
Abstract
Hybrid materials composed of metal nanoparticles and metal-organic frameworks have attracted attention for various applications because of the synergistic functionality between their constituent materials. Interfacial interaction is expected however the mechanism remains ambiguous. Here we report the valence bands of palladium nanocubes covered by copper(II) 1, 3, 5-benzenetricarboxylate (HKUST-1), denoted as Pd@HKUST-1, and the charge transfer from the palladium nanocubes to HKUST-1 at the Pd/HKUST-1 interface is investigated quantitatively. Interfacial density of states are different from those of internal constituents and imply that the Cu–O group in HKUST-1 acts as a charge accepter. The role of Cu–O group in charge transfer behaviour is also observed experimentally. Finally, we reveal the charge transfer mechanism from the Pd 4d bands to the Cu 3d (4sp) – O 2p hybridization bands of HKUST-1 at the Pd/HKUST-1 interface, which explains the enhanced hydrogen storage capacity in Pd@HKUST-1.
Similar content being viewed by others


Introduction
Hybrid materials composed of transition-metal nanoparticles and metal-organic frameworks (MOFs) have attracted interests increasingly because of their potential applications in gas storage, catalysis and chemical sensing1. However, the design of new hybrid materials remains a great challenge in chemistry, physics and materials science on such a small scale. The mechanism of interaction between the constituent materials remains unclear. Recently, charge-transfer behaviour has been shown to play an important role in the catalytic activity and hydrogen storage properties of these materials2,3,4. For example, the charge transfer behaviour of palladium (Pd) nanocubes covered by copper(II) 1, 3, 5- benzenetricarboxylate (HKUST-1), denoted by Pd@HKUST-1, has been shown to improve its hydrogen storage properties5,6. Hydrogen is an essential component in many industrial processes and also a potential clean energy source that does not emit air pollution or greenhouse gases on burning7. As a representative metal/MOF hybrid material, Pd@HKUST-1 is an important example for studying the interaction between the constituent materials linked with improved hydrogen storage, thus aiding the design of other functional hybrid materials through rational modification of their electronic structures.
Palladium nanoparticles can absorb large amounts of hydrogen at ambient pressure and room temperature, which is related to their electronic structures with unfilled bands8,9. The density of states (DOS) of palladium is composed of Pd 4d bands and 5s bands10,11. Theoretically, the hydrogen concentration in palladium is only related to the number of holes in the 4d band, because the transfer of H 1s electrons into Pd 4d holes creates Pd–H bonds12,13. As a representative MOF, HKUST-1 has a richly porous structure consisting of copper atoms interlinked organic 1, 3, 5- benzenetricarboxylate (BTC) frame ligands14,15. The electronic structure of HKUST-1 is mainly composed of two partial DOS: one from Cu and the other from O16. In previous studies, the core-level spectra of Pd@HKUST-1 were investigated with laboratory X-ray photoelectron spectroscopy (XPS)5,13. The core-level Pd 3d peak of Pd@HKUST-1 was shifted to a higher binding energy compared with that of palladium nanocubes. The core-level Cu 2p peak was shifted to a lower energy compared with that of HKUST-113. Recently, a density functional theory calculation has suggested that atomic charge displacement occurs between palladium atom and copper atom at the interface of palladium (100) surface and Cu-edged HKUST-117. These results clearly imply the possibility of the charge transfer between Pd and Cu at the Pd/HKUST-1 interface in Pd@HKUST-1. To reveal the charge transfer behaviour, the valence bands and the conduction bands near the Fermi level are more informative than the core-level spectra. However, the electronic structure of Pd@HKUST-1 near the Fermi level has not yet been experimentally investigated.
In studying the electronic structure of Pd@HKUST-1 near the Fermi level based on charge transfer, it is important to investigate the valence bands of the transition metals, palladium and copper, in bulk materials and at Pd/HKUST-1 interface. However, photoelectron spectroscopy using UV light or soft X-rays provides only a surface-sensitive electronic structure because of the short probing depth. By contrast, with the use of hard X-rays photoelectron spectroscopy (HAXPES), the kinetic energy of photoelectrons can be enhanced, and the probing depth is much greater18. Additionally, near-edge X-ray absorption fine structure (NEXAFS) are equally applicable for determining the conduction bands19. Both the total electron yield from surface electrons from a nanometre-thick layer and the fluorescence yield from bulk electrons from a layer hundreds of nanometres thick can be used to distinguish the intrinsic signal from the surface contamination.
To reveal intrinsic electronic structure, we use HAXPES and NEXAFS to analyse the electronic structures of Pd, HKUST-1 and Pd@HKUST-1. Furthermore, we compare the observed valence band of Pd@HKUST-1 with the simple physical mixing of the spectra of Pd and HKUST-1 using the Kerkhof–Moulijn model to analyse these spectra quantitatively20. To the best of our knowledge, this work is the first study using HAXPES and NEXAFS to investigate the electronic structure of Pd@HKUST-1, as opposed to previous studies of the core-level spectra. The findings show that the hydrogen storage properties of Pd@HKUST-1 can be explained through the electronic structure near the Fermi level, which arises from an interfacial charge transfer.
Results
Interfacial charge transfer from Pd 4d to Cu 3d in Pd@HKUST-1 investigated by HAXPES
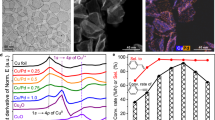
We used the same samples as reported in the previous work5. Pd@HKUST-1 was prepared by a facile reactive seeding method. As observed by transmission electron microscopy (TEM), shown in Fig. 1a, Pd nanocubes had an average edge length of 10 nm (±2 nm, error bar from size analysis of TEM image). The flat {100} facets of Pd nanocubes acted as the seeds for the HKUST-1 growth, such that all the Pd nanocubes were completely covered by HKUST-1 MOF. The Pd nanocubes were dispersed homogeneously in HKUST-1 of 3 nm (±2 nm) thickness, which was measured from the distances between Pd nanocubes in Fig. 1a. To obtain the intrinsic electronic structure with minimal influence from any surface contamination, we used HAXPES at a photoelectron energy of Ek = 5.95 keV to study Pd@HKUST-1, and compared with the results of Pd nanocubes and HKUST-1. The probing depths of Pd and HKUST-1 were 17 and 36 nm, respectively, which are three times of the values of the inelastic mean free path (IMFP) evaluated by the TPP-2M (Tanuma, Powel and Penn) formula21. The deep penetration enabled us to obtain the intrinsic electronic information from the interior of Pd@HKUST-1. After subtracting a background intensity, the intensities were normalized by the spectral area. Figure 1b shows the normalized HAXPES valence band (VB) spectra of the Pd nanocubes, HKUST-1 and Pd@HKUST-1. The valence-band spectrum centre (using gravity centre) of the Pd nanocubes is closer to the Fermi level than that of HKUST-1. The spectrum centre of Pd@HKUST-1 is located between those of Pd nanocubes and HKUST-1. This is related to the d-band centre and explained after the following quantitative analysis. The VB spectrum of Pd nanocubes shows three main peaks at 4.2 and 2.5 eV, and near the Fermi level, corresponding to the DOS of Pd 4d in the fcc palladium bulk10. However, despite the identical DOS, the sharpness of the peaks shrinks in the Pd nanocubes. This is possibly because size effects in the Pd nanocubes do not present in the corresponding bulk materials22. The VB spectrum of HKUST-1 shows features similar to CuO as shown in Fig. 1b (The similarity of HKUST-1 to CuO is explained in Supplementary Methods and Supplementary Figs. 1–3). The features come mainly from Cu 3d bands23,24. The peak located at 7.5 eV in the HKUST-1 spectrum, which is a typical feature of CuO bulk24, indicates that Cu2+ of the HKUST-1 MOF has a similar electronic structures to that of CuO bulk. This is also consistent with the divalent copper state Cu2+ of the HKUST-1 core-level spectra as observed by laboratory XPS16,25. This is related to the formation of bonds between copper and the organic groups of ligands, such as Cu-OCO and Cu-OH2, in HKUST-114. In the Pd@HKUST-1 VB spectrum, the feature of the Cu2+ peak at 7.5 eV becomes less distinct, which suggests that the electronic states of Cu2+ are altered. This feature indicates that Pd@HKUST-1 is not merely a simple physical mixture of Pd nanocubes and HKUST-1, but a synergy of these two materials.

Charge transfer at the Pd/HKUST-1 interface investigated by HAXPES valence band. a TEM image of Pd@HKUST-1, where Pd nanocubes are covered by HKUST-1. (scale bar: 20 nm) b Normalized HAXPES valence-band (VB) spectra of Pd nanocubes (green solid line), HKUST-1 (blue solid line) and Pd@HKUST-1 (red solid line) referring to CuO (violet dashed line) at an incident energy of 5.95 keV, which show the Pd 4d states in Pd nanocubes and Cu 3d states in HKUST-1. c Kerkhof–Moulijn model of the monolayer stacks used to calculate the intensity of hard X-ray photoelectron spectroscopy for Pd@HKUST-1. d Deviation (cyan area) of Pd@HKUST-1 HAXPES VB spectrum (red solid line) from the calculated VB spectrum (black dot-dashed line) created by summing the partial HAXPES VB spectra of Pd nanocubes (green dotted line) and the partial HAXPES VB spectrum of HKUST-1 (blue dotted line) according to the Kerkhof–Moulijn model
To evaluate whether the properties of Pd@HKUST-1 arise from the simple physical mixing of the Pd nanocubes and HKUST-1, or from interaction between them, we first summed the two separate spectra of the components, the Pd nanocubes and HKUST-1. Then we compared the summed result with the observed Pd@HKUST-1 spectrum to determine the extent of deviation linked to the interaction between Pd and HKUST-126. In order to apply appropriate weights to the separate Pd and HKUST-1 spectra before summing them, it is necessary to determine the relative intensities of Pd atoms and all the atoms of HKUST-1 probed by the HAXPES measurement. Generally, the intensity of the electrons emitted from the surface of a material is expressed as follows20:
Here, S is the surface area of the materials in the beam spot. n is the atomic number of the measured elements. σ is the photoionization cross section (PICS). λ is the IMFP of the electrons. t is the thickness of the materials. Because of the dispersion of the Pd nanocubes in HKUST-1, Pd@HKUST-1 can be assumed to consist of sheets of HKUST-1 with the monolayers of Pd nanocubes between them. Hence, it is suitable to use the Kerkhof–Moulijn model of stacks of monolayers for the physical summation of the respective spectra of the Pd nanocubes and HKUST-1, shown in Fig. 1c. According to this model, the Pd@HKUST-1 hybrid material can be considered to be layers of the Pd nanocubes covered by the HKUST-1 sheets. The ratio of intensities of the electrons from the Pd nanocubes to those from HKUST-1 can be expressed as follows20:
Here, all the parameters are labelled as Pd and HK (Cu) to distinguish the Pd nanocubes and HKUST-1. All the values of these parameters for the Pd nanocubes and HKUST-1 are listed in Table 127,28. The atomic ratios are estimated from the weight ratio (23.7 wt.%) of Pd in Pd@HKUST-1, using the relative molecular mass 658.9 for HKUST-1 (C18H12O15Cu3)14. For the Pd nanocubes, the VB is mainly dominated by the Pd 4d bands. For the HKUST-1, we evaluated all the elements according to Eq. 1, which shows that the intensity is proportional to the atomic ratio and the PICS (nσ). As shown in Table 1, the value of Cu 3d is much greater than those of other states from the organic BTC ligand of HKUST-1. Hence, the observed spectra for the Pd nanocubes and HKUST-1 are predominantly from the Pd 4d and Cu 3d electrons. Theoretically, the d-band centres of Cu 3d and Pd 4d are 2.67 and 1.83 eV, respectively29. Experimentally, we observed the valence-band centre of 4.37 and 3.02 eV in the Pd spectra and HKUST-1 spectra, respectively, presented in the spectra of Fig. 1b, which implies the dominated roles of Pd 4d and Cu 3d in the spectra. Using these parameter values and Eq. 2, the intensity ratio contributed by the Pd nanocubes and HKUST-1 is 0.66 (±0.03):0.34 (∓0.03). The error bar of the intensity ratio depends on tPd and tHK. The intensity ratio ranges from 0.69:0.31 to 0.63:0.37 for 8 ≤ tPd ≤ 12 nm and 1 ≤ tHK ≤ 5 nm (Supplementary Methods and Supplementary Table 1).
The summed spectrum according to the ratio above is shown as a black dotted dash line in Fig. 1d. The observed Pd@HKUST-1 spectrum and the partial spectra of the Pd nanocubes or HKUST-1 are also plotted for comparison. Then, the deviation of the simply summed spectrum from the experimental spectrum is shown in the cyan region, which indicates an electronic interaction between the Pd nanocubes and HKUST-1, instead of a simple physical mixture of the two. The deviation can be separated into three parts, marked as A, B, and C, which is robust even considering the error bar of sample size (Supplementary Methods and Supplementary Fig. 4). As discussed above, the Pd@HKUST-1 spectrum is dominated by Pd 4d near the Fermi level. Here, the negative deviation marked by A means a reduced contribution from the electronic states of the Pd nanocubes compared with the simple summation, which implies a lower occupancy of Pd 4d near the Fermi level due to a charge transfer. If the electron density is transferred from donor Pd nanocubes, HKUST-1 is possible to act as an electron accepter, which should show the features of Cu+. Actually, two features arise in the deviation curve. One is a clear feature marked by C, which is consistent with the Cu 3d9 states in CuO (Cu2+)24. The other is the positive deviation of B near the Fermi level, which indicates the presence of a Cu+ (3d10) configuration24, due to the charge transfer from the palladium atoms to the copper atoms. The above-mentioned results demonstrate that the electrons are transferred from the Pd nanocubes to HKUST-1 and absorbed by the Cu–O couple.
Interfacial states of Pd@HKUST-1 revealed by the DOS calculation
To investigate the reason for this deviation of VB spectra, we calculated the DOS for all the elements of the Pd@HKUST-1 using a density functional theory (DFT) method. We used a periodic palladium (100) slab model with three layers and simplified the HKUST-1 MOF ligand BTC to 1,4-benzenedicarboxylate (BDC), denoted by s-HKUST-1, and then set up an interface model of these two as shown in Fig. 2a (The detailed process is explained in Supplementary Methods and Supplementary Figs. 5–6). The atomic arrangement at the interface was set to be Pd-Cu-O, which is more stable than the Pd-O interface17. All the total DOS of the Pd@s-HKUST-1, Pd (100) slab and s-HKUST-1 are shown in Fig. 2b. On the basis of the calculated work function, the respective positions of the DOS for the three materials are similar to those in the observed spectra. The Pd@s-HKUST-1 DOS shifts 0.454 eV further from the Fermi level. The shift is greater than 0.2 eV that was observed in the experimental spectrum, because the model of Pd@s-HKUST-1 used in the calculations only includes the atoms near the interface. Focusing on the Pd@HKUST-1 interface, we calculated DOS of the atoms at the Pd/s-HKUST-1 interface and compared the results from atoms in each material. The partial DOS for Pd 4d, Cu 3d and O 2p is plotted in Fig. 2c–e, with the deviation of interfacial states from internal states. For Pd 4d, in Fig. 2c, the profile shape shows little change. However, there is a clear decrease of density of states near the Fermi level. The negative deviation near the Fermi level corresponds to the region A in Fig. 1d. Comparing to the internal Cu states in Fig. 2d, the interfacial Cu states deviate positively in the range of −3 ~ −1 eV, consistent with region B in Fig. 1d. Unfortunately, we could not find a deviation similar to region C here. Because there are unfilled states in Pd 4d near the Fermi level, but Cu 3d is fully filled, it is impossible for electrons to transfer from Pd to isolated Cu. We have to consider the linked oxygen. Actually, we found the shape of partial DOS of interfacial O 2p is different from that of internal O 2p in Fig. 2e, which can be caused by the interaction of Pd, Cu, and O. Considering the interaction of Cu and O, we merged the two partial DOS in Fig. 2f, which provides more unfilled states in the hybridized states. Then, we found the similar deviation between the interfacial and internal states at −1 and −7.5 eV, corresponding to the region B and C in Fig. 1d. Now, we confirm the deviation we observed in the HAXPES VB derives from the interface and the necessity of oxygen in the behaviour.

Interfacial and internal DOS calculated using DFT method. a Unit cell of interface model used for calculation, which consists Pd (100) slab of three atomic layers and a simplified HKUST-1 (s-HKUST-1) lattice. b Calculated total DOS for Pd@s-HKUST-1, Pd (100) slab and s-HKUST-1. Partial DOS of c Pd 4d, d Cu 3d and e O 2p. f The sum of the partial DOS of Cu 3d and O 2p. In c–f, the green dashed line is the states of the periodic Pd(100) slab without s-HKUST-1 covering, the violet solid lines indicate the states of the interfacial atoms, and the navy dashed lines indicate the states of the internal atoms of s-HKUST-1. The regions corresponding to the region A, B or C in Fig. 1d are also marked in circles and labelled as A, B or C
The role of oxygen in charge transfer revealed by NEXAFS
As discussed above, oxygen also participates in the charge transfer behaviour. To determine the relationship between copper and oxygen, here we measured the NEXAFS of the Cu L edge and O K edge in Fig. 3 for Pd@HKUST-1 and HKUST-1. The Cu L edge was obtained while Cu 2p electrons absorbed the photon energy and were then excited into unoccupied 3d states and the O K edge corresponds to the 1s electrons excited to empty 2p states. Because Cu directly binds to O atoms14, here we chose CuO and Cu2O as references in Fig. 3c, d. In Fig. 3a, the observed Cu L-edge features of Pd@HKUST-1 suggest the strong hybridization between the Cu 3d and O 2p orbitals19, which reduces the d-electron number from the Cu 3d10 configuration. The L3 and L2 peaks of HKUST-1 has the same peak position as CuO (Fig. 3c), consistent with a previous report of HKUST-1 with a divalent Cu2+16. Similar to CuO with a typical octahedron structure, the Cu and O atoms in HKUST-1 form as two connected distorted octahedra (Fig. 3a inset). While the Pd nanocubes was surrounded by the HKUST-1 in the structure, only the interfacial Cu2+ was reduced to Cu+, roughly speaking, as the result of the Cu-O hybridization change. The coexistence of Cu2+ and Cu+ states in Pd@HKUST-1 leads to a peak located between the peak of Cu2+ and that of Cu+. According to the interfacial structure, 3 nm-thick HKUST-1 (lattice constant is 2.6 nm) is only one layer with more than half amount of copper atoms at the interface. However, the peak shift of Pd@HKUST-1 is less than half between Cu2+ and Cu+. This may be related to the weak reduction of the Cu-O bond because of the transferred electrons on the hybridization bands. In other words, the copper is slightly less positive and the oxygen is slightly less negative. In Fig. 3b, both O K-edge spectra of Pd@HKUST-1 and HKUST-1 contain two regions, the sharp π* region around 525.5 eV and the broad σ* region around 540.5 eV. Considering the local atomic structure around O, the π* region derives mainly from electrons excited into the O 2p orbitals that are hybridized with Cu 3d and the σ* region derives from electrons excited into the O 2p orbitals that are hybridized with Cu 4sp and C 2sp. On the basis of previous researches19,30,31,32, the peaks are distinguished and marked by red arrows for those related to Cu and by grey arrows for the peaks related to C. The main features at 525.5 and 540.5 eV are also explained in Supplementary Methods and Supplementary Fig. 7. Compared with the CuO and Cu2O references (Fig. 3d), the peak position of the HKUST-1 O K edge was different from that of CuO because O connects not only with Cu but also with C (Fig. 3a inset). For Pd@HKUST-1, the energy value of the first peak shifts by 0.4 eV comparing with HKUST-1, which is much lower than the shift value of CuO to Cu2O, 2.1 eV. This is attributed to the amount of the interfacial oxygen binding with copper. Thus, the charge transfer from Pd 4d to the Cu–O hybridization bands is only at the interface region. Considering the atomic structures of HKUST-1, there are probably two kinds of atomic connections at the interface, Pd–Cu–O and Pd–O–Cu. This means there are Cu–O features and/or Pd–O features appearing in the NEXAFS spectra. We also measured the O K edge of the standard PdO reference (Supplementary Methods and Supplementary Fig. 8), which is the same as the calculated result33. The O K edge NEXAFS spectra of Pd@HKUST-1 showed only features from Cu-O, without the features of Pd-O. Finally, the NEXAFS spectra confirmed the involvement of oxygen in the charge transfer behaviour of interfacial Pd–Cu–O atomic structure.

Cu and O conduction band observed by NEXAFS. a Cu L edge and b O K edge NEXAFS spectra of HKUST-1 and Pd@HKUST-1. c Cu L edge and d O K edge of the CuO and Cu2O powder are plotted at the bottom of the figure. a, b for reference. The inset of a shows the atomic structure related to Cu, O and C atoms of HKUST-1
Discussion
As shown above, the electronic states of the Pd nanocubes near the Fermi level are mainly composed of Pd 4d band. The HKUST-1 electronic structures indicate the occurrence of Cu 3d-O 2p hybridization. Different from the Cu 3d band with fully filling, the Cu-O hybridization bands are not fully filled, which produces unsaturated bands near the Fermi level. While the two materials are composited to form the Pd@HKUST-1 system, some electrons of the Pd 4d bands near the Fermi level transfer to the Cu-O hybridization bands at the Pd/HKUST-1 interface, as illustrated in Fig. 4. This transfer generates holes in the Pd 4d band. When hydrogen is absorbed by the Pd nanocubes, the holes together with unfilled Pd 4d bands are filled by the H 1s electrons, and then the Pd-H chemical bonds form8,34. Experimentally, Pd@HKUST-1 absorbs much more hydrogen than the Pd nanocubes9, which has unfilled bands only above the Fermi level.

Schematic of charge transfer from Pd nanocubes to HKUST-1 at the interface. a Pd@HKUST-1 structure and the magnification of the interfacial charge transfer behaviour. b Change of electronic structures at the composite interface corresponding to the charge transfer process and the hydrogen abosorption
The electron transfer from the Pd 4d band creates holes in the electronic structure of the Pd nanocubes, which are essential to the hydrogen absorption behaviour. The hole ratio was evaluated quantitatively by comparing the amount of electron transferred from Pd 4d with the total amount of Pd 4d electrons. The amount of transferred electrons from Pd 4d was calculated by integrating the deviation region marked as A in Fig. 1d. The total amount of Pd 4d electrons can be calculated by integrating the entire Pd VB (green dotted line in Fig. 1d). Then, the value of the transferred electron (or created hole) ratio is approximately 2% (Supplementary Methods and Supplementary Table 2). Using the same method for the B region and C region and comparing the entire HKUST-1 VB (blue dotted line in Fig. 1d), the values related to B and C are similar, 6% (Supplementary Methods and Supplementary Table 2), which confirms the acceptance of electrons in HKUST-1 and the reduction of Cu2+ to Cu+. Considering the fluctuations arising from the standard deviation of the spectrum and the energy resolution, it is reasonable that roughly 4 ± 2% of the electrons are transferred from Pd 4d bands to Cu–O hybridization bands. The hydrogen capacity of Pd@HKUST-1, improved due to the charge transfer behaviour, can be evaluated semi-quantitatively using the calculated Pd 4d DOS (Supplementary Methods and Supplementary Fig. 9). The integrated DOS is considered to correspond to the ten electrons in the 4d bands for each palladium atom11. Once one electron is transferred from the Pd 4d band, corresponding to a 10% reduction in the DOS, the donor palladium atom performs as Pd+. In Pd@HKUST-1, the 4 ± 2% electrons transferring from Pd 4d suggest the Pd valence in Pd@HKUST-1 as Pd0.4+, which creates holes in Pd 4d bands for the additional hydrogen absorption of 0.4H per Pd atom. It is similar to the amount of hydrogen absorption, 0.5H per Pd atom5. The summed two contributions from the bands below and above the Fermi level in Pd@HKUST-1 lead to the increased hydrogen capacity that is approximately twice that of Pd nanocubes.
This mechanism implies that we can improve the hydrogen storage of hybrid materials by enlarging the interfacial charge transfer between the d-electron transition metal and the organic MOF, the latter of which contains transition metal-oxygen bands. Increasing the specific interface area will favour the hydrogen storage by enhancing the charge transfer at the interface. Moreover, charge transfer behaviour is not only relevant to hydrogen storage, but also to other properties such as catalytic activity. For example, the binding energy shift of Zr 3d in a Cu/Zr-based MOF catalyst implies that the enhanced the conversion of CO2 to methanol with 100% selectivity is related to the similar charge transfer behaviour3. The temperature at half conversion (T50) of composites of Pt and various kinds of MOFs for CO was found to be related to the different binding energy shift of Pt 4f7/24. A Pt/Fe-based MOF composite with organic groups of different selectivities featured the binding energy shifts of Pt 4f, Fe 2p and O 1s2, which indicated the same mechanism as that of this work. This charge transfer mechanism confirms the considerable potential of transition metals and MOFs as new heterogeneous catalytic materials. For the hydrogen storage or catalyst behaviour, there are two advantages in metal/MOF composites: (1) the new interface between a d-electron transition metal and a metal oxide provides more flexible unoccupied bands for chemical reactions; (2) the pore structure of MOF makes the interface area exposing to the reaction gas directly because of no solid materials covering on the interface. On the basis of these features, we can develop the new metal/oxide interface for the hydrogen storage and catalyst activity. For this purpose, more advanced technologies are required to realize the sharp interfacial structures, the smaller size of metal nanoparticles, the thinner MOF between nanoparticles, and the homogeneous coverage. Such nanostructures would enable to improve effective synergistic functionality of the metal/MOF materials, such as activation energy of hydrogen, carbon dioxide emission and so on. This understanding will drive progress in the creation of new functional materials for hydrogen storage, catalysis and other applications.
In summary, we compared the observed VB of Pd@HKUST-1 with the physical mixing spectrum of the Pd nanocubes and HKUST-1 using the Kerkhof–Moulijn model for quantitative analysis. There is a deviation between the observed and the simulated spectrum. This implies the interaction between the Pd nanocubes and MOF (in particular, Cu and O in MOF) at the interface. To understand deviation, we calculated the DOS of Pd@HKUST-1 composite in the framework of the density functional theory and evaluated the deviation of the interfacial Pd 4d, Cu 3d and O 2p DOS from the internal DOS, which were consistent with the HAXPES results. Furthermore, NEXAFS results provided evidence for an interaction between Cu 3d and O 2p. In conclusion, our results imply that charge transfer occurs from Pd 4d bands to the hybridization bands of Cu and O in HKUST-1 at the Pd/HKUST-1 interface, which causes the enhanced hydrogen storage in the hybrid material Pd@HKUST-1 compared with that of the Pd nanocubes.
Methods
HAXPES
HAXPES measurements were performed at the National Institute for Materials Science (NIMS) beamline BL15XU at SPring-8. A high-resolution hemispherical electron analyser (VG Scienta R4000) was used. The photon energy was set at 5.95 keV. The angle between the objective lens-axis of the analyser and photon propagation direction was fixed at 90° in the horizontal plane. The total energy resolution was estimated to be 0.24 eV by measuring the Fermi cut-off of an evaporated Au thin film. The binding energy referred to the Fermi level of the Au film. For spectral measurements of the nanomaterials, a commercial carbon powder was mixed with the samples to enhance the conductivity. The commercial carbon was also measured as referenced background and subtracted from the spectra later.
NEXAFS
NEXAFS data were recorded at BL12, SAGA Light Source. Commercial equipment (ULVAC–PHI ESCA 1600) with a total electron yield mode and a fluorescence yield mode was used to collect spectrum. The total electron yield mode probed the materials of the order of several nanometres and was used for studying of nanomaterials. The probing depth for the fluorescence yield mode was of the order of 100 nm, and this was used for the bulk reference samples. The photon energy resolution was ~0.1 eV.
Calculation
Calculations were performed with the DMol3 software package based on density functional theory35. The generalized gradient approximation with Perdew-Burke-Ernzerhof functional was used to describe the exchange-correlation interaction36. Double numerical plus polarization basis sets were employed. Core electrons were treated by the effective core potential of Stuttgart-Dresden group37. The convergence criteria for energy, maximum force, and displacement were set as 2 × 10−5 Ha, 0.004 Ha/Å, and 0.005 Å, respectively. Atomic charges were estimated using Hirshfeld population method38. A periodic Pd slab model with three layers of (100) planes with a (4 × 5) unit cell including sixty palladium atoms was used. The ligand, 1,3,5-benzenetricarboxylate (BTC), in the HKUST-1 was simplified to 1,4-benzenedicarboxylate (BDC). Then, HKUST-1 was simplified to C32H16O16Cu6. For the interface model of Pd@s-HKUST-1, there was a Pd:Cu:O:C ratio equal to 12:4:8:4 at the interface, unlike the Cu:O:C ratio of 2:8:4 in HKUST-1.
Samples
Except for the main samples, there are also samples as reference included for the data analysis. During HAXPES experiment, we observed Cu bulk, CuO and Cu2O powders as the reference samples. During NEXAFS experiment, we observed CuO, Cu2O, PdO, and benzene-1,3,5-tricarboxylate (BTC) powders as the reference samples. The Cu bulk was standard sample prepared by the synchrotron beamline BL15XU, SPring-8. CuO, Cu2O and PdO, and BTC powders were all purchased from Aldrich company. CuO powders are nanoparticles with particle size below 50 nm. The purity of the anhydrous Cu2O powders is ≥99.99%. The purity of PdO powders is ≥99.97%. The purity of BTC is above ≥99.99%.
Data availability
The datasets that support the findings of the current study are available from the corresponding author on reasonable request.
Change history
30 October 2018
The previously published version of this Article contained an error in Fig. 1. In panel c, the thickness of the HKUST-1 layer (3 nm) was incorrectly labelled. This error has been corrected in both the PDF and HTML versions of the Article.
References
Zhu, Q.-L. & Xu, Q. Metal–organic framework composites. Chem. Soc. Rev. 43, 5468–5512 (2014).
Zhao, M. et al. Metal–organic frameworks as selectivity regulators for hydrogenation reactions. Nature 539, 76–80 (2016).
Rungtaweevoranit, B. et al. Copper nanocrystals encapsulated in Zr-based metal–organic frameworks for highly selective CO2 hydrogenation to methanol. Nano Lett. 16, 7645–7649 (2016).
Yoshimaru, S., Sadakiyo, M., Staykov, A., Kato, K. & Yamauchi, M. Modulation of catalytic activity on Pt nanoparticles through charge-transfer interaction with metal–organic frameworks. Chem. Commun. 53, 6720–6723 (2017).
Li, G. et al. Hydrogen storage in Pd nanocrystals covered with a metal–organic framework. Nat. Mater. 13, 802–806 (2014).
Falcaro, P. et al. Application of metal and metal oxide nanoparticles@MOFs. Coord. Chem. Rev. 307, 237–254 (2016).
Schlapbach, L. & Züttel, A. Hydrogen-storage materials for mobile applications. Nature 414, 353–358 (2001).
Kobayashi, H. et al. On the nature of strong hydrogen atom trapping inside Pd nanoparticles. J. Am. Chem. Soc. 130, 1828–1829 (2008).
Li, G. et al. Shape-dependent hydrogen-storage properties in Pd nanocrystals: which does hydrogen prefer, octahedron (111) or cube (100)? J. Am. Chem. Soc. 136, 10222–10225 (2014).
Lu, Z., Wei, S.-H. & Zunger, A. Electronic structure of random Ag0.5Pd0.5 and Ag0.5Au0.5 alloys. Phys. Rev. B 44, 10470 (1991).
Turchi, P., Drchal, V. & Kudrnovsky, J. Stability and ordering properties of fcc alloys based on Rh, Ir, Pd, and Pt. Phys. Rev. B 74, 064202 (2006).
Wicke, E., Brodowsky, H. & Züchner, H. Hydrogen in palladium and palladium alloys. In Hydrogen in metals II. 73-155 (Springer, Berlin Heidelberg 1978).
Kobayashi, H., Mitsuka, Y. & Kitagawa, H. Metal nanoparticles covered with a metal–organic framework: from one-pot synthetic methods to synergistic energy storage and conversion functions. Inorg. Chem. 55, 7301–7310 (2016).
Chui, S. S.-Y., Lo, S. M.-F., Charmant, J. P., Orpen, A. G. & Williams, I. D. A chemically functionalizable nanoporous material [Cu3(TMA)2(H2O)3]n. Science 283, 1148–1150 (1999).
Furukawa, H., Cordova, K. E., O’Keeffe, M. & Yaghi, O. M. The chemistry and applications of metal-organic frameworks. Science 341, 1230444 (2013).
Duke, A. S. et al. Active sites in copper-based metal–organic frameworks: understanding substrate dynamics, redox processes, and valence-band structure. J. Phys. Chem. C 119, 27457 (2015).
Nanba, Y., Tsutsumi, T., Ishimoto, T. & Koyama, M. Theoretical study of the hydrogen absorption mechanism into a palladium nanocube coated with a metal–organic framework. J. Phys. Chem. C 121, 14611–14617 (2017).
Takata, Y. Hard X-ray photoemission spectroscopy. In Very high resolution photoelectron spectroscopy. 373-397. (Springer, Berlin Heidelberg 2007).
Chen, J. G. NEXAFS investigations of transition metal oxides, nitrides, carbides, sulfides and other interstitial compounds. Surf. Sci. Rep. 30, 1–152 (1997).
Kerkhof, F. & Moulijn, J. Quantitative analysis of XPS intensities for supported catalysts. J. Phys. Chem. 83, 1612–1619 (1979).
Tanuma, S., Powell, C. & Penn, D. Calculations of electron inelastic mean free paths. IX. Data for 41 elemental solids over the 50 eV to 30 keV range. Surf. Inter. Anal. 43, 689–713 (2011).
Miho, Y., Kobayashi, H. & Kitagawa, H. Hydrogen storage mediated by Pd and Pt nanoparticles. Chem. Phys. Chem. 10, 2566–2576 (2009).
Heinemann, M., Eifert, B. & Heiliger, C. Band structure and phase stability of the copper oxides Cu2O, CuO, and Cu4O3. Phys. Rev. B 87, 115111 (2013).
Wang, Y. et al. Electronic structures of Cu2O, Cu4O3, and CuO: a joint experimental and theoretical study. Phys. Rev. B 94, 245418 (2016).
Chen, H., Wang, L., Yang, J. & Yang, R. T. Investigation on hydrogenation of metal–organic frameworks HKUST-1, MIL-53, and ZIF-8 by hydrogen spillover. J. Phys. Chem. C 117, 7565–7576 (2013).
Yang, A. et al. The valence band structure of AgxRh1−x alloy nanoparticles. Appl. Phys. Lett. 105, 153109 (2014).
Trzhaskovskaya, M., Nefedov, V. & Yarzhemsky, V. Photoelectron angular distribution parameters for elements Z = 1 to Z = 54 in the photoelectron energy range 100–5000 eV. At. Data Nucl. Data Tables 77, 97–159 (2001).
Scofield, J. H. Theoretical photoionization cross sections from 1 to 1500 keV. No. UCRL–51326, (California University, Livermore, Lawrence Livermore Lab, 1973).
Hammer, B. & Nørskov, J. K. Theoretical surface science and catalysis–calculations and concepts. Adv. Catal. 45, 71–129 (2000).
Xu, J. et al. X-ray absorption spectra of graphene and graphene oxide by full-potential multiple scattering calculations with self-consistent charge density. Phys. Rev. B 92, 125408 (2015).
Li, C. et al. The organic-moiety-dominated Li+ intercalation/deintercalation mechanism of a cobalt-based metal–organic framework. J. Mater. Chem. A 4, 16245–16251 (2016).
Han, R. et al. Toward interfacing organic semiconductors with ferromagnetic transition metal substrates: enhanced stability via carboxylate anchoring. Chem. Commun. 52, 9805–9808 (2016).
Mogi, M. et al. Near-edge X-ray absorption fine structure of PdO at O K-edge. Jpn. J. Appl. Phys. 44, 4057–4059 (2005).
Yamauchi, M., Ikeda, R., Kitagawa, H. & Takata, M. Nanosize effects on hydrogen storage in palladium. J. Phys. Chem. C 112, 3294–3299 (2008).
Delly, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 113, 7756 (2000).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Dolg, M., Wedig, U., Stoll, H. & Preuss, H. Energy-adjusted Ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 86, 866 (1987).
Hirshfeld, F. L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 44, 129–138 (1977).
Acknowledgements
The HAXPES measurements were performed at the NIMS Synchrotron X-ray Station at SPring-8 under proposal nos. 2014B4906, 2016B4910 and 2017A4910 as part of the project NIMS Nanotechnology Platform (Project Nos. A-14-NM-0116, A-16-NM-0131 and A-17-NM-0011). We would like to thank Dr. Shigenori Ueda for the helpful discussion. We would also like to thank Hiroshima Synchrotron Orbital Radiation, Hiroshima University, and JAEA/SPring-8 for the use of the HAXPES setup at the NIMS BL15XU beamline of SPring-8. The NEXAFS measurements were performed at BL12, SAGA Light Source (No. 1711115S) with help from Eiichi Kobayashi and at BL SINS, Singapore Synchrotron Light Source with help from Xiaojiang Yu and Xiao Chi. This work was supported by Kakenhi Hojyokin (OS: Grants-in-Aid for BasicResearch (C), No. 15K04616). This work was also supported by CoreResearch for Evolutional Science and Technology (CREST) and ACCEL (JPMJAC1501) from the Japan Science and Technology Agency (JST).
Author information
Authors and Affiliations
Contributions
Y. C. and O. S. managed the project. G. L., H. Kobayashi, and H. Kitagawa prepared the samples. O. S., L. S. R. K., A. Y., and C. S. performed the HAXPES experiments. Y. C. and O. S. performed the NEXAFS experiments. Y. N. and M. K. performed the DOS calculation. Y. C. analysed all the experimental data. Y. C. and O. S. wrote the manuscript with contribution from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Sakata, O., Nanba, Y. et al. Electronic origin of hydrogen storage in MOF-covered palladium nanocubes investigated by synchrotron X-rays. Commun Chem 1, 61 (2018). https://doi.org/10.1038/s42004-018-0058-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-018-0058-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
