity with increased levels of atmospheric oxidation. The first set of samples was collected in a remote forest at the PROPHET research tower (University of Michigan Biological Station, 45.33°N, 84.42°W) during PROPHET 2016 (Program for Research on Oxidants: PHotochemistry, Emissions and Transport). The campaign ran from 01 July 2016 to 31 July 2016, and samples were collected from 11 July 2016 to 31 July 2016. This remote, mixed forest contains a variety of coniferous and deciduous trees, including types of aspen, northern hardwood, and pine48. It is well isolated from major urban areas; Pellston, MI is the nearest town (5.5 km from the PROPHET tower, population < 900), and major metropolitan areas such as Detroit, Milwaukee, and Toronto are more than 400 km away. This site was selected because of its prominent biogenic emissions with minimal anthropogenic influence, allowing a focused study of biogenic volatile organic (BVOC) oxidation products. This was a collaborative field campaign, with a variety of other measurements collected, including online particle-phase characterization data via AMS that are used in this study.
Samples were also collected near downtown Atlanta (33.78°N, 84.41°W), at the Jefferson Street SEARCH site (Southeastern Aerosol Research and Characterization). These measurements were collected from 26 July 2017 to 18 August 2017. The Atlanta site was selected because of its mix of anthropogenic and biogenic precursors and conditions; there is significant anthropogenic influence expected with high NOx concentrations and other anthropogenic pollutants (e.g., SO2), as well as high concentrations of biogenic organic compounds such as isoprene and monoterpenes, observed during similar field campaigns in the Southeast US49. Using the established SEARCH site infrastructure, these measurements were once again part of a collaborative mission, and we use online particle-phase measurements collected via AMS in this study.
Finally, samples were collected in Queens, New York, at the New York State Department of Environmental Conservation Air Monitoring Station (40.74°N, 73.83°W). Measurements were collected from 01 September 2017 to 21 September 2017. This site is located in a densely populated area, and anthropogenic emissions are expected to be dominant. These measurements are supported by PM2.5 and other trace gas pollutant measurements made by the New York State Department of Environmental Conservation.
Sampling
Particle-phase samples were collected with a custom built passivated stainless steel sampler for simultaneous collection of offline gas- and particle-phase samples with a very low surface area inlet (minimal or no upstream tubing) to minimize sampling artifacts and losses. The sampler was designed using passivated stainless steel filter holders (Pall Corporation) and an 84 mesh stainless steel screen (McMaster Carr). The screen was installed over the opening of the filter holders to limit particle size to a PM10 size cut (mesh size determined by aerosol penetration efficiency at 1 m3/hour, accounting for losses to the screen by diffusion, impaction, and interception). Particle-phase samples were collected on PTFE membrane filters (47 mm diameter, 1.0 μm pores, Tisch Environmental) at 1 m3/hour according to federal reference methods (Electronic Code of Federal Regulations (2018), available at: https://www.ecfr.gov/cgi-bin/retrieveECFR?gp = &SID = 0f3bfa16342b3e5b858743bbbbdcfa4f&r = PART&n = 40y6.0.1.1.1). Field blanks were collected with all samples. All filters were spiked with deuterated standards (containing benzene-d6, ethylbenzene-d10, diethyl phthalate-d4 (all from AccuStandard), N-dodecanol-d25, octanoic acid-d15, N-hexadecane-d34, and N-octane-d18 (all from Cambridge Isotope Laboratories)), stored at −30 °C to −80 °C, and transported using expedited shipping in insulated coolers with multiple ice packs to ensure good sample quality upon analysis.
At PROPHET (forested field site), particle-phase samples were collected on the research tower, 28 m above the ground and 6 m above the canopy. The sampler was facing west on the tower, and no inlet tubing was used. Samples were collected both during the day (9:00am–5:00 pm) and at night (9:30pm-5:30am), avoiding sunrise and sunset periods to capture distinct daytime and nighttime chemistry. At the Atlanta site, the inlet was positioned 4.5 m above the ground, facing west. Minimal inlet tubing was used in this sampling setup (18” of 5/8” stainless steel tubing), so that the sampler could be housed out of direct sunlight, in an air conditioned trailer at the site. Samples were collected during the day (9:00am-7:00 pm) and overnight (10:30pm-5:00am), once again avoiding transitional periods. At the New York site, the sampler was positioned 4.5 m above the ground, facing west. Minimal inlet tubing was used here (9” of 5/8” stainless steel tubing), also to house the sampler away from direct sunlight and with temperature control. Samples were collected during the day only (7:00am–4:00 pm, referred to as “all day” samples, or 1:00pm–5:30 pm, referred to as “afternoon” samples).
At PROPHET, a high-resolution time-of-flight aerosol mass spectrometer (HR-TOF-AMS, Aerodyne Research Inc.) was used to measure non-refractory submicron aerosols50. In brief, particles are sampled through a 100-micron critical orifice and are focused into a particle beam using an aerodynamic lens. After traversing a vacuum chamber, the particles in the beam are impacted onto a porous tungsten vaporizer heated to 600 °C. Vapors are subsequently ionized by electron ionization (70 eV). The resulting ions are detected using a high-resolution time-of-flight mass spectrometer. The HR-TOF-AMS was operated in a high sensitivity mode, referred to as V-mode. Sampling inlets for HR-TOF-AMS measurements were located at two different heights, 6 m and 30 m, and data from the 30 m inlet on the PROPHET tower are considered in this study. Both inlets were fitted with PM2.5 cyclones to remove dust and prevent instrument clogging. The results presented here are of PM1, due to the transmission efficiency of PM1 through the AMS aerodynamic lens. Measurements were alternated between the 6 m and 30 m heights at 10 minute intervals. Ionization efficiency (IE) HR-TOF-AMS calibrations were performed at the beginning and end of this field campaign, according to standard protocols51,52. The IE calibrations were performed using monodisperse 300-nm ammonium nitrate particles.
Another Aerodyne HR-ToF-AMS was deployed at the Atlanta Jefferson Street site. The sampling was done continuously with an inlet facing west at 4.5 m above the ground, and the inlet was coupled with a PM2.5 cyclone to remove coarse particles. However, as above, the data presented here is for PM1. IE calibrations were performed every week.
For the AMS instruments at both sites, a nafion dryer was attached upstream of the AMS to dry the particles to below a relative humidity of 20%, because humidity has the potential to affect particle collection efficiency at the vaporizer. The composition-dependent collection efficiency (CDCE) was applied to HR-TOF-AMS data from both sites, according to Middlebrook et al53. Data analysis was performed in Igor Pro v6.37 (Wavemetrics, Inc.) using the Squirrel v1.16 H and Pika v1.57I packages (Sueper, D. ToF-AMS Data Analysis Software, available at: http://cires1.colorado.edu/jimenez-group/wiki/index.php/ToF-AMS_Analysis_Software). High-resolution mass spectral fitting was performed on the HR-TOF-AMS V-mode data. Elemental analyses were performed to find the atomic ratios of oxygen to carbon (O/C) according to the improved-ambient method by Canagaratna et al17.
Four environmental laboratory chamber experiments were performed in the Georgia Tech Environmental Chamber facility54. Experiments were performed at 70% relative humidity at 25 °C with seed aerosols generated by atomization of a sulfuric acid (0.01 M) and magnesium sulfate (0.005 M) solution. α-pinene and β-pinene (12 ppb) were used as precursor VOCs and were oxidized by NO3•. These experiments were designed to study nighttime VOC oxidation as it has been shown that nitrate radical oxidation of monoterpenes can contribute substantially to nighttime aerosol production in the southeastern U.S55. These experiments are included in this study because they present an opportunity to explore the effects of precursor identity and chemical conditions on compositional variability in a controlled manner. Here, α-pinene and β-pinene were selected as precursors because they are prevalent monoterpenes observed at the forested site and Atlanta site. Their difference in structure presents an opportunity to study the effects that these structural differences have on oxidation chemistry. Future research will include a broader selection of biogenic and anthropogenic VOC precursors. Two experiments were run for each precursor, with RO2• fate controlled primarily either by NO3• (“RO2• + NO3• experiments”) or by HO2• (“RO2• + HO2• experiments”), following experimental protocols similar to those in Boyd et al54. Briefly, seed aerosols and a precursor VOC were injected into the chamber prior to the introduction of N2O5, which serves as a source of NO3•. For RO2• + HO2• experiments, 25 ppm formaldehyde was also injected. N2O5 was premade in a flow tube by mixing O3 and NO2 and was introduced to the chamber, which marked the beginning of an experiment. Approximately 60 ppb N2O5 was added to the chamber for RO2• + NO3• experiments, and 80 ppb N2O5 was added for RO2• + HO2• experiments. Filter samples were collected on PTFE membrane filters (47 mm diameter, 0.45 μm pores, Maine Manufacturing LLC) 3–4 h after the beginning of each experiment for 100–120 min at 28–30 L/min.
Model simulations use GECKO-A, an explicit chemical mechanism generator that applies observed structure-activity relationships (SARs) to predict as fully as possible the atmospheric chemistry of precursor hydrocarbons5,56. SOA formation is described via gas-particle partitioning57, using vapor pressure relationships58,59. The completed mechanisms are implemented in a box model. To maintain a tractable mechanism size, the following standard thresholds are imposed: (1) for any given reaction, pathways with branching ratio < 5% are ignored and the reaction rates of the other pathways are incremented proportionally so that the overall reaction rate is unchanged; (2) we lump together products contributing < 5% to the total yield from a reaction, by substituting them with their highest-yield positional isomer (GECKO-A does not account for stereoisomers). The model has been tested with these thresholds for the oxidation of α-pinene60,61. In this study we also apply the GECKO-A SARs to the oxidation of β-pinene. Here, α-pinene and β-pinene were selected for consistency with the chamber experiments discussed above. The resulting combined mechanism contains ~83,700 unique non-radical species, including ~18,500 lumped species which represent the contributions of an additional ~128,000 very low-yield isomers. These species should be regarded as potential products. Not all possible reaction pathways are relevant to any given set of experimental conditions. The effective diversity of the model is best assessed by applying experimentally-relevant detection limits to the model output.
Six different single precursor modeling experiments were performed, either as chamber simulations (α- or β-pinene precursor with NO3• oxidation, and subsequent RO2• fate controlled by NO3• or HO2•) or as ambient (forest) simulations (α- or β-pinene precursor, OH• oxidation). Chamber simulations were run for 2–4 h of simulated oxidant exposure, and ambient simulations were run for 8 h of simulated exposure under constant noontime conditions. All runs were performed at 295 K, with 70% relative humidity. Initial precursor concentration was 12 ppb in all cases, to match the chamber experiments discussed previously. For the GECKO-A chamber simulations, runs started with 60 ppb N2O5 (as NO3• source), with 25 ppm of formaldehyde added for the RO2• + HO2• experiment. In the ambient simulations, 30 ppb O3 and 0.1 ppb of NOX were added. After applying an abundance threshold of 0.5 ppq, as previously discussed, an average of 2296 ± 2911 compounds remained for each simulation at the final simulation time.
Analysis
All filters were extracted in 2 mL of methanol ( ≥ 99.9% purity, Sigma-Aldrich). Various solvents were considered and tested for filter extraction (e.g., acetonitrile, dichloromethane), but methanol was selected because of its well-documented use in extraction of oxidized organics, organonitrates, and organosulfates20,28,62,63. It is important to note that only reasonably polar organic compounds will dissolve in methanol, so non-polar compounds may be left behind. However, non-polar compounds do not ionize well via electrospray ionization (discussed below), and are therefore not observed during analysis anyway.
Extracts were sonicated at room temperature for 60 min, and were exposed to gentle nitrogen flow to reduce their volume to 200 μL methanol (≥99.9% purity, Sigma-Aldrich), thereby increasing analyte concentration. Extracts were stored at −30 °C until analysis. Field and laboratory blanks were extracted using these same methods. All filters from each campaign were extracted at the same time and subsequently analyzed together.
Samples were analyzed on an Agilent 1260 Infinity HPLC with electrospray ionization (ESI), and an Agilent 6550 Q-TOF tandem mass spectrometer. Separation was performed with an Agilent Poroshell 120 SB-Aq reverse phase column column (2.1 × 50 mm, 2.7 μm particle size). Formic acid at 0.1% (98–100%, Sigma-Aldrich) was used as a modifier in the PROPHET sample set to promote ionization in the ESI source. In the Atlanta and New York City sample sets, 0.1% acetic acid (for HPLC, Sigma-Aldrich) was used as a modifier. Methanol (≥99.9% purity, Sigma-Aldrich) and water (Milli-Q, 18.2 MΩ·cm, <3 ppb TOC) at ambient temperature were used as LC solvents.
In all analyses, 5 μL aliquots of each sample were injected to the LC column. For the PROPHET sample set, the following solvent gradient was applied: from 0 to 2 min, 95% water (A) and 5% methanol (B); from 2–10 min, increase B to 90%; from 10–17 min, hold at 90% B; then decrease to 5% B to prepare for the next run, similar to previously established methods64. For the Atlanta and New York City sample sets, the above gradient was slightly extended: from 0 to 2 min, 95% water (A) and 5% methanol (B); from 2–22 min, increase B to 90%; from 22–27 min, hold at 90% B; then decrease to 5% B to prepare for the next run. A flow rate of 0.2 mL/min was used in all cases.
The electrospray source was run in both positive and negative ionization mode at the following source conditions, optimized for small molecule identification with our Q-TOF: drying gas temperature and flow of 225 °C and 13 L/min (PROPHET) or 17 L/min (Atlanta/New York City), respectively; fragmentor voltage at 365 V and capillary voltage at 4000 V; sheath gas flow and temperature at 400 °C and 12 L/min, respectively; nebulizer pressure at 20 psig. The fragmentor voltage was lowered to investigate possible compound fragmentation caused by this voltage, leading to the observation of IVOCs (as discussed above). However, there was no difference in the volatility distribution observed when this value was lowered.
ESI allows for measurement of compounds that are efficiently ionized in the source. Oxygen-containing compounds are prominent, as are nitrogen-containing compounds (e.g., amines, as they are sufficiently basic and thus readily ionized via ESI)23,65, and compounds containing a combination of oxygen, nitrogen, and sulfur atoms. We observe nitrogen- and sulfur-containing compounds without oxygen, though they contribute minimally to observed abundance. Sulfur-containing compounds without oxygen are difficult to detect without specialized pre-treatment, and thus omitted here23,24. We note that the organic compounds we observe during this analysis do not reflect the full range of organic compounds present as particles in the atmospheres we sampled, but rather a fraction that is functionalized with oxygen, nitrogen, and/or sulfur.
To ensure accurate mass results, a solution of reference masses was introduced to the ESI source throughout analysis, containing compounds with extremely high purity and predictable ionization in positive and negative mode: 5 mM purine and 2.5 mM HP-0921 (in 95% acetonitrile (≥99.9% purity, Honeywell), 5% water (Milli-Q, 18.2 MΩ·cm, <3 ppb TOC), Agilent Technologies). The Q-TOF was tuned and calibrated for optimal performance in its low mass range (m/z < 1700), and data were collected for ions ranging from m/z 50–1000.
The data were initially processed with Agilent Mass Hunter software. Mass Hunter was selected for automated peak finding and identification because it has been optimized to work with the Agilent LC-Q-TOF data output, and because of its strengths in feature identification and formula generation (see below). We carefully evaluated Mass Hunter’s performance at multiple points in our analyses with analytical standards, to verify its efficacy at feature and formula identification. This software has been used extensively in previously published work, in the field of atmospheric chemistry (e.g., Zhang et al64.) and in others (e.g., Chen et al.66).
For all samples, ions from m/z 50–600 were extracted and assigned masses, allowing for [M + H]+ and [M + Na]+ addition in ESI positive mode, [M-H]− and [M + HCOO]− or [M + CH3COO]− in negative mode, as well as neutral water loss. To ensure high quality data, only ions with strong signal (isotope height of 1000 counts and compound height of 5000 counts to confidently surpass instrument noise) and strong peak quality score (>70%, a score produced by Mass Hunter, including criteria such as signal-to-noise ratio, retention time peak width, retention time peak shape, and isotope pattern) were selected. Formulas were assigned to the resulting masses, constraining elements according to C3–60H4–122O0–20N0–3S0–1. The formula with the best formula match score for each compound was selected. Only formulas with formula quality scores greater than 75% were retained for subsequent non-targeted analysis (formula quality score is a score produced by Mass Hunter, incorporating mass match, isotope spacing, and isotope abundance). Samples and blanks were processed identically. The high mass accuracy (<1–2 ppm) and mass resolution (>25,000) of the Q-TOF, paired with accurate adduct assignment and the use of isotopic distribution (pattern and spacing) ensures accurate molecular formula identification for the analytes observed in this non-targeted analysis, similar to previous work67,68. Compared to online methods, our instrumentation has the advantage of isolating analyte peaks in LC space for mass detection and formula assignment without interference from other mass spectral peaks (as is typical with direct-MS analysis), therefore increasing the likelihood of a high mass accuracy formula assignment well beyond the mass resolution (i.e., M / ∆M) of the MS.
We performed extensive data quality control and quality assurance (QC/QA) using external code written in R, following best practices established in the literature67,69.
Masses appearing in both a sample and its corresponding blank (using a conservative 5 ppm tolerance) with an abundance ratio of less than 5:1 were eliminated from the results. Masses appearing in both a sample and its corresponding blank with a ratio larger than 5 were retained, and the abundance of the blank was subtracted from the abundance of the sample in these cases. Positive and negative mode data were then combined, and abundances of any compounds appearing in both ionization modes were averaged.
For further data quality control, all molecular formulas were screened to identify possible incorrect assignments. The number of C, H, O, N, and S atoms were further limited according to values presented by Kind and Fiehn (e.g., for masses below 500 Da, to no more than 39 C, 72 H, 20 O, etc67.). Compounds were additionally flagged based on a disagreement with the nitrogen rule (molecules with an odd number of N-atoms must have an odd nominal mass67), non-integer double bond equivalent values, large mass differences between the observed mass and proposed mass for a particular formula, and outlier H/C ratios (H/C < 0.2 or H/C > 3.1)67. Alternative molecular formulas proposed by the Agilent software for these flagged compounds were evaluated, and if no reasonable alternative formula existed, the compounds were omitted from analysis.
An additional screening step was added to reduce variability from sample to sample caused by low abundance impurities in the LC solvents. Here, frequently occurring impurity ions were excluded and a conservative abundance threshold was implemented to far surpass the instrument LOD (ion height >15,000 counts or peak abundance >100,000 counts); see Supplementary Note 1 for additional details. This threshold is unrelated to the threshold used in the GECKO-A analysis, discussed above. All together, our LC-ESI-Q-TOF’s capabilities, in combination with strict quality control on peak assignment, formula assignment, and blank subtraction enable confident mass and formula identification for compounds in complex aerosol mixtures and follows best practices from the literature.
To study sample-to-sample compositional variability, molecular formulas and retention times were compared. The fraction of overlapping compounds was computed by comparing the number of compounds occurring in both samples (with a formula and retention time match, NShared) in a set of two by the number of compounds present in each sample in the set (N1 and N2 for samples 1 and 2 in the set), and averaging the two values to provide the percent of distinct or overlapping compounds in a given sample comparison, as show in Equations 1 and 2. Compound-specific model results were treated in the same way. However, instead of tracking isomers with molecular formula and LC retention time for the model results, chemical structures were used to compare across modeling experiments.
Additional data analyses performed in this study include a sensitivity analysis and a statistical backward trajectory analysis using HYSPLIT. Briefly, a sensitivity analysis was performed to evaluate the effect of LOD thresholds on the results of the sample-to-sample compositional variability analysis presented here. In addition, a backward trajectory analysis with 24-hour HYSPLIT trajectories was performed to evaluate the effect of air parcel history (and associated transport, chemistry, and meteorology) on the compositional variability observed at each field site. Additional details for both analyses can be found in above and in Supplementary Notes 3 and 5.
Data availability
The data that support the findings presented here are available online at https://figshare.com/projects/Supplemental_Data_for_An_omnipresent_diversity_and_ variability_in_the_chemical_composition_of_atmospheric_functionalized_organic_aerosol_/37487. Basic R code for data processing and sample comparison are described in the Methods and Supplemental Information, but are available upon request. The chemical mechanism from GECKO-A modeling is also available upon request.
References
Jimenez, J. L. et al. Evolution of organic aerosols in the atmosphere. Science 326, 1525–1529 (2009).
Hallquist, M. et al. The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos. Chem. Phys. 9, 5155–5236 (2009).
Pöschl, U. & Shiraiwa, M. Multiphase chemistry at the atmosphere-biosphere interface influencing climate and public health in the anthropocene. Chem. Rev. 115, 4440–4475 (2015).
Goldstein, A. H. & Galbally, I. E. Known and unexplored organic constituents in the earth’s atmosphere. Environ. Sci. Technol. 41, 1515–1521 (2007).
Aumont, B., Szopa, S. & Madronich, S. Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach. Atmos. Chem. Phys. 5, 2497–2517 (2005).
O’Brien, R. E. et al. Molecular characterization of organic aerosol using nanospray desorption/electrospray ionization mass spectrometry: CalNex 2010 field study. Atmos. Environ. 68, 265–272 (2013).
Pope, C. A. & Dockery, D. D. Health effects of fine particulate air pollution: lines that connect. J. Air Waste Manag. Assoc. 56, 709–742 (2006).
Jerrett, M. et al. Long-term ozone exposure and mortality. N. Engl. J. Med. 360, 1085–1095 (2009).
Tao, F., Gonzalez-Flecha, B. & Kobzik, L. Reactive oxygen species in pulmonary inflammation by ambient particulates. Free Radic. Biol. Med. 35, 327–340 (2003).
Gentner, D. R. et al. Elucidating secondary organic aerosol from diesel and gasoline vehicles through detailed characterization of organic carbon emissions. Proc. Natl Acad. Sci. 109, 18318–18323 (2012).
Hunter, J. F. et al. Comprehensive characterization of atmospheric organic carbon at a forested site. Nat. Geosci. 10, 748–753 (2017).
Krechmer, J. E. et al. Ion mobility spectrometry-mass spectrometry (IMS-MS) for on- and offline analysis of atmospheric gas and aerosol species. Atmos. Meas. Tech. 9, 3245–3262 (2016).
Isaacman-VanWertz, G. et al. Using advanced mass spectrometry techniques to fully characterize atmospheric organic carbon: current capabilities and remaining gaps. Faraday Discuss. 200, 579–598 (2017).
Nozière, B. et al. The molecular identification of organic compounds in the atmosphere: state of the art and challenges. Chem. Rev. 115, 3919–3983 (2015).
Stark, H. et al. Methods to extract molecular and bulk chemical information from series of complex mass spectra with limited mass resolution. Int. J. Mass. Spectrom. 389, 26–38 (2015).
Heald, C. L. et al. A simplified description of the evolution of organic aerosol composition in the atmosphere. Geophys. Res. Lett. 37, L08803 (2010).
Canagaratna, M. R. et al. Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications. Atmos. Chem. Phys. 15, 253–272 (2015).
O’Brien, R. E. et al. Molecular characterization of S- and N-containing organic constituents in ambient aerosols by negative ion mode high-resolution Nanospray Desorption Electrospray Ionization Mass Spectrometry: CalNex 2010 field study. J. Geophys. Res. Atmos. 119, 706–12,720 (2014). 12.
Budisulistiorini, S. H. et al. Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site. Atmos. Chem. Phys. 15, 8871–8888 (2015).
Riva, M., Barbosa, T. D. S., Lin, Y., Stone, E. A. & Gold, A. Characterization of organosulfates in secondary organic aerosol derived from the photooxidation of long-chain alkanes. Atmos. Chem. Phys. 16, 11001–11018 (2016).
O’Brien, R. E. et al. Probing molecular associations of field-collected and laboratory-generated SOA with nano-DESI high-resolution mass spectrometry. J. Geophys. Res. Atmos. 118, 1042–1051 (2013).
Romonosky, D. E., Laskin, A., Laskin, J. & Nizkorodov, S. A. High-resolution mass spectrometry and molecular characterization of aqueous photochemistry products of common types of secondary organic aerosols. J. Phys. Chem. A 119, 2594–2606 (2015).
Oss, M., Kruve, A., Herodes, K. & Leito, I. Electrospray ionization efficiency scale of organic compound. Anal. Chem. 82, 2865–2872 (2010).
Purcell, J. M. et al. Sulfur speciation in petroleum: Atmospheric pressure photoionization or chemical derivatization and electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Energy Fuels 21, 2869–2874 (2007).
Hayes, P. L. et al. Organic aerosol composition and sources in Pasadena, California, during the 2010 CalNex campaign. J. Geophys. Res. Atmos. 118, 9233–9257 (2013).
Aiken, A. C., DeCarlo, P. F., Kroll, J. H., Huffman, J. A. & Docherty, K. S. O/C and OM/OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry. Environ. Sci. Technol. 42, 4478–4485 (2008).
Wang, X. K. et al. Molecular characterization of atmospheric particulate organosulfates in three megacities at the middle and lower reaches of the Yangtze River. Atmos. Chem. Phys. 16, 2285–2298 (2016).
Surratt, J. D. et al. Organosulfate formation in biogenic secondary organic aerosol. J. Phys. Chem. A 112, 8345–8378 (2008).
Lin, P., Yu, J. Z., Engling, G. & Kalberer, M. Organosulfates in humic-like substance fraction isolated from aerosols at seven locations in East Asia: A study by ultra-high-resolution mass spectrometry. Environ. Sci. Technol. 46, 13118–13127 (2012).
Li, Y., Pöschl, U. & Shiraiwa, M. Molecular corridors and parameterizations of volatility in the chemical evolution of organic aerosols. Atmos. Chem. Phys. 16, 3327–3344 (2016).
D’Ambro, E. L. et al. Molecular composition and volatility of isoprene photochemical oxidation secondary organic aerosol under low and high NOx conditions. Atmos. Chem. Phys. 17, 159–174 (2017).
Gentner, D. R. et al. Emissions of terpenoids, benzenoids, and other biogenic gas-phase organic compounds from agricultural crops and their potential implications for air quality. Atmos. Chem. Phys. 14, 5393–5413 (2014).
Gentner, D. R. et al. Review of urban secondary organic aerosol formation from gasoline and diesel motor vehicle emissions. Environ. Sci. Technol. 51, 1074–1093 (2017).
Kroll, J. H. et al. Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol. Nat. Chem. 3, 133–139 (2011).
Polissar, A. V., Hopke, P. K. & Harris, J. M. Source regions for atmospheric aerosol measured at Barrow, Alaska. Environ. Sci. Technol. 35, 4214–4226 (2001).
Gentner, D. R. et al. Emissions of organic carbon and methane from petroleum and dairy operations in California’s San Joaquin Valley. Atmos. Chem. Phys. 14, 4955–4978 (2014).
Budisulistiorini, S. H. et al. Real-time continuous characterization of secondary organic aerosol derived from isoprene epoxydiols in downtown atlanta, georgia, using the aerodyne aerosol chemical speciation monitor. Environ. Sci. Technol. 47, 5686–5694 (2013).
Karambelas, A., Pye, H. O. T., Budisulistiorini, S. H., Surratt, J. D. & Pinder, R. W. Contribution of isoprene epoxydiol to urban organic aerosol: evidence from modeling and measurements. Environ. Sci. Technol. Lett. 1, 278–283 (2014).
Ervens, B., Turpin, B. J. & Weber, R. J. Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies. Atmos. Chem. Phys. 11, 11069–11102 (2011).
Ng, N. L. et al. Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry. Atmos. Chem. Phys. 10, 4625–4641 (2010).
Russell, L. M., Bahadur, R. & Ziemann, P. J. Identifying organic aerosol sources by comparing functional group composition in chamber and atmospheric particles. Proc. Natl Acad. Sci. 108, 3516–3521 (2011).
Kostenidou, E. et al. Organic aerosol in the summertime Southeastern United States: components and their link to volatility distribution, oxidation state and hygroscopicity. Atmos. Chem. Phys. 18, 5799–5819 (2018).
Koop, T., Bookhold, J., Shiraiwa, M. & Pöschl, U. Glass transition and phase state of organic compounds: dependency on molecular properties and implications for secondary organic aerosols in the atmosphere. Phys. Chem. Chem. Phys. 13, (19238–19255 (2011).
Tuet, W. Y. et al. Chemical oxidative potential of secondary organic aerosol (SOA) generated from the photooxidation of biogenic and anthropogenic volatile organic compounds. Atmos. Chem. Phys. 17, 839–853 (2017).
Ye, J., Gordon, C. A. & Chan, A. W. H. Enhancement in secondary organic aerosol formation in the presence of preexisting organic particle. Environ. Sci. Technol. 50, 3572–3579 (2016).
Rickards, A. M. J., Miles, R. E. H., Davies, J. F., Marshall, F. H. & Reid, J. P. Measurements of the sensitivity of aerosol hygroscopicity and the κ parameter to the O/C ratio. J. Phys. Chem. A 117, 14120–14131 (2013).
Xu, L. et al. Wintertime aerosol chemical composition, volatility, and spatial variability in the greater London area. Atmos. Chem. Phys. 16, 1139–1160 (2016).
Carroll, M. A., Bertman, S. B. & Shepson, P. B. Overview of the program for research on oxidants: photochemistry, emissions, and transport (PROPHET) summer 1998 measurements intensive. J. Geophys. Res. 106, 24275–24288 (2001).
Xu, L. et al. Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States. Proc. Natl Acad. Sci. 112, 37–42 (2014).
DeCarlo, P. F. et al. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 78, 8281–8289 (2006).
Jayne, J. T. et al. Development of an aerosol mass spectrometer for size and composition analysis of submicron particles. Aerosol Sci. Technol. 33, 49–70 (2000).
Jimenez, J. L. Ambient aerosol sampling using the aerodyne aerosol mass spectrometer. J. Geophys. Res. 108, 8425 (2003).
Middlebrook, A. M., Bahreini, R., Jimenez, J. L. & Canagaratna, M. R. Evaluation of composition-dependent collection efficiencies for the Aerodyne aerosol mass spectrometer using field data. Aerosol Sci. Technol. 46, 258–271 (2012).
Boyd, C. M. et al. Secondary organic aerosol (SOA) formation from the β-pinene+NO3 system: effect of humidity and peroxy radical fate. Atmos. Chem. Phys. 15, 7497–7522 (2015).
Xu, L., Suresh, S., Guo, H., Weber, R. J. & Ng, N. L. Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: Spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates. Atmos. Chem. Phys. 15, 7307–7336 (2015).
Lee-Taylor, J. et al. Multiday production of condensing organic aerosol mass in urban and forest outflow. Atmos. Chem. Phys. 15, 595–615 (2015).
Camredon, M., Aumont, B., Lee-Taylor, J. & Madronich, S. The SOA/VOC/NOx system: an explicit model of secondary organic aerosol formation. Atmos. Chem. Phys. 7, 5599–5610 (2007).
Nannoolal, Y., Rarey, J., Ramjugernath, D. & Cordes, W. Estimation of pure component properties: Part 1. Estimation of the normal boiling point of non-electrolyte organic compounds via group contributions and group interactions. Fluid Phase Equilib. 226, 45–63 (2004).
Nannoolal, Y., Rarey, J. & Ramjugernath, D. Estimation of pure component properties: Part 3. Estimation of the vapor pressure of non-electrolyte organic compounds via group contribution and group interactions. Fluid Phase Equilib. 269, 117–133 (2008).
Valorso, R. et al. Explicit modelling of SOA formation from α-pinene photooxidation: Sensitivity to vapour pressure estimation. Atmos. Chem. Phys. 11, 6895–6910 (2011).
McVay, R. C. et al. SOA formation from the photooxidation of α-pinene: Systematic exploration of the simulation of chamber data. Atmos. Chem. Phys. 16, 2785–2802 (2016).
Ng, N. L. et al. Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3). Atmos. Chem. Phys. 8, 4117–4140 (2008).
Riva, M. et al. Chemical characterization of secondary organic aerosol from oxidation of isoprene hydroxyhydroperoxides. Environ. Sci. Technol. 50, 9889–9899 (2016).
Zhang, H., Surratt, J. D., Lin, Y. H., Bapat, J. & Kamens, R. M. Effect of relative humidity on SOA formation from isoprene/NO photooxidation: enhancement of 2-methylglyceric acid and its corresponding oligoesters under dry conditions. Atmos. Chem. Phys. 11, 6411–6424 (2011).
Pudenzi, M. A. & Eberlin, M. N. Assessing Relative electrospray ionization, atmospheric pressure photoionization, atmospheric pressure chemical ionization, and atmospheric pressure photo- and chemical ionization efficiencies in mass spectrometry petroleomic analysis via pools and pairs. Energy Fuels 30, 7125–7133 (2016).
Chen, Q. et al. Untargeted plasma metabolite profiling reveals the broad systemic consequences of xanthine oxidoreductase inactivation in mice. PLoS ONE 7, e37149 (2012).
Kind, T. & Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinforma. 8, 105 (2007).
Thurman, E. M. & Ferrer, I. The isotopic mass defect: a tool for limiting molecular formulas by accurate mass. Anal. Bioanal. Chem. 397, 2807–2816 (2010).
Zielinski, A. T. et al. A new processing scheme for ultra-high resolution direct infusion mass spectrometry data. Atmos. Environ. 178, 129–139 (2018).
Acknowledgements
We thank the University of Michigan Biological Station and the New York State Department of Environmental Conservation for enabling us to collect samples at these two locations. We gratefully acknowledge the NOAA Air Resources Laboratory (ARL) for the provision of the HYSPLIT transport and dispersion model and READY website (http://www.ready.noaa.gov) used in this publication. We also thank Dr. Harald Stark and Dr. Lynn Mazzoleni for CIMS and ultra-high-resolution data shown in Fig. 3, Dr. Jason Surratt for synthesizing standards used in this work, and Steve Geringer for his help with Fig. 5. D.R. Gentner acknowledges support from National Science Foundation Award 1622389. J. Lee-Taylor and J.L. Jimenez are grateful for support under EPA STAR 83587701-0 and DOE (BER/ASR) DE-SC0016559. This publication has not been formally reviewed by EPA. The views expressed in this document are solely those of the authors and do not necessarily reflect those of the Agency. EPA does not endorse any products or commercial services mentioned in this publication. The National Center for Atmospheric Research is sponsored by the National Science Foundation. A.A.T. Bui and R. J. Griffin acknowledge support of the National Science Foundation through AGS-1552086. M. Takeuchi, T. Joo, G. Eris, Y. Chen, N.L. Ng acknowledge support from National Science Foundation CAREER AGS-1555034.
Author information
Authors and Affiliations
Contributions
J.C.D. and D.R.G. designed the study and led analyses. J.C.D. collected filter samples at the forested site. A.A.T.B. and R.J.G. collected and analyzed the AMS data at the forested site. T.J. collected filter samples and T.J., Y.C., and N.L.N. collected and analyzed the AMS data in Atlanta. P.K. collected filter samples in NYC. M.T. and G.E. ran chamber experiments and collected samples. J.L-T., J.L.J., and B.A. developed and ran GECKO-A simulations. E.B.B. contributed to initial filter data analysis, method development, and observations. J.C.D. analyzed all the filter data and compiled all results. J.C.D. and D.R.G. wrote the paper, with relevant methods contributions from co-authors. All authors commented on and discussed the manuscript to help refine the interpretation and presentation of results.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ditto, J.C., Barnes, E.B., Khare, P. et al. An omnipresent diversity and variability in the chemical composition of atmospheric functionalized organic aerosol. Commun Chem 1, 75 (2018). https://doi.org/10.1038/s42004-018-0074-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-018-0074-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
