
Abstract
Maternal care acts as a strong environmental stimulus that can induce phenotypic plasticity in animals and may also alter their microbial communities through development. Here, we characterize the developmental metatranscriptome of the small carpenter bee, Ceratina calcarata, across developmental stages and in the presence or absence of mothers. Maternal care had the most influence during early development, with the greatest number and magnitude of differentially expressed genes between maternal care treatments, and enrichment for transcription factors regulating immune response in motherless early larvae. Metatranscriptomic data revealed fungi to be the most abundant group in the microbiome, with Aspergillus the most abundant in early larvae raised without mothers. Finally, integrative analysis between host transcriptome and metatranscriptome highlights several fungi correlating with developmental and immunity genes. Our results provide characterizations of the influence of maternal care on gene expression and the microbiome through development in a wild bee.
Similar content being viewed by others


Introduction
Parental care is an evolutionary adaptation in many species that has a profound impact on an individual’s development and species evolution1. Various forms and levels of parental care exist across mammals2,3, birds4,5, fish6,7, and insects8,9, directly enhancing the fitness of offspring by providing protection from predators and parasites, prolonged access to food supply, shelter, and social interactions10,11. Moreover, parental care has marked impacts on immunity and microbiome, such as the transfer of core microbes essential for offspring survival as observed in earwigs12, burying beetles13, African clawed frogs14, and mice15. The neurobiological and behavioral effects of parental care have been widely researched in social species, as it plays an essential role in the evolution of social behavior16,17. Abnormalities in behavior and brain chemistry have also been documented in other social mammals. Studies in rats have shown that low maternal care levels (characterized by low licking and grooming behavior) are linked to higher levels of stress reactivity in offspring18. In birds, where biparental care is prevalent, even the deprivation of one parent may result in permanent effects on the hypothalamic–pituitary–adrenal (HPA) axis, responsible for stress response and regulation of physiological processes5.
In particular, social insects are powerful model systems to explore the intricacies of parental care in the development of offspring. Bees demonstrate a highly diverse range of social behaviors, from obligately eusocial colonies with reproductive queens and foraging workers to solitary nests in which mothers do all tasks alone19. Such differences in social behavior can exert considerable influence on an organism’s microbiome and gene expression. Eusocial species typically receive their microbiome through horizontal transmission from nestmates, whereas solitary species acquire theirs horizontally from the wider environment20. Furthermore, facultatively social bee offspring reared with or without parental care display changes in gene expression patterns indicating that parental care can drive regulatory processes at the molecular level in offspring. For example, the adult small carpenter bee Ceratina calcarata showed more aggression and avoidance when reared without a mother, a behavioral change associated with changes in DNA methylation and gene expression patterns21.
Social and solitary insects also display differences in development. Obligately eusocial insects possess distinct castes, produced during development through differences in larval diet and developmental gene regulatory networks, ultimately resulting in different microbiome compositions for each caste22,23. While developmental castes are not present in facultatively social and solitary bees, recent studies have found that they exhibit clear changes in microbiome composition and richness across developmental stages24,25. Additionally, bees undergo complete metamorphosis and experience drastic changes in microbiome composition during defecation at the prepupal stage, which empties the gut, leading to an altered adult microbiome25. Gut microbiomes host a suite of taxa belonging to diverse groups such as bacteria, fungi, protists, viruses, and parasites (e.g., nematodes and arachnids) that individually and collectively impact host health and recently discovered important associations to host behavior via the gut–brain axis26. Microbiomes are, in turn, shaped by the environment and stressors associated with the host, which includes the influence of maternal care. For instance, breastfed infants develop healthy microbiomes supporting stronger immunological defenses and improved neurodevelopment27. In insects, earwigs reared without mothers had a reduced microbiome and suffered greater mortality than offspring obtaining essential anti-fungal bacteria from their mothers12. Hence, studies on the bee’s gut microbiome demonstrate an important role of microfauna in the healthy development of bee immunity28, digestion and nutrition29, behavior and cognitive ability30, reflecting the importance of host–microbiome species interactions25,31.
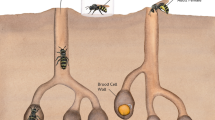
Our model species, C. calcarata is a subsocial bee, which is characterized by prolonged maternal care, overlapping generations, and parent-offspring interactions8,32,33. The methylome, genome, and multiple transcriptomes of this species have been sequenced34,35,36. C. calcarata mothers are nest loyal, typically creating a single nest each year after emerging from overwintering in natal nests to mate in the spring32. Mothers excavate a linear tunnel in the pith of dead plant stems and sequentially lay one egg at a time on a freshly collected pollen ball before closing a cell by creating a wall from pith shavings32. As offspring develop, mothers continue to guard the nest and inspect the developing brood by breaking down and reconstructing cell walls and are, therefore subsocial32. This is done until offspring reach adult maturity, then they will continue to guard and feed adult offspring until late summer or fall36. Maternal care via guarding prevents fungal and bacterial outbreaks throughout offspring development (Fig. 1). While maternal care effects were explored in this species at the adult stage via gene expression patterns in the brain21, and its microbiome was explored through development25, we do not know what the changes in gene expression patterns are across this species’ development, nor do we know how maternal care impacts its developmental microbiome. This model species offers a natural experiment for studying the implications of maternal care for the connected phenomena of development and microbiome diversity.

a Mothers remain in nests throughout offspring development until adulthood, providing cleaning and nest guarding (care). b Absence of mothers results in dirtier nests due to overabundance of fungi (no-care). Illustration created in full by Katherine Odanaka.
Here we characterize the developmental transcriptome of C. calcarata and experimentally compare how the presence or absence of maternal care impacts gene expression patterns across development. Second, we examine these same offspring’s microbiomes across development and between maternal care groups. Finally, we integrate gene regulatory networks and co-expression with metatranscriptomic data across development and care groups to identify key candidate genes in offspring of various ages impacted by the absence of maternal and modified microbiomes. We predict dysregulation of offspring gene expression and altered metagenomic microbial communities in the absence of maternal care. This study provides unique insights into our understanding of offspring development, maternal care, stress, and immune response not only important for offspring health but also parental care in animals, including intergenerational inheritance more broadly.
Results
Developmental transcriptome
Four generalized clusters of time course gene expression across C. calcarata offspring development were identified, summarizing the 19 individual stages into four overall developmental stages: (1) early larvae, (2) late larvae, (3) pupae, and (4) callows (Figs. 2 and S1; Supplementary Data 4). Overall, in 1957 unique differentially expressed genes (DEGs) were identified based on developmental stage. Early larval development had 340 DEGs uniquely upregulated when compared to other stages (Supplementary Data 5; Fig. S2). We also implemented multiple analyses (WGCNA37,38, DESeq239,40, and MaSigPro41) to further identify recurrently well-supported DEGs (i.e., robust DEGs) for each stage as these DEGs likely play a critical role in development, which resulted in 156 robust DEGs in early larvae that include nucleolar GTP-binding protein 1 (non1), nucleolar protein 58 (nop58), and eukaryotic translation initiation factor 3 subunit C (eIF3-S8) (Supplementary Data 4). Enriched GO terms for all early larvae upregulated genes related to cell growth and processes, such as TORC2 signaling, regulation of the cell cycle, physiological development, and morphogenesis (Supplementary Data 5). Significant transcription factor (TF) enrichment determined by STREME42 found no significant motif enrichment in early larvae when compared to other developmental stages (Supplementary Data 27). Out of the 383 upregulated DEGs unique to late larvae, 37 robust DEGs encoded kinesin-like proteins, such as KLP61F and KIF18A, essential cytoskeletal motor proteins (Supplementary Data 5 and 11). DEGs upregulated in late larvae were enriched for several cell cycle regulatory pathways and digestive and metabolic processes (Supplementary Data 6). Further analysis using WGCNA network clustering revealed late larval DEGs formed three significant clusters enriched for physiological developmental processes and metabolic processes, but also stress response pathways (e.g., detoxification, cellular response to stress; Supplementary Data 12). Late larval DEGs were enriched for motifs accommodating TFs mainly belonging to C2H2 zinc finger and SMAD factors (Supplementary Data 27), three of which were also identified as DEGs and include mothers against dpp (mad), TF GAGA (trl), and pair-rule protein odd-paired (opa) (Fig. 3). GO enrichment of late larval motifs were mainly involved with cell differentiation and gene regulatory processes whereas TF DEGs were mainly involved with embryogenesis, development, and neurodevelopment (Fig. 3, Supplementary Data 28).

The four overall developmental stages include early larvae, late larvae, pupae, and callow (see Fig. S1 for reference).

PCA plot of the four main developmental stages of C. calcarata in the presence of maternal care (care) and in the absence of maternal care (no-care). Ellipses for care and no-care groups are shown for each developmental stage.
The pupal stage had 1010 uniquely upregulated DEGs, with 113 robust DEGs that include genes involved with nervous system development such as leucine-rich repeat-containing protein 24 (LRRC24), neural-cadherin (CadN), and Down syndrome cell adhesion molecule-like protein (Dscam2; Supplementary Data 5). GO enrichment for pupal development is related primarily to protein modification and neurodevelopmental processes such as central nervous system neuron development and neuron fate determination (Supplementary Data 6 and 12). Pupal DEGs were enriched for a single TF motif that binds a C2H2 zinc finger, ZNF610 (Supplementary Data 27), which harbors diverse biological roles such as segmentation and oogenesis, and several molecular functions including protein binding and exonuclease activity (Supplementary Data 28). Callows had 224 uniquely upregulated DEGs and 78 robust DEGs, some of which encode mitochondrial enzymes, which include isocitrate dehydrogenase subunit betaC mitochondrial (IDH3B), NADH dehydrogenase iron-sulfur protein 42 C mitochondrial (NDUFS4), and isocitrate dehydrogenase subunit gamma C mitochondrial (IDH3G) (Supplementary Data 5). GO enrichment all upregulated DEGs in callows related to eye development and structural organization (Supplementary Data 6), wing disc and head development, sensory perception, and several biosynthetic and metabolic processes (Supplementary Data 12). Unique callow DEGs were focused on RNA metabolism (e.g., tRNA metabolic processes) and protein transport (Supplementary Data 6).
Effects of maternal care on the developmental transcriptome
To experimentally assess the effects of maternal care on the developmental transcriptome, we performed transcriptional analysis on care and no-care developing brood. Overall, when care and no-care conditions were compared across all stages, 558 DEGs were unique to care, and 685 were unique to no-care (Supplementary Data 7). Cared-for bees were enriched for immunity, metabolism, and developmental processes, whereas no-care bees were mostly related to cellular development, cellular responses, and regulatory pathways (Supplementary Data 8). Comparisons between all care and no-care individuals revealed motif enrichment for a significant motif enriched only in cared individuals that binds the TF button-head (btd), which is involved with head segmentation (Supplementary Data 27). Clustering of two principal components (PC1 49% and PC2 17%) explained 66% of the variance in expression across the developmental stages in the presence and absence of maternal care (Fig. 3). No-care treatments resulted in differential gene expression (>4000 DEGs) between care and no-care conditions at each developmental stage (Figs. 4 and S3; Supplementary Data 9). In total, 4953 unique DEGs (Supplementary Data 9) were identified across all four stages and maternal care groups, of which 898 were uniquely identified as hub genes in our WGCNA network analysis (Supplementary Data 13).

a Early larvae, b late larvae, c pupae, and d callow stage of C. calcarata in the presence and absence of maternal care. The differentially expressed genes with log2 fold change greater than 1 are shown in red.
Early larvae had 3755 upregulated DEGs due to differences in maternal care, with more DEGs identified in no-care early larvae (Fig. 4a). Negative binomial and network analyses identified venom allergen 3 (involved in the immune response) and digestion inhibitory genes such as chymotrypsin inhibitor and lysosomal aspartic protease to be upregulated in no-care early larvae (Supplementary Data 9 and 13). Inhibition of peptidases such as chymotrypsin has been shown to interfere with insect metabolism43, whereas proteases are involved with insect immunity against pathogenic invasions44. Conversely, defensin 1 was upregulated in care early larvae, an important gene for antimicrobial peptides in insects45 (Supplementary Data 9). Gene Ontology (GO) revealed genes upregulated in the care early larvae were enriched for immune processes (e.g., natural killer cell activation, phagocytosis, and T cell activation). No-care early larvae were instead enriched for protein movement and reproductive purposes (e.g., meiotic cell cycle, embryo development, and protein targeting; Supplementary Data 10). GO enrichment for the top three significant modules from WGCNA results for each maternal care group, and stage showed that maternal care presence leads to regulatory pathways focused on cellular development and gene regulation, but also immune defenses and metabolism (e.g., response to bacterium and monosaccharide metabolic process; Supplementary Data 14). WGCNA modules in no-care early larvae were conversely enriched for antigen processing, epithelial cell proliferation, and several catabolic processes (Supplementary Data 14). Furthermore, among all maternal care+stage group comparisons, significant TFs were only detected in no-care early larvae DEGs which were enriched for a motif that binds to the chromatin-linked adapter for MSL proteins (CLAMP) and protein hairy (h) (Table 1; Supplementary Data 27). GO enrichment of this motif is related to several developmental processes, such as brain development and chromatin assembly (Supplementary Data 28).
No-care late larvae had 206 upregulated DEGs (Fig. 4b), including genes involved with homeostatic compensation mechanisms such as potassium voltage-gated channel protein (shab). DEGs upregulated in late larvae were enriched for reproductive processes and physiological development, including mesoderm development and rhythmic process (Supplementary Data 10). DEGs upregulated in no-care late larvae were enriched for ion transmembrane transport, protein import, and regulation of transporter activity (Supplementary Data 10). Care and no-care late larvae shared biological processes enriched for light detection, catabolic processes, and stress response (Supplementary Data 14).
Pupae reared without mothers had 506 upregulated DEGs, including LRRC15, involved in insect immune response46 (Fig. 4c). GO enrichment of upregulated DEGs included protein phosphorylation in care pupae or protein dephosphorylation in no-care pupae (Supplementary Data 10), whereas network clustering found identical modules in care and no-care pupae enriched for compound eye development, digestive system development, and regulation of mitotic cell cycle (Supplementary Data 14).
Among callow offspring, 622 DEGs were upregulated in no-care, including hexamerin, which is known to be involved in immune response47, and calcium/calmodulin-dependent protein kinase II (CAMKII), known to be involved with learning and memory48 (Fig. 4d). GO enrichment for care callows include protein modification processes and several physiological developmental processes such as mesoderm development/formation, exocrine system development, salivary gland development, and gland morphogenesis (Supplementary Data 10). No-care callows GO enrichment include terms such as RNA modification, tRNA modification, response to hormone, and secretion (Supplementary Data 10). Furthermore, GO enrichment of WGCNA network clustering for care and no-care callow DEGs revealed major physiological development, such as wing disc development, and several metabolic and biosynthetic processes (Supplementary Data 14). However, WGCNA analysis also revealed that care callows were enriched for unique physiological processes such as locomotion and metamorphosis, whereas no-care callows were uniquely enriched for cell response pathways such as cAMP-mediated signaling and sensory perception of pain (Supplementary Data 14). Interestingly, no significant motif enrichment was identified in callows in any of the developmental stage comparisons or stage + care group comparisons. Finally, RFC models using transcriptomic data revealed that gene expression data was better at predicting offspring based on their developmental stage (average accuracy of 94%; Supplementary Data 19), rather than the maternal care group (average accuracy of 71.6%; Fig. 5a; Supplementary Data 18).

RFCs assign samples either to the overall developmental stage or maternal care group using either a gene expression data or b metatranscriptomic abundance data. RFC training was done from 10% to 90% of samples withheld. Models using gene expression data, on average, performed better in classifying samples to developmental stages or care groups compared to models using metatranscriptomic data (all domains included). Error bars were generated using the loess smoothing curve method. Full results for gene expression data are shown in Supplementary Data 18 and 19 for the maternal care group and overall developmental stage, respectively. Full results for metatranscriptomic data are shown in Supplementary Data 20 and 21 for the maternal care group and developmental stage, respectively.
Effects of development and maternal care on the microbiome
Metatranscriptomic analyses using metaSPADES49 identified 842 microbial genera across developmental stages and care groups. The most dominant microbial group is fungi, contributing to approximately 85% of the microbiome composition, followed by bacteria (8%) based on contig abundance (Supplementary Data 15). Absence of mothers resulted in a significant difference in taxa composition (Bray–Curtis PERMANOVA F = 7.72, df = 3, p = 0.001, Fig. 6) and alpha diversity across developmental stages (Shannon ANOVA F = 1.26, df = 3, p = 8.52e−09; Fig. 6; Supplementary Data 16). Metatranscriptomic data performed better in predicting the maternal care group (average accuracy of 70%; Supplementary Data 20) rather than the developmental stage (average accuracy of 61%; Fig. 5b; Supplementary Data 21), even when using bacterial, fungal, or bacterial+fungal genera only (Fig. S4; Supplementary Data 20 and 21). Furthermore, there is a striking difference in microbiome composition from fully grown larvae and prepupae stages across all individuals (Fig. 6) and within groups (Fig. S5).

See Supplementary Data 2 individual for developmental stage names.
Maternal presence significantly affected alpha diversity (Shannon ANOVA F = 16.75, df = 1, p < 0.01) and community composition (Bray–Cutis PERMANOVA F = 10.43, df = 1, p = 0.001) (Supplementary Data 17) during early larvae development, with greater diversity in no-care offspring. Aspergillus was the top fungal genus that dominated the community composition in early larvae in both care and no-care groups (Supplementary Data 22). However, Aspergillus’ relative abundance was significantly greater in no-care individuals (28%) relative to offspring reared with mothers (16%; two-tailed t-test t = −4.58, df = 38, p < 0.01). Aspergillus was also identified in SIMPER50 analysis as a top 10 contributor to metagenomic composition dissimilarity (Supplementary Data 23) and was the only fungus identified as a top 10 feature during random forest classification (RFC) model testing for maternal care influence when using all taxon groups using the randomForest package in R51 (Supplementary Data 20). Other top genera based on relative abundance and identified by SIMPER were also fungi, including Ascosphaera and Paecilomyces, which also had greater abundances in no-care early larvae (Supplementary Data 22 and 23). Negative binomial distribution analysis (NBDA) obtained from DESeq2 analysis corroborated our earlier findings, with significant overrepresentation of fungi in early larvae (Supplementary Data 24), as well as a significant WGCNA cluster of hub fungi in no-care early larvae (Supplementary Data 26). In late larvae, taxa composition and richness did not show significant differences between care groups (Supplementary Data 17). Indeed, the top genus, Aspergillus, was not significantly different in abundance between no-care and care individuals (two-tailed t-test t = 1.27, df = 48, p = 0.212).
Pupal taxa richness was significantly different from the larval stages but were not different from callows (Supplementary Data 16). Interestingly, while pupae defecate and lose most of their gut microbiota, there was a significant difference in taxa richness and composition between care groups (Supplementary Data 17), with different top genera depending on maternal care presence (Supplementary Data 22). Aspergillus and Ascosphaera were significantly overrepresented in care pupae and suggest maternal presence may be reintroducing fungal taxa during the pupal stage (Supplementary Data 24). WGCNA network analysis found the top fungal cluster from the larval stages also evident in pupae (Fig. 7, Supplementary Data 25).

a Shows each developmental stage (two modules for early larvae, one for late larvae, one for pupal, and two for callow). In b, hub genera are highlighted in dark gray, and c the module compositions are colored by domain. Nodes are sized according to whether they are a hub genus (but also colored in diagram B).
The top genera in callows were more diverse in taxa richness, and composition differed significantly between care groups (ANOVA, F = 11.69, df = 1, p < 0.001; Bray–Curtis PERMANOVA F = 2.36, df = 1, p = 0.015; Supplementary Data 17). For instance, callows in the presence of mothers were dominated by a mix of fungi (e.g., Aspergillus and Ascosphaera), viruses (e.g., Nepovirus and Betacoronavirus), and protists (e.g., Gregarina and Plasmodium; Supplementary Data 22). Conversely, no-care callows were mostly dominated by bacteria, including notable genera such as Pseudomonas, Acinetobacter, and Burkholderia. There was no significant overrepresentation of taxa between callow care groups or between callows and other developmental stages (Supplementary Data 24). Interestingly, WGCNA network analysis found unique but smaller modules for callows, which included clusters of primarily bacteria and protists (Fig. 7; Supplementary Data 26). As such, callows differed the most in module clustering and composition (Fig. 7). The presence of mothers contributes to slightly greater taxa abundances, as suggested by hub genera identified in cared callows but not in uncared callows (Supplementary Data 25).
Comparison and integration of gene expression and metatranscriptomic data
We used the mixOmics52,53 R package to run an integrative correlation analysis of gene expression and metatranscriptomic abundance data which revealed that Ascosphaera held the majority of top positive correlations in cared-for offspring, primarily in pupal and callow developmental stages (Fig. 8a). Ascosphaera correlated with mitochondrial respiratory genes and enzymes (i.e., UQCRFS1, IDH3B, IDH3G) in cared pupae, and correlated with ribosomal developmental genes (i.e., non1, nop16, and nop58) in callows. Similarly, top positive correlations in no-care individuals also identified Ascosphaera as a central fungus correlating with several developmental genes, but primarily across the larval and pupal stages (Fig. 8b). Surprisingly, instead of several strong correlations between Aspergillus and developmental genes in no-care early larvae, Aspergillus only correlated with a few genes involved with mitotic development (i.e., FBXL7, ppan, and fam136a) in pupae, and the mitochondrial enzyme IDH3B and the glycotransferase PIGM in early larvae. Callows were the most different, with genes correlating with the viruses Betacoronavirus and Nepovirus, and the protist Plasmodium, reiterating previous results that a change in microbiome composition is most notable at the callow stage.

Correlations shown are at least the top 15 positive or negative correlations throughout the developmental stages and are separated into a positive correlations for care, b positive correlations for no-care, c negative correlations for care, and d negative correlations for no-care offspring. Connections between nodes are colored to indicate the developmental stage, and the thickness of the line indicates the strength of the correlation.
Among the top negative correlations in cared individuals, Paecilomyces was the top fungus with the most negative correlations across all four developmental stages (Fig. 8c). Similar to Paecilomyces, Fusarium also negatively correlated with several genes across the four stages, whereas the remaining fungi primarily correlated with only one or two developmental stages. In contrast, Ascosphaera held the most negative correlations in no-care offspring, with correlations to ribosomal and mitochondrial genes mainly in early larvae (Fig. 8d). All fungi negatively correlated with genes from no-care early to pupal individuals, whereas protists and viruses negatively correlated with developmental genes primarily in no-care callows.
Discussion
This study characterized the regulation of gene expression and the metatranscriptome of C. calcarata across development and in the presence or absence of maternal care. The developmental transcriptome reveals gene expression according to development and maternal care, with a stronger baseline gene expression profile attributed to development. We found that early and late larval stages exhibit a diverse microbiome with fungi predominating. In the absence of maternal care, early larvae are most susceptible to fungal and bacterial overabundance, whereas later developmental stages have a much-reduced microbiome. Finally, we found interesting correlations between fungal taxa and candidate bee developmental genes that are potentially involved with immunity. To our knowledge, our study is the first to provide metatranscriptomic insights into the relative role of maternal care on offspring development and a foundational framework for the developmental microbiome, a critical component of bee health.
Time course developmental gene expression of C. calcarata
Genes encoding ribosomal protein subunits, nucleolar proteins, and translation initiation TFs were found to be upregulated during the early larval stage of C. calcarata. Ribosome genesis is central to larval development in insects and has an important role in early developing endoderm of zebrafish54,55. Ribosomal proteins are essential during periods of cell proliferation and development56. Additionally, kinesin-like protein-encoding genes central to normal mitotic processes (KLP61F) are upregulated in late larvae. The disruption of such genes has been shown to result in larval lethality57.
Neurodevelopmental genes are upregulated in the pupal stage, such as NLGN1, LRRC24, and cadN, which are central to synapse function and cognition58,59,60. Additionally, the upregulated genes include an essential TF (sox15), which is mainly expressed in external sensory organs’ socket cells and has a role in the proper electrophysiological function of Drosophila melanogaster’s mechanosensory organs61. Tramtrack (Ttk), which is an important transcriptional repressor for pupal Drosophila neurodevelopment and has a role in caste differentiation of ants, is upregulated in pupal C. calcarata62,63. Overall, pupal gene expression found across C. calcarata is mainly involved in neurodevelopment and mechanosensation.
During the callow stage, there are several upregulated genes involved in oxidative phosphorylation, including cox6a1 (cytochrome c oxidase subunit 6A1) and NDUFA7 (NADH mitochondrial complex I dehydrogenase 1 alpha subcomplex subunit 7). Flight muscles of D. melanogaster are rich in mitochondria64; the upregulation of mitochondrial enzyme-encoding genes indicates the essential role of wing development in newly emerged C. calcarata callow. Overall, upregulated callow developmental genes are mainly involved in wing muscle development.
Across all developmental stages, TF enrichment was most often associated with the late larval stage and consisted mainly of C2H2 zinc finger TFs. C2H2 zinc finger TFs are involved with growth and play a role in stress response65, which may be indicative of rapid development that occurs prior to pupation32 and a sensitivity to external stimuli as demonstrated by GO enrichment of late larval motifs relating to signal transduction, cell communication, and several physiological developmental processes.
Pupae were enriched for ZNF610, which has a putative role in DNA methylation66,67. Insect pupae are sensitive to external stimuli (e.g., temperature), which can epigenetically alter gene expression resulting in polyphenism. While DNA methylation has been linked to caste differentiation in social bees (i.e., honeybee queens and workers), the lack of caste differentiation in solitary bees suggests that DNA methylation plays a more prominent role in the regulation of gene expression and transcription68. Finally, the lack of TF enrichment in callows compared to other developmental stages or between callow maternal care groups advocates for similar, basic biological processes that occur at the adult stage post-development.
Maternal care has the strongest effect on gene expression during early larvae development
Our results indicate that maternal care deprivation affects gene expression across the development of C. calcarata, most significantly during early larval development. In this stage, with the absence of maternal care, upregulated genes encode trehalase, venom acid phosphatase (acph-1), and venom allergen 3. Trehalase (which converts trehalose to glucose) is upregulated in the absence of maternal care, as found in response to increased oxidative stress in mice69. Additionally, venom acid phosphatase is a major allergen in Apis mellifera and is a component of bee venom, which has a protective property in bees70,71. These results suggest that the absence of mothers during early development is associated with genes involved in mediating immune and oxidative stress responses. Parental care has been shown to play an important role in egg survivorship and early larval development in other insects with facultative parental care. For instance, European earwig mothers will increase their care load depending on pathogen exposure within their nests, accordingly, such as returning to nests more often for egg care72. In burying beetles (Nicrophorus vespilloidesi), larval survivorship and growth rates were significantly higher in the presence of parental care13.
On the other hand, among downregulated genes is eukaryotic translation initiation factor 3 subunit E (eIF3-S6) and various ribosomal protein subunits. These results indicate that maternal care deprivation could be linked to downregulating genes essential for translation initiation and potentially affect cell proliferation and development. Additionally, no-care early larvae were the only group+stage with DEGs enriched for a significant motif that binds to a C2H2 zinc finger and a zipper-type TF, with enriched roles in TF activity, brain, and eye development. While we might expect stressed offspring to show term enrichment directed to immunity, regulation of transcription activity is itself a response to stress and serves as a defensive mechanism by regulating several genes in order to maintain cellular integrity, prioritize development, or changes in cellular behavior73.
Several other genes demonstrated changes in expression due to maternal care, but a notable change in expression occurred in a potassium voltage-gated channel protein (shab) which was upregulated in no-care late larval samples. It has been shown to be upregulated as a compensatory mechanism in response to loss of Ca2+-dependent K+ channel conductance in D. melanogaster74. It could be that shab upregulation is part of a homeostatic mechanism because of maternal care deprivation. Pupae reared without mothers had upregulated genes, such as LRRC15, whose transcription has been shown to be enhanced in response to neonicotinoids in honeybees75. There were also many more significantly upregulated DEGs in no-care callows, which include hexamerin, a gene found to be upregulated in the leafhopper Circulifers haematoceps as part of an immune response and bacterial infection controlling mechanism47. Additionally, CAMKII is upregulated in no-care conditions; upregulation of this gene is known to result in memory impairment in Drosophila48.
RFC testing demonstrated that gene expression was highly accurate in predicting the developmental stage of offspring when compared to predicting the maternal care group, suggesting that while mothers do play a role in gene regulation during early larval development, the absence of mothers does not arrest the full development of C. calcarata offspring.
Fungi pose a risk of developing early larvae without maternal care
We expected a change in metatranscriptomic composition across development and between maternal care groups, and we observed a significant difference in taxa richness, specifically between early and late larvae versus pupal stages. Interestingly, we found that while transcriptomic data groups prepupae with late larvae based on gene expression, microbiome composition shows a greater similarity between prepupae and the pupal stages. This difference is likely due to prepupal gene expression patterns most similar to late larval stages as it is a pre-metamorphic state25, however, defecation begins in prepupae32, and emptying of the gut would result in a similar, decreased microbiome composition in prepupae and in pupae.
Our RFC models showed that the metagenome is slightly better at predicting the maternal care group rather than the developmental stage for offspring, highlighting the importance of nest cleaning and rebuilding of nest cell walls by mothers32 to keep fungi and bacteria at a minimum. Studies have shown that Ceratina nests without mothers had a greater percentage of infested cells and greater brood mortality76.
The fungi Aspergillus consistently showed up as a highly abundant and persistent genus in C. calcarata in the early larval stages. Aspergillus was dominant in early larvae samples reared without mothers, along with other plant-associated and potentially pathogenic fungi such as Paecilomyces and Ascosphaera. Aspergillus is associated with environmental diseases such as plant molding and food rot77. The most well-known disease by Aspergillus in bees is stonebrood disease, in which Aspergillus flavus infects honeybee larvae causing mortality and mummification78. Ascosphaera, a fungus that causes chalkbrood disease in honey bees79, was also found overrepresented in no-care early larvae. Although direct effects of Aspergillus or other fungal pathogens on C. calcarata larval mortality remain to be tested, Aspergillus is known to cause pollen spoilage and increased mortality in solitary ground-nesting Nomia alkali bees80 and Diadasia chimney bees81, and the strain Ascosphaera proliperda caused chalkbrood disease in Megachile leaf-cutting bees and Osmia mason bees82. It is evident that Ceratina mothers actively keep their brood cells clean to avoid the spread of fungal pathogens and pollen spoilage32,76 and that this active cleaning appears to be most important in the early developmental stages (Fig. 6).
Key correlating genes with fungi
Integrative analyses revealed that Ascosphaera and Paecilomyces consistently harbored the most associations with host offspring gene expression across the four developmental stages and between care groups. Ascosphaera was the central fungus with the most positive correlations in both care and no-care treatments, but interestingly there were many more correlations between Ascosphaera and developmental genes when mothers were present. This suggests that even in the presence of mothers, offspring are reacting to a fungal and highlights the need for future studies to explore the pathogenic effects of Ascosphaera on offspring survivorship in C. calcarata. In callows, Ascosphaera correlated with pitchoune (pit), potentially involved with ribosomal biogenesis and protein synthesis83, and in pupae with isocitrate dehydrogenase IDH3B, involved with amino acid metabolism84; these genes were not positively correlated with Ascosphaera under no-care conditions. Although the effects of Ascosphaera are unclear in solitary wild bees, it is known to infect the later larval and pupal stages of honey bees by germinating in their digestive tract and mummifying larvae and pupae85. We speculate that Ascosphaera induces a stress response in C. calcarata in cared and no-cared larvae and pupae, leading to increased energy production and mitotic and ribosomal development to offset developmental stress. Conversely, Ascosphaera positively correlated with the cellular respiratory gene UQCRFS1 in pupal stages in cared treatments, but a lack of mothers resulted in the same correlation during the late larval stages. The UQCRFS1 gene is involved with energy production, a process that is often increased under stress conditions86, and this corroborates the results found in honey bee larvae infected with Ascosphaera apis, where energy metabolism was elevated87. Furthermore, in no-care offspring, Ascosphaera negatively correlated with several ribosomal developmental genes in the early larval stage. This finding suggests that in a stressed environment, ribosomal biogenesis—which requires abundant energy metabolism - is diminished. This might be due to increased energy expenditure in other processes in no-care early larvae, such as the positive correlation between Ascosphaera and RBKS, a gene involved with carbohydrate metabolism. Indeed, bees feed on a carbohydrate rich diet, and C. calcarata early larvae are known to feed constantly on their pollen mass provision allowing them to develop into pupae in little under a month32. Future functional work on solitary bee offspring is needed to better understand how offspring respond genetically and physiologically when faced with Ascosphaera infections, with or without maternal presence, as our results suggest that certain fungi may be adept at surviving in nests despite cleaning and guarding by mothers.
Paecilomyces is a fungus found in soil and decaying plant material and produces secondary metabolites that can be toxic to insects88. As such, the presence of Paecilomyces still induces a stress response even in brood reared with mothers. For instance, while Paecilomyces negatively correlated with Dscam2—which has a role in neuronal development89 and immunity90—in no-care late larvae, it also negatively correlated with Dscam2 in cared-for late larvae, suggesting that irrespective of maternal care, regulation of immunity genes in offspring are faced with a cost due to fungal stressors. Additionally, Paecilomyces negatively correlated with other neurodevelopmental genes in care and no-care late larvae suggesting that neurodevelopment is deficient in the presence of Paecilomyces and may lead to nervous system dysfunction, as was found to occur in ghost moth larvae infected with P. hepiali91. Future functional studies on fungal infections during development will be necessary to fully understand how these fungi impact non-apid bee survival and how maternal presence may alleviate the stress of fungal stressors.
Whereas previous studies focused only on the effects of maternal care in adult Ceratina gene expression21 or only on the Ceratina developmental microbiome without maternal absence25, our study uniquely analyzes the changes in gene expression patterns and microbiome composition across developmental stages in the presence and absence of maternal care. Arsenault et al.21 found that the removal of mothers during the larval stage resulted in more aggression and avoidance in adults as a result of changes in gene expression patterns, with notable changes in genes related to neuronal function and metabolism. Although we did not explore behavior in our study, we found that maternal care had the greatest impact during early larvae development with the greatest number and magnitude of DEGs, also involved primarily with neurodevelopment, but also involved ribosome biogenesis. Microbiome analyses corroborated the finding that maternal care plays an important role during early larvae development, as fungal pathogens such as Aspergillus, had the greatest abundance in the absence of maternal care, but was much reduced in offspring with mothers present. This differs from Nguyen et al.25, which did not find Aspergillus as a dominant fungus in their Ceratina samples but did find other fungi to be prominent, which were also found in high abundance in our study, such as Ascosphaera and Penicillium. Machine learning analyses of the full metatranscriptomic dataset found that gene expression was better able to predict offspring developmental stage than maternal care group, whereas the opposite was true for microbial communities, reinforcing the utility of hologenomic datasets to examine the relative roles of genes and environment. Potential interdependence between host gene expression and microbiome datasets highlights important interactions, such as the fungi Ascosphaera and Paecilomyces, as prominent stressors and the role of maternal care in maintaining development. This study provides foundation developmental and microbiome characterizations of a wild bee and network syntheses to develop hologenomic insights into early childhood development and health.
Methods
Brood rearing and sample collection
C. calcarata nests were collected from sumac and raspberry stems in Toronto, Canada (43.7735° N, 79.5019° W). Care and no-care treatments were raised in the respective presence and absence of maternal care to each of the 19 individual developmental stages based on Rehan and Richards32 (Supplementary Data 2). Following the protocol design in Arsenault et al.21, care and no-care offspring were reared in the lab at 23 °C and 50% relative humidity to each of the focal developmental stages before assay with and without the presence of mothers, respectively. All samples were flash-frozen in liquid nitrogen and stored in a −80 °C freezer.
RNA extractions, library construction, and sequencing
RNA was extracted using Qiagen RNeasy mini kit from whole body samples of five individuals from each stage in both care and no-care treatments. In total, 190 samples of larvae, pupae, and adults (from 140 nests) were sequenced in this study. For this experiment, there were 95 care group samples and 95 no-care group samples used. The pupae and adult samples used were all females. RNA samples were then sent to Genome Quebec for library preparation using the TruSeq DNA PCR-free method and paired-end sequencing using the Illumina NovaSeq 6000 platform. All samples were sequenced to an average depth of approximately 33× coverage to 100 bp (Supplementary Data 3). The reads were then aligned to the C. calcarata reference genome (PRJNA791561) using STAR92. Adapter contamination was less than 0.2% in reads and STAR accounts for poor quality ends of reads during alignment.
Differential gene expression analysis
Normalized counts and DEGs across developmental stages and between care and no-care treatments were obtained using DESeq239,40. Heatmaps were generated using pheatmap92,93 in R based on variance-stabilizing transformed data from the DESeq2 package. PCA plots were generated based on variance-stabilized transformed data. To explore changes in gene expression for DEGs between care and no-care groups, volcano plots were generated using EnhancedVolcano package94. As recommended in maSigPro user’s guide41, EdgeR was used to obtain normalized library counts without any CPM filtering in order to keep all genes for analysis95. To survey gene expression over the developmental stages, these data were then inputted into maSigPro to obtain generalized linear models in which the adjusted p-value is less than 0.05. Additionally, maSigPro uses a linear step-up Benjamini–Hochberg false discovery rate procedure, in which the adjusted p-value is less than 0.0541,96. A higher regression model was used (degree = 18) to account for the 19 individual developmental stages over time, with a backward stepwise regression to identify gene profile differences. Gene clusters obtained from maSigPro were used to generate generalized linear plots, which allowed the 19 individual developmental stages to be merged into four overall developmental stages (early larvae, late larvae, pupae, and callows) that will be used for further analysis (Supplementary Data 2, Figs. 2 and S1). The GO enrichment of the gene lists obtained using DESeq2 and maSigPro were then functionally annotated using TopGO with enriched terms identified by Fisher’s exact tests with a p-value less than 0.0597.
Gene overrepresentation and cluster analysis
To identify key co-occurring DEGs, normalized gene expression data for the complete dataset (characterized by stage and maternal care group) from DESeq2 was used for weighted gene co-expression analysis (WGCNA) using the wgcna R package37,38. The samples were clustered using the hclust distance method and using a soft thresholding power of 6, module size of 50, and unsigned network type, WGCNA analysis was conducted, and no outlier samples were detected37,38 (Fig. S6A). In this study, we have focused on the top three significant modules (based on the highest positive correlation values and p < 0.05) for each stage and group. GO enrichment for these genes was obtained as well. Genes were classified as a hub gene in the module if the absolute gene significance value (GS) was≥0.2 and absolute module membership values (MM) were ≥0.9 according to cut-off values implemented in previous studies utilizing WGCNA98,99; the MM cut-off was increased to 0.9 from 0.8.
Taxonomic classification
Unmapped transcriptomic reads were used to characterize the holobiome of each offspring and for the taxonomic classification of microbial communities. First, metaSPADES (version 3.10.1), a package from the SPAdes toolkit49 was used to obtain contigs from the reads. Contig assembly was done for paired-end reads using default kmer settings (-k 21, 33, 55, 77). Contig files for 190 samples were produced, then BLASTed using blastn100 (version 2.12.0) against the non-redundant nucleotide nt database, setting parameters -max_target_seqs and -max_hsps to 1 in order to extract only the top alignment hit for each contig query. We removed contigs shorter than 200 bp and contigs mapping to environmental bacterial contaminating genera Sodalis or Wolbachia101 (3). Next, NCBI Entrez Direct E-Utilities efetch (version 15.3) (https://www.ncbi.nlm.nih.gov/books/NBK25499/) was used to extract full lineage information for the remaining contigs (e.g., kingdom, phylum, class, order, family, genus, species) based on the BLAST hit taxonomy ids (i.e., ‘taxids’). Contigs were kept if they matched any one of the six groups of interest at the genus level: arachnida, bacteria, fungi, nematoda, protista, and viruses. Then, we further filtered the contigs using a minimum relative abundance threshold of 0.1% within a sample for each group of interest. Genera were considered top taxa for each category (i.e., developmental stage + care group) if they were above 1% relative contig abundance for that category.
Diversity analyses
We tested for significant differences in genus dissimilarity and richness using Bray–Curtis dissimilarity and Shannon-Wiener diversity indices, respectively. First, we used the R package vegan102 to test Bray–Curtis dissimilarity on the metatranscriptomic dataset, transformed using the total method in DECOSTAND, to infer any significant differences across the four developmental stages (early larvae, late larvae, pupal, callow). PERMANOVA via the ADONIS2 method from vegans was used to determine if the resulting dissimilarity was significant between stages. To ensure PERMANOVA was not violated due to unequal variation among groups, the BETADISPER method was used to test for the significance of the PERMANOVA result; it is violated if an ANOVA test on the BETADISPER result is significant. If valid, Tukey’s Honest Significant Difference (HSD) via the TUKEYHSD method was implemented to identify groups that were significantly different from each other. Next, we ran Shannon-Wiener to test for significant differences in alpha diversity across all developmental stages. ANOVA via the AOV method was used to determine the significance of alpha diversity, followed by Tukey’s HSD to identify significantly different groups. We also ran the Kruskal–Wallis test on Shannon–Wiener alpha indices as a second non-parametric test to identify significantly different groups. First, Bartlett’s test of variance using the BARTLETT.TEST method was implemented to ensure variance across samples was equal. If not significant, the Kruskal–Wallis using the KRUSKAL.TEST method was applied. If significant, Dunn’s test using DUNNTEST from the R package FSA103 was used to identify significantly different groups. Next, we ran the above analyses to test for significant differences in dissimilarity and alpha diversity for each stage between maternal care groups (e.g., care: early larvae vs. no-care: early larvae). Finally, we ran a similarity percentage (SIMPER) using Bray–Curtis dissimilarities in PAST50 (version 4.06) to identify the top 10 taxa contributing to observed differences between overall developmental stage, maternal care groups, and between stage and maternal care groups.
Taxa overrepresentation and cluster analysis
We used an NBDA from the R package DESeq2 to identify genera that were significantly overabundant between care groups for each developmental stage (e.g., care: early larvae vs. no-care: early larvae), or between developmental stages only (e.g., early larvae vs. late larvae). This resulted in four comparisons for each stage and care group and six comparisons between stages only; this totaled 20 comparisons. Prior to analysis, DESeq2 requires that there be at least one variable with no zeros present in any of the samples. Every taxon in our metatranscriptomic data had at least one zero abundance value in at least one sample. To circumvent this, a pseudo taxon with a value of 1 across all 190 samples was added to allow DESeq2 to properly run and normalize the dataset. Next, the normalized dataset was used for WGCNA. Briefly, missing data or zero-variance data are checked and/or removed; this resulted in the removal of the pseudo taxon. Next, outlier samples were removed if they were more than five standard deviations from other samples via hierarchal clustering of samples; this resulted in the removal of three samples (J11-4, K11-2, and H120-9); the remaining 187 samples we kept for further analysis. Next, analysis was done on developmental stage (e.g., early larvae) or developmental stage + maternal care group (e.g., care: early larvae), which were one-hot coded to convert categorical terms into binary code where 1 indicates a sample is represented by the category and 0 indicating the sample is absent from the category. A range of powers was tested, and a soft-threshold power of 10 was selected, along with a network type set as signed and a minimum module size of 20 (Fig. S6B). All modules that had significant, positive correlations were recorded and analyzed further for hub taxa based on absolute GS ≥ 0.2 and absolute MM ≥ 0.8 using cut-offs obtained from studies98,99.
Random forest classification
The R package randomForest51 was used for random forest classification to test if gene expression and metagenomic data can clearly predict the developmental stage or maternal care group of offspring. For the metagenomic data, we trained several random forest classifiers (RFCs), which included the complete metatranscriptomic data (i.e., all six domains), only the bacterial domain, only the fungal domain, and a combined bacterial+fungal domain dataset, to predict either overall developmental stage or care group, RFC testing was also done on the normalized gene expression dataset. Prior to RFC on gene expression (20,825 genes), we filtered out DEGs with 15 or fewer counts, shrinking the data to 16,023 genes. The RFCs were trained using 9 training set sizes (10–90%, increasing by 10%), with each training set size run 10 times to obtain an average classification accuracy. This resulted in 90 trials per RFC. Before each RFC, we ran the TUNERF method to determine an optimal mtry value (used to determine the number of features to select for the RFC to minimize error) for 5000 runs (ntree = 5000). The R package caret104 was used for the confusion matrix, which summarizes the overall performance of the RFC. The R package randomForestExplainer105 was used to determine the top 10 important features (i.e., taxa or genes) that influenced the RFC model accuracy and was done for each trial run at the 80% training set size. Results summarized and compared between RFCs will focus on 80% of samples withheld.
TF enrichment
To identify candidate TF binding sites enriched upstream of DEGs, we implemented STREME42 from the MEME (Multiple Em for Motif-Elicitation) package (version 5.4.1) to find significantly enriched motifs 5kbp upstream of all unique DEGs for each stage, all upregulated DEGs for each stage comparison, all unique DEGs for each maternal care group, and finally, for all upregulated DEGs for each stage + group using an E-value cut-off of 0.05. Significant motifs from STREME were used in subsequent MEME suite packages for motif identification and enrichment. We used the TOMTOM106 webserver to identify matched motifs using the JASPAR107 non-redundant core 2022 database for vertebrates and insects separately using default settings. Then we implemented GOMO108 (Gene Ontology for Candidate Motifs) to assess the functional role of identified significant motifs using default settings.
Integration of gene expression and metatranscriptomic data
Finally, to detect any patterns or associations between the metagenome and host gene expression, we used the R package mixOmics52,53 to integrate metagenomic abundance data with gene expression data using sparse partial least squares (sPLS) regression analysis. The sPLS function integrates two datasets measured on the same individuals (e.g., host gene expression and metatranscriptomic data) to detect highly correlated (positive or negative) variables. We filtered the normalized host gene expression data (20,825 genes) to include only DEGs that had a log2foldC > 1. Next, we further filtered these DEGs to focus on hub DEGs from WGCNA that had a GS value > 0.5. This resulted in 32 top genes to be further cross-analyzed with metatranscriptomic data. Next, we filtered the metatranscriptomic genus abundance data to focus only on the top contributors based on SIMPER analyses. This totaled 11 genera. Then we split our filtered gene expression and metatranscriptomic datasets into samples for each developmental stage and group setting (e.g., care; early larvae). We performed sPLS on the filtered datasets for each development and group setting using ncomp = 2 (for two components), mode = regression (to fit a linear relationship between the variables), and near.zero.var = FALSE (since our filtered data is a small portion of the entire dataset and would have fewer zeros throughout). Correlation matrices between genes and taxa were obtained, and only the top 15 positive and negative correlations were extracted for network visualization using Cytoscape109 (version 3.9.1).
Statistics and reproducibility
This study examines transcriptomic and metatranscriptomic sequence data extracted from 190 C. calcarata individuals reared with or without maternal presence from early larval to callow stages. For comparisons between maternal presence and absence, in total, 95 individuals were reared with mothers, and 95 individuals were reared without mothers. A sample size of 5 individuals was used for each developmental stage and maternal care group (5 replicates per individual stage and care group), resulting in a total of 40 individuals reared for early larval development, 50 reared for late larval development, 90 reared for pupal development, and 10 reared to the callow stage. Methods for RNA extraction, library preparation, and sequencing, mapping to reference genome, extraction of transcriptomic and metatranscriptomic data, taxonomy classification, and TF enrichment are explained in the methods section.
All statistical analyses were performed in R. Testing the difference in Aspergillus abundance between care groups for early larvae, and late larvae was done using a two-tailed t-test, with the t.test function in R and var.equal set to TRUE. PERMANOVA for Bray–Curtis dissimilarity beta diversity was performed, followed by ANOVA for beta dispersion and Tukey HSD to determine significantly different stages and care groups. ANOVA for Shannon–Weiner alpha diversity estimates were also performed, followed by post hoc Tukey HSD to identify significantly different groups. In addition, Bartlett’s test was not significant on Shannon–Wiener alpha indices, so the Kruskal–Wallis test was used, followed by post hoc Dunn’s test to determine significantly different groups. Data were deemed significant when P values were less than 0.05.
Integration of gene expression and microbiome composition data identified important positive and negative correlations between genes and taxa for individuals reared with and without mothers across the developmental stages, using correlations obtained from the mixOmics R package, with details explained in Section “Integration of gene expression and metatranscriptomic data.”
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw reads for RNA sequencing are available on the NCBI Sequence Read Archive under BioProject PRJNA926970. Source data used to generate figures and results in this study are available within the paper in Supplementary Data 1 file.
Code availability
The code used to obtain results is available upon request.
References
Kuijper, B. & Hoyle, R. B. When to rely on maternal effects and when on phenotypic plasticity? Evolution 69, 950–968 (2015).
Rilling, J. K. & Young, L. J. The biology of mammalian parenting and its effect on offspring social development. Science 345, 771–776 (2014).
Stockley, P., & Hobson, L. Parental care and litter size coevolution in mammals. Pro. R. Soc. B https://doi.org/10.1098/rspb.2016.0140 (2016).
Cockburn, A. Prevalence of different modes of parental care in birds. Proc. R. Soc. B https://doi.org/10.1098/rspb.2005.3458 (2006).
Banerjee, S. B., Asterbery, A. S., Fergus, D. J., & Adkins-Regan, E. Deprivation of maternal care has long-lasting consequences for the hypothalamic–pituitary–adrenal axis of zebra finches. Proc. R. Soc. B https://doi.org/10.1098/rspb.2011.1265 (2011).
Goldberg, R. L., Downing, P. A., Griffin, A. S., & Green, J. P. The costs and benefits of paternal care in fish: a meta-analysis. Proc. R. Soc. B https://doi.org/10.1098/rspb.2020.1759 (2020).
Satoh, S., Hotta, T. & Kohda, M. Maternal care-providing cichlid Neolamprologus furcifer selectively focuses on high-threat carnivorous intruders, limiting attention to other threats. Front. Ecol. Evolut. https://doi.org/10.3389/fevo.2021.616810 (2021).
Tallamy, D. W. & Wood, T. K. Convergence patterns in subsocial insects. Annu. Rev. Entomol. 31, 369–390 (1986).
Wong, J. W. Y., Meunier, J. & Kölliker, M. The evolution of parental care in insects: the roles of ecology, life history and the social environment. Ecol. Entomol. 38, 123–137 (2013).
Royle, N. J., Smiseth, P. T. & Kölliker, M. The Evolution of Parental Care (eds N. J. Royle, P. T. Smiseth and M. Kölliker). (Oxford University Press, Oxford, UK, 2012).
Smiseth, P. T., Kölliker, M. & Royle, N. J. What is Parental care? The Evolution of Parental Care (eds N. J. Royle, P. T. Smiseth and M. Kölliker), pp. 1–17. (Oxford University Press, Oxford, UK, 2012).
Greer, J. A., Swei, A., Vredenburg, V. T. & Zink, A. G. Parental care alters the egg microbiome of maritime earwigs. Microb. Ecol. 80, 920–934 (2020).
Eggert, A., Reinking, M. & Muller, J. K. Parental care improves offspring survival and growth in burying beetles. Anim. Behav. 55, 97–107 (1998).
Poorten, T. J. & Kuhn, R. E. Maternal transfer of antibodies to eggs in Xenopus laevis. Develop. Comp. Immunol. 33, 171–175 (2009).
Myles, I. A., Pincus, N. B., Fontecilla, N. M. & Datta, S. K. Effects of parental omega-3 fattya cid intake on offspring microbiome and immunity. PLoS ONE 9, e87181 (2014).
Alexander, R. D. The evolution of social behavior. Annu. Rev. Ecol. Syst. 5, 325–383 (1974).
West-Eberhard, M. J. Wasp societies as microcosms for the study of development and evolution. In (eds West-Eberhard MJ, Turillazzi S). Natural History and Evolution of Paper-Wasps. (Oxford University Press, New York, 1996).
Champagne, F. A., Francis, D. D., Mar, A. & Meaney, M. J. Variations in maternal care in the rat as a mediating influence for the effects of environment on development. Physiol. Behav. 79, 359–371 (2003).
Michener, C. D. The Bees of the World. 2nd Edition. (John Hopkins University Press, Baltimore, 2007).
Kowallik, V. & Mikheyev, A. S. Honey bee larval and adult microbiome life stages are effectively decoupled with vertical transmission overcoming early life perturbations. mBio 12, e02966–21 (2021).
Arsenault, S. V., Hunt, B. G. & Rehan, S. M. The effect of maternal care on gene expression and DNA methylation in a subsocial bee. Nat. Commun. 9, 3468 (2018).
Kapheim, K. M. et al. Caste-specific differences in hindgut microbial communities of honey bees (Apis mellifera. PloS ONE 10, e0123911 (2015).
Smith, C. R., Anderson, K. E., Tillberg, C. V., Gadau, J., & Suarez, A. V. Caste determination in a polymorphic social insect: nutritional, social, and genetic factors. Am. Nat. https://doi.org/10.1086/590961 (2008)
Kapheim, K. M., Johnson, M. M. & Jolley, M. Composition and acquisition of the microbiome in solitary, ground-nesting alkali bees. Sci. Rep. 11, 1–11 (2021).
Nguyen, P. N. & Rehan, S. M. Developmental microbiome of the small carpenter bee, Ceratina calcarata. Environ. DNA 4, 808–819 (2022).
Weiner, A., Turjeman, S. & Koren, O. Gut microbes and host behaviour: the forgotten members of the gut-microbiome. Neuropharmacology 227, 109453 (2023).
Granger, C. L. et al. Maternal breastmilk, infant gut microbiome and the impact on preterm infant health. Acta Paediatr. 110, 450–457 (2020).
Lang, H. et al. Specific strains of honeybee gut Lactobacillus stimulate host immune system to protect against pathogenic Hafnia alvei. Am. Soc. Microbiol. 10, e0189621 (2022).
Kwong, W. K., Engel, P., Koch, H., & Moran, N. A. Genomics and host specialization of honey bee and bumble bee gut symbionts. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.1405838111 (2014).
Li, L. et al. Gut microbiome drives individual memory variation in bumblebees. Nat. Commun. 12, 6588 (2021).
Shell, W. A. & Rehan, S. M. Comparative metagenomics reveals expanded insights into intra- and interspecific variation among wild bee microbiomes. Commun. Biol. 5, 603 (2022).
Rehan, S. M. & Richards, M. H. Nesting biology and subsociality in Ceratina calcarata (Hymenoptera: Apidae). Can. Entomol. 142, 65–74 (2010).
Nalepa, C. A. Altricial development in subsocial cockroach ancestors: foundation for the evolution of phenotypic plasticity in termites. Evolut. Dev. 12, 95–105 (2010).
Rehan, S. M., Glastad, K. M., Lawson, S. P. & Hunt, B. G. The genome and methylome of a subsocial small carpenter bee, Ceratina calcarata. Genome Biol. Evolut. 8, 1401–1410 (2016).
Withee, J. R. & Rehan, S. M. Social aggression, experience, and brain gene expression in a subsocial bee. Integr. Comp. Biol. 57, 640–648 (2017).
Shell, W. A. & Rehan, S. M. Social modularity: conserved genes and regulatory elements underlie caste-antecedent behavioural states in an incipiently social bee. Proc. Biol. Sci. 286, 20191815 (2019).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008).
Langfelder, P. & Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 46, 1–17 (2012).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 500 (2014).
Zhu, A., Ibrahim, J.G., Love, M.I. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics https://doi.org/10.1093/bioinformatics/bty895 (2018).
Conesa, A., Nueda, M. J., Ferrer, A. & Talón, M. maSigPro: a method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics 22, 1096–1102 (2006).
Bailey, T. L. STREME: accurate and versatile sequence motif discovery. Bioinformatics 37, 28343–2840 (2021).
Samiksha, Singh, D., Kesavan, A. K. & Sohal, S. K. Exploration of anti-insect potential of trypsin inhibitor purified from seeds of Sapindus mukorossi against Bactrocera cucurbitae. Sci. Rep. 9, 17025 (2019).
Sojka, D., Hartmann, D., Bartošová-Sojková, P. & Dvořák, J. Parasite cathepsin D-like peptidases and their relevance as therapeutic targets. Trends Parasitol. 32, 708–723 (2016).
Erler, S., Popp, M. & Lattorff, H. M. G. Dynamics of immune system gene expression upon bacterial challenge and wounding in a social insect (Bombus terrestris). PLoS ONE 6, e18126 (2011).
Dolan, J. et al. The extracellular leucine-rich repeat superfamily; a comparative survey and analysis of evolutionary relationships and expression patterns. BMC Genom. 8, 320 (2007).
Eliautout, R. et al. Immune response and survival of Circulifer haematoceps to Spiroplasma citri infection requires expression of the gene hexamerin. Develop. Comp. Immunol. 54, 7–19 (2016).
Malik, B. R., Gillespie, J. M. & Hodge, J. J. CASK and CaMKII function in the mushroom body α‘/β‘ neurons during Drosophila memory formation. Front. Neural Circuits 7, 52 (2013).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Hammer, Ø., Harper, D. A. T. & Ryan, P. D. PAST: paleontological statistics software package for education and data analysis. Paleontol. Electron. 4, 1–9 (2001).
Liaw, A. & Wiener, M. Classification and regression by randomForest. R. N. 2, 18–22 (2002).
Lê Cao, K.-A., González, I. & Déjean, S. integrOmics: an R package to unravel relationships between two omics datasets. Bioinformatics 25, 2855–2856 (2009).
Rohart, F., Gautier, B., Singh, A. & Lê Cao, K.-A. mixOmics: an R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 13, e1005752 (2017).
Texada, M. J., Koyama, T. & Rewitz, K. Regulation of body size and growth control. Genetics 216, 269–313 (2020).
Provost, E., Weier, C. A. & Leach, S. D. Multiple ribosomal proteins are expressed at high levels in developing zebrafish endoderm and are required for normal exocrine pancreas development. Zebrafish 10, 161–169 (2013).
Warner, J. R. The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24, 437–440 (1999).
Heck, M. M. et al. The kinesin-like protein KLP61F is essential for mitosis in Drosophila. J. Cell Biol. 123, 665–679 (1993).
Südhof, T. C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911 (2008).
Katzman, A. & Alberini, C. M. NLGN1 and NLGN2 in the prefrontal cortex: their role in memory consolidation and strengthening. Curr. Opin. Neurobiol. 48, 122–130 (2018).
Schroeder, A. & de Wit, J. Leucine-rich repeat-containing synaptic adhesion molecules as organizers of synaptic specificity and diversity. Exp. Mol. Med. 50, 1–9 (2018).
Miller, S. W., Avidor-Reiss, T., Polyanovsky, A. & Posakony, J. W. Complex interplay of three transcription factors in controlling the tormogen differentiation program of Drosophila mechanoreceptors. Develop. Biol. 329, 386–399 (2009).
Badenhorst, P., Finch, J. T. & Travers, A. A. Tramtrack co-operates to prevent inappropriate neural development in Drosophila. Mech. Dev. 117, 87–101 (2002).
Glastad, K. M., Ju, L. & Berger, S. L. Tramtrack acts during late pupal development to direct ant caste identity. PLoS Genet. 17, e1009801 (2021).
Rhooms, S.-K., Murari, A., Goparaju, N. S. V., Vilanueva, M. & Owusu-Ansah, E. Insights from Drosophila on mitochondrial complex I. Cell. Mol. Life Sci. 77, 607–618 (2020).
Guo, H.-J. et al. Identification of an Apis cerana zinc finger protein 41 gene and its involvement in the oxidative stress response. Insect Biochem. Physiol. 108, e21830 (2021).
Patel, A., Hashimoto, H., Zhang, X. & Cheng, X. Chapter seventeen: characterization of how DNA modifications affect DNA binding by C2H2 zinc finger proteins. Methods Enzymol. 573, 387–401 (2016).
Zhou, C. et al. DNA methylation biomarkers for head and neck squamos cell carcinoma. Epigenetics 13, 398–409 (2018).
Duncan, E. J. Phenotype plasticity: what has DNA methylation got to do with it? Insects 13, 110 (2022).
Mizunoe, Y. et al. Trehalose protects against oxidative stress by regulating the Keap1-Nrf2 and autophagy pathways. Redox Biol. 15, 115–124 (2018).
Hossen, M. S., Shapla, U. M., Gan, S. H. & Khalil, M. I. Impact of bee venom enzymes on diseases and immune responses. Molecules 22, 25 (2016).
Grunwald, T. et al. Molecular cloning and expression in insect cells of honeybee venom allergen acid phosphatase (Api m 3). J. Allergy Clin. Immunol. 117, 848–854 (2006).
Diehl, J. M. C. & Meunier, J. Surrounding pathogens shape maternal egg care but not egg production in the European earwig. Behav. Ecol. 29, 128–136 (2017).
de Nadal, E., Ammerer, G. & Posas, F. Controlling gene expression in response to stress. Nat. Rev. Genet. 12, 833–845 (2011).
Kim, E. Z., Vienne, J., Rosbash, M. & Griffith, L. C. Nonreciprocal homeostatic compensation in Drosophila potassium channel mutants. J. Neurophysiol. 117, 2125–2136 (2017).
Di Prisco, G. et al. Neonicotinoid clothianidin adversely affects insect immunity and promotes replication of a viral pathogen in honey bees. Proc. Natl Acad. Sci. USA 110, 18466–18471 (2013).
Sakagami, S. F. & Maeta, Y. Some presumably presocial habits of Japanese Ceratina bees, with notes on various social types in Hymenoptera. Insectes Sociaux 24, 319–343 (1977).
Amaike, S. & Keller, N. P. Aspergillus flavus. Annu. Rev. Phytopathol. 49, 107–133 (2011).
Vojvodic, S., Jensen, A. B., James, R. R., Boomsma, J. J. & Eilenberg, J. Temperature dependent virulence of obligate and facultative fungi pathogens of honeybee brood. Vet. Microbiol. 149, 200–205 (2011).
Aronstein, K. A. & Murray, K. D. Chalkbrood disease in honey bees. J. Invertebr. Pathol. 103, S20–S29 (2010).
Batra, S. W. T. & Bohart, G. E. Alkali bees: response of adults to pathogenic fungi in brood cells. Science 165, 607–607 (1969).
Linsley, E. G. & MacSwain, J. W. Notes on some effects of parasitism upon a small population of Diadasia bituberculata (Cresson). Pan-Pac. Entolomol. 28, 131–135 (1952).
Wynns, A. A., Jensen, A. B. & Eilenberg, J. Ascosphaera callicarpa, a new species of bee-loving fungus, with a key to the genus for Europe. PLoS ONE 8, e73419 (2013).
Zaffran, S. et al. A Drosophila RNA helicase gene, pitchoune, is required for cell growth and proliferation and is a potential target of d-Myc. Development 125, 3571–3584 (1998).
Zhang, S. et al. Effects of Lysiphlebia japonica (Ashmead) on cotton–melon aphid Aphis gossypii Glover lipid synthesis. Insect Mol. Biol. 24, 348–357 (2015).
Qin, X., Evans, J. D., Aronstein, K. A., Murray, K. D., & Weinstock, G. M. Genome sequences of the honey bee. pathogens Paenibacillus larvae and Ascosphaera apis. Insect Mol. Biol. https://doi.org/10.1111/j.1365-2583.2006.00694.x (2006).
Killiny, N., Hijaz, F., Ebert, T. A. & Rogers, M. E. A plant bacterial pathogen manipulates its insect cector’s energy metabolism. Appl. Environ. Microbiol. 83, e03005–e03016 (2017).
Li, L. et al. Changes in antioxidant enzymes activity and metabolomic profiles in the guts of honey bee (Apis mellifera) larvae infected with Ascosphaera apis. Insects 11, 419 (2020).
Asaff, A., Cerda-García-Rojas, C. & de la Torre, M. Isolation of dipicolinic acid as an insecticidal toxin from Paecilomyces fumosoroseus. Appl. Microbiol. Biotechnol. 68, 542–547 (2005).
Zhu, K., Xu, Y., Liu, J., Xu, Q. & Ye, H. Down syndrome cell adhesion molecule and its functions in neural development. Neurosci. Bull. 27, 45–52 (2011).
Armitage, S. A. O. et al. Dscam1 in pancrustacean immunity: current status and a look to the future. Front. Immunol. 8, 662 (2017).
Rao, Z. et al. Comparative transcriptome analysis of Thitarodes armoricanus in response to the entomopathogenic fungi Paecilomyces hepiali and Ophiocordyceps sinensis. Insects 11, 4 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Kolde, R. Pheatmap: pretty heatmaps. R Package Version 1.0.12. https://CRAN.R-project.org/package=pheatmap (2012).
Blighe, K., Rana, S., & Lewis, M. EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling. R package version 1.14.0. https://github.com/kevinblighe/EnhancedVolcano (2022).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300 (1995).
Alexa, A., & Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. R package version 2.46.0. (2021).
Wang, W. et al. Weighted gene co-expression network analysis of expression data off monozygotic twins identifies specific modules and hub genes related to BMI. BMC Genom. 18, 872 (2017).
Liu, Y. et al. Identification of hub genes and key pathways associated with bipolar disorder based on weighted gene co-expression network analysis. Front. Physiol. 10, 1081 (2019).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Graystock, P., Rehan, S. M. & McFrederick, Q. S. Hunting for healthy microbiomes: determining the core microbiomes of Ceratina, Megalopta, and Apis bees and how they associate with microbes in bee collected pollen. Conserv. Genet. 18, 701–711 (2017).
Oksanen, J. et al. Package ‘vegan’. Commun. Ecol. Package version 2, 5–7 (2020).
Ogle, D. H., Doll, J.C., Wheeler, P., & Dinno, A. FSA: Fisheries Stock Analysis. R package version 0.9.3, https://github.com/fishR-Core-Team/FSA (2022).
Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 28, 1–26 (2008).
Paluszynska, A., Biecek, P., Jiang, Y. randomForestExplainer: Explaining and Visualizing Random Forests in Terms of Variable Importance. R package version 0.10.1. https://CRAN.R-project.org/package=randomForestExplainer (2020).
Gupta, S., Stamatoyannopolous, J. A., Bailey, T. L. & Noble, W. S. Quantifying similarity between motifs. Genome Biol. 8, R24 (2007).
Castro-Mondragon, J. A. et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173 (2022).
Buske, F. A., Bodén, M., Bauer, D. C. & Bailey, T. L. Assigning roles to DNA regulatory motifs using comparative genomics. Bioinformatics 26, 860–866 (2010).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Wimmer, E. A., Jäckle, H., Pfeifle, C. & Cohen, S. M. A Drosophila homologue of human Sp1 is a head-specific segmentation gene. Nature 366, 690–694 (1993).
Bakke, J. et al. Transcription factor ZNF148 is a negative regulator of human muscle differentiation. Sci. Rep. 7, 8138 (2017).
Schneider, B. G. et al. Virulence of infecting Helicobacter pyloristrains and intensity of mononuclear cell infiltration are associated with levels of DNA hypermethylation in gastric mucosae. Epigenetics 8, 1153–1161 (2013).
Wiersdorff, V., Lecuit, T., Cohen, S. M. & Mlodzik, M. Mad acts downstream of Dpp receptors, revealing a differential requirement for dpp signaling in initiation and propagation of morphogenesis in the Drosophila eye. Development 122, 2153–2162 (1996).
Kim, J., Johnson, K., Chen, H. J., Carroll, S. & Laughon, A. Drosophila Mad binds to DNA and directly mediates activation of vestigial by Decapentaplegic. Nature 388, 304–308 (1997).
Urban, J. A., Urban, J. M., Kuzu, G. & Larschan, E. N. The Drosophila CLAMP protein associates with diverse proteins on chromatin. PLoS ONE 12, e0189772 (2017).
Omelina, E. S., Baricheva, E. M., Oshchepkov, D. Y. & Merkulova, T. I. Analysis and recognition of the GAGA transcription factor binding sites in Drosophila genes. Comput. Biol. Chem. 35, 363–370 (2011).
Hursh, D. A. & Stultz, B. G. Odd-paired: the Drosophila zic gene. Adv. Exp. Med. Biol. 1046, 41–58 (2018).
Zhou, D. et al. Mechanisms underlying hypoxia tolerance in Drosophila melanogaster: hairy as a metabolic switch. PLoS Genet. 10, e1000221 (2008).
Zhan, Y., Maung, S. W., Shao, B. & Myat, M. M. The bHLH transcription factor, hairy, refines the terminal cell fate in the Drosophila embryonic trachea. PLoS ONE 5, e14134 (2010).
Acknowledgements
This work was funded by a Mitacs Elevate Fellowship to KDC, NSERC USRA to M.S., Ontario Graduate Scholarship and Vernon Stong Fellowship to J.H., Weston Family Foundation Microbiome Initiative, NSERC EWR Memorial Steacie Fellowship and Discovery Grants to SMR. We thank Katherina Odanaka for the illustrations used in Fig. 1, Farida Samad-Zada for assistance with RNA extractions, and members of the Rehan lab for helpful feedback on earlier versions of this manuscript.
Author information
Authors and Affiliations
Contributions
S.M.R. conceived of the study. S.M.R. and M.S. designed the study. M.S. collected specimens, performed rearing experiments, and prepared samples for sequencing. K.D.C. and J.H. created the bioinformatics pipelines, M.S. analyzed all mapped reads for transcriptomic analysis, and K.D.C. analyzed all unmapped reads for metatranscriptomic analysis. J.H. analyzed mapped reads for transcription factor enrichment. K.D.C. and M.S. drafted the first version of the paper with contributions and edits from J.H. and S.M.R. All authors read and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Manuel Breuer and Luke Grinham.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chau, K.D., Shamekh, M., Huisken, J. et al. The effects of maternal care on the developmental transcriptome and metatranscriptome of a wild bee. Commun Biol 6, 904 (2023). https://doi.org/10.1038/s42003-023-05275-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-05275-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
