
Abstract
Environmental degradation has the potential to alter key mutualisms that underlie the structure and function of ecological communities. How microbial communities associated with fishes vary across populations and in relation to habitat characteristics remains largely unknown despite their fundamental roles in host nutrition and immunity. We find significant differences in the gut microbiome composition of a facultative coral-feeding butterflyfish (Chaetodon capistratus) across Caribbean reefs that differ markedly in live coral cover (∼0–30%). Fish gut microbiomes were significantly more variable at degraded reefs, a pattern driven by changes in the relative abundance of the most common taxa potentially associated with stress. We also demonstrate that fish gut microbiomes on severely degraded reefs have a lower abundance of Endozoicomonas and a higher diversity of anaerobic fermentative bacteria, which may suggest a less coral dominated diet. The observed shifts in fish gut bacterial communities across the habitat gradient extend to a small set of potentially beneficial host associated bacteria (i.e., the core microbiome) suggesting essential fish-microbiome interactions may be vulnerable to severe coral degradation.
Similar content being viewed by others
Introduction
Environmental degradation associated with the Anthropocene is threatening the persistence of mutualistic relationships that are key to the stability of ecological functioning1. The increasingly severe degradation of coral reefs from both local and climatic stressors has led to novel habitat states with conspicuously altered fish and invertebrate communities2,3,4,5, making them a model system for studying ecological responses to environmental change. A potentially pervasive but largely overlooked response to habitat degradation is the change to host-associated microbiomes—the communities of bacteria, archaea, fungi, unicellular eukaryotes, protozoa, and viruses that live on internal and external surfaces of reef organisms. Host microbiomes potentially respond faster than their hosts to changing environmental conditions and can promote acclimatization processes as well as genetic adaptation6,7,8,9,10. Thus, microbial communities could play a key role in mediating a host’s resilience and ability to adapt to environmental change. However, it remains to be explored whether mutualisms between fish hosts and gut microbiomes can shift to alternative beneficial relationships to provide a mechanism of resilience to habitat change, or whether the mutualism breaks down and simply reflects a cascading effect of degradation at all levels of ecological organization.
The importance of gut microbial communities in maintaining host health is well recognized in mammals and other vertebrates11,12, including a wealth of research into the importance of microbes in fish in aquaculture settings13,14,15,16. Fish harbor microbiomes that are unique from the microbial communities in their surrounding environment17,18. As the gut microbiome diversifies throughout the development of the fish host, a relatively stable gut microbiome is typically established within the first months of the fish’s life13. These resident (autochthonous) microbes, which are consistently found associated with the fish population across space and time and potentially provide critical functions for the host, are referred to as the “core microbiome”15,19,20. In contrast, the numerous microbes occurring in the gastrointestinal tract after being ingested are transient (allochthonous) and may vary intraspecifically with the developmental stage and potentially include opportunistic pathogens. Because of their importance in maintaining host metabolic homeostasis, the degree of stability of the core microbiome across a range of environmental conditions emerges as a key trait for predicting the resilience of host populations in aquatic animals21,22,23.
In coral reef fishes, recent studies have suggested that intestinal microbiomes influence key physiological functions associated with nutrient acquisition, metabolic homeostasis, and immunity24,25,26,27. For example, gut bacteria provide many herbivorous fish hosts with the ability to digest complex algal polymers24,26,27 and appear susceptible to human disturbances such as eutrophication28. The gut microbiome is also a major factor in the innate immune responses to a wide variety of pathogenic microorganisms and other stressors in the surrounding environment29,30. Given the rapid physical, chemical, and biotic changes affecting coral reefs, especially in the light of increasing mass coral bleaching events31, it is essential to gain a better understanding of how fish gut microbiome assemblages respond to environmental variation so that we can assess how these mutualisms govern host health and resilience to habitat change.
Here, we examined the variability and composition of the gut microbiome of the facultative coral-feeding foureye butterflyfish, Chaetodon capistratus (Linnaeus, 1758), inhabiting a set of reefs that differ markedly in coral cover and diversity across a tropical coastal lagoon (Bahía Almirante) at Bocas del Toro on the Caribbean coast of Panamá. The Chaetodontidae family (butterflyfishes) is among the largest and most iconic families of coral reef-associated fishes32 and an ideal group for studying microbiome responses to habitat degradation. Chaetodontids range from extreme diet specialists to facultative corallivores and generalists capable of consuming different types of prey such as corals, algae, polychaetes or crustaceans33,34,35. Due to their intimate link to the reef benthos, specialized coral-feeding species of Indo-Pacific butterflyfishes are highly sensitive to reductions in coral cover36,37,38. Chaetodon capistratus is the only one of the four Western Atlantic Chaetodon species with a relatively high proportion of anthozoans in its diet (mainly hard and soft corals)39,40,41. Because of this relative specialization, we chose it as a model species to study relationships between reef habitats and fish host gut microbiomes.
The Bahía Almirante encompasses an inner bay of protected reefs subjected to seasonally high temperatures and a watershed delivering nutrients from agriculture and sewage. In 2010, the bay faced an unprecedented hypoxic event, which led to massive coral bleaching and mortality on some sheltered reefs while others located near the bay’s mouth remained unaffected42. We capitalized on this gradient of habitat states across the bay to detect variation across fish gut microbiomes in relation to coral degradation. We hypothesized that fish residing on more degraded reefs (i.e., low live coral cover) have a more diverse and variable microbiome as a result of alternative feeding behaviors and potentially increased stress43. In contrast, given its role in sustaining host biological functions, we expected that the core microbiome would remain consistent across the habitat gradient.
Results
Benthic habitat and fish density
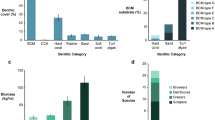
Reefs located within the three zones classified a priori as outer bay, inner bay, and inner bay disturbed (Fig. 1a), differed in terms of their benthic composition (PCoA; Fig. 2a) with marked differences in the level of live coral cover (Fig. 2b and Table S1). Live hard coral cover (Fig. 2b and Table S1) and coral diversity (Shannon diversity; Fig. S1) were highest on reefs of the outer bay. Both stony coral species (i.e., Acropora cervicornis and Agaricia tenuifolia) and fire corals (i.e., Millepora alcicornis, Millepora complanata) dominated at outer bay reefs. In the inner bay zone, reefs displayed an intermediate level of live coral cover (Fig. 2b and Table S1), largely dominated by the lettuce coral Agaricia tenuifolia. Sponges represented more than a quarter of the benthic cover on these reefs (Fig. S2 and Table S1). Live coral cover was lowest in the inner bay disturbed zone (Fig. 2b) where dead coral skeleton was prevalent together with sponges (Fig. S2 and Table S1). Our focal species Chaetodon capistratus showed significantly lower mean density levels at the outer bay than in the two inner bay zones (Fig. S3). Density levels were similar (1–5 individuals per 100 m2 transect) across all surveyed reefs inside of the bay apart from Cayo Hermanas (SIS, inner bay zone) where up to 25 individuals were recorded in one of the transects (Fig. S3).

a Map of the Bahía Almirante (Bocas del Toro, Panamá) indicating the position of the nine reefs where samples were collected (generated using GSHHG version 2.3.7 https://www.soest.hawaii.edu/pwessel/gshhg/). Data: Friederike Clever. b Outer bay reefs with highest levels of live coral cover, c inner bay reefs with intermediate levels of coral cover, and d reefs located in the inner bay disturbed zone were highly impacted by a hypoxic event in 2010. e The study species foureye butterflyfish (Chaetodon capistratus). Photographs by Matthieu Leray.

Composition and percent coral cover of benthic communities across nine reefs and three reef zones illustrating a habitat gradient: a PCoA representing dissimilarities in benthic community composition based on Bray–Curtis. Reefs are color-coded by reef zone, substrate groups are depicted in black; b percent live coral cover across reef zones from high coral cover at the outer bay to very low cover on disturbed reefs at the inner bay. Diamond shapes depict means.
Composition of the whole gut microbiome
A total of 5,245,987 high-quality reads were retained for subsequent statistical analyses. The number of reads per sample ranged from 10,369 to 79,466, with a mean ± SD of 41,307 ± 10,990 reads. We identified 10,711 different ASVs in the total dataset. The number of ASVs per sample ranged from 13 to 1,281, with a mean ± SD of 179 ± 210 ASVs. This dataset primarily comprised ASVs belonging to 15 bacterial phyla (Fig. S4a). As predicted, C. capistratus’ gut microbiome composition was distinct from the microbiome in seawater and the microbiome of potential prey items (sessile invertebrates) (Fig. S4a, b). Chaetodon capistratus’ overall gut microbiome was dominated by Proteobacteria (mainly Gammaproteobacteria, 68.6%) followed by Firmicutes (16.1%), Spirochetes (9.27%), Cyanobacteria (3.98%) (Fig. S4a). Bacteria in the phylum Proteobacteria (Alpha-, Delta-, and Gammaproteobacteria) were dominated by a single genus (Endozoicomonas) in the gut of C. capistratus (93.9%) (Fig. S4b). Endozoicomonas were also abundant in hard- and soft coral samples (23.36 and 41.25% respectively. Firmicutes was abundant in fish guts (16.1% of fish gut bacteria) but representatives of this phylum were nearly absent from potential prey and seawater (Fig. S4a, b). Venn diagrams revealed that fish gut microbiomes shared largely similar proportions of ASVs with coral and sponge microbiomes in each zone (Fig. S5a–c). Fish shared a slightly higher proportion of ASVs with corals (hard and soft coral microbiomes combined) in the inner bay (6.6%; Fig. S5b) and inner bay disturbed zones (6.38%; Fig. S5c) than at the outer bay (5.43%; Fig. S5a). The proportion of shared ASVs between fish gut- and sponge microbiomes was lowest at the inner bay (2.58%; Fig. S5b) and highest at the inner bay disturbed zone (3.41%; Fig. S5c).
Composition of the core gut microbiome
Indicator analysis identified 27 ASVs in eight families (i.e., Endozoicomonadaceae, Brevinemataceae, Ruminococcaceae, Lachnospiraceae, Vibrionaceae, Peptostreptococcaceae, Clostridiaceae, Thermaceae) as part of the “core” microbiome associated with the fish intestinal tract (Fig. S6 and Table S2). The genus Endozoicomonas (phylum Proteobacteria, class Gammaproteobacteria), described as a symbiont of marine invertebrates44, comprised 71.3% of the ASVs in the core followed by genus Brevinema (phylum Spirochetes, class Spirochaetia) (10.7%) and anaerobic fermentative bacteria in the families Ruminococcaceae (9.7%), Lachnospiraceae (5.6%), and Clostridiaceae (1.7%) (phylum Firmicutes, class Clostridia) (Fig. S6).
Blastn searches against nr/nt NCBI database revealed that ASVs identified as part of the core gut microbiome (i.e., Endozoicomonas) were previously found in scleractinian and soft coral tissue at our study area and in Curaçao among other locations (Table 1). Some Endozoicomonas ASVs were closely related to sequences identified previously in sponges, clams, ascidians, tunicates, and coral mucus as well as the intestinal tract of a coral reef fish species (Pomacanthus sexstriatus). Sequences assigned to Ruminococcaceae closely resembled bacteria reported from herbivorous marine fishes (Kyphosus sydneyanus, Naso tonganus, Acanthurus nigrofuscus, and Siganus canaliculatus), the omnivorous coral reef fish Pomacanthus sexstriatus and a freshwater fish. An Epulopiscium ASV matched to a sequence detected in the guts of two coral reef fishes, the omnivore Naso tonganus and the carnivore Lutjanus bohar and to sequences found in the coral Orbicella faveolata. Other Lachnospiraceae bacteria found in this study resembled sequences known from cattle rumen, hot springs, farm waste, human and other animal feces. Within Ruminococcaceae in Firmicutes, ASVs assigned to the genus Flavonifractor closely resembled bacteria reported from the hindgut of the temperate herbivorous marine fish Kyphosus sydneyanus in New Zealand. Brevinema sequences similar to ours have been previously isolated from the gut of the coral reef fish Naso tonganus as well as freshwater and intertidal fish intestinal tracts. Retrieved Vibrionaceae (genus Vibrio) were similar to sequences found in a coral reef fish gut of Zebrasoma desjardinii. A Romboutsia ASV (family Peptostreptococcaceae), a recently described genus of anaerobic, fermentative bacteria associated with the intestinal tract of animals including humans45,46,47, which also occurs in mangrove sediments48, matched a sequence found in the tissue of the sea fan Gorgonia ventalina at our study site Bocas del Toro (Table 1).
Alpha diversity of the whole gut microbiome
We estimated alpha diversity using Hill numbers of three different orders of diversity (Hill numbers, {q = 0, 1, 2}) that place more or less weight on the relative abundance of ASVs. This approach allowed for balancing the representation of rare ASVs that might be the result of sequencing errors. Diversity of the whole gut microbiome was lower in fish of the outer bay zone than in fish of the inner bay and inner bay disturbed zones (Fig. 3a–c). Diversity differed significantly among the three zones when taking into account ASV frequency with the Shannon index (Fig. 3b) and when emphasizing abundant ASVs with the Simpson index (Fig. 3c and Table S3). However, observed ASV richness did not significantly differ among zones (Fig. 3a and Table S3). Benjamin–Hochberg corrected post hoc tests showed significantly higher Shannon diversity in fish guts of the inner bay zone versus the outer bay zone (Table S4). Fish of the inner bay disturbed zone had a higher microbial diversity than fish of the outer bay zone based on both Shannon and Simpson (Table S4). Pairwise comparisons of alpha diversity between reefs revealed that fish that resided on the reef with the highest level of coral cover overall (37.07%), Salt Creek (SCR, outer bay), had a significantly lower diversity of microbes in their guts than fish from all three inner bay disturbed reefs (RNW, PST, and PBL) for both Shannon and Simpson diversity (Table S5).

Differences in diversity (mean ± SE) of ASVs between the whole gut microbiome (a–c) and the core gut microbiome (d–f) of Chaetodon capistratus across reefs. Alpha diversity was measured based on Hill numbers using three metrics that put more or less weight on common species. The observed richness (a, d) does not take into account relative abundances. Shannon exponential (b, e) weighs ASVs by their frequency. Simpson multiplicative inverse (c, f) overweighs abundant ASVs. Significance depicts differences in alpha diversity among reef zones (Kruskal–Wallis test with post hoc Dunn test). Diamonds depict means.
Alpha diversity of the core gut microbiome
Diversity of ASVs in the core microbiome was lowest at the outer bay when comparing ASV richness among fish of the outer bay, inner bay, and inner bay disturbed zones and was highest in fish in the inner bay disturbed zone with both the Shannon index and Simpson index (Fig. 3d–f). Alpha diversity differed significantly among the three zones (Table S3) and pairwise testing revealed that this was largely due to differences between fish of the outer bay and inner bay disturbed zones (Table S4). When compared by reef, lower core microbial diversity in fish from Salt Creek (SCR, outer bay) than fish from other reefs across all zones was responsible for the most significant comparisons (Table S5).
Beta diversity of the whole gut microbiome
Permutational Analysis of Multivariate Dispersion (PERMDISP2) indicated no difference in variability in the whole fish gut microbiome across zones and reefs using dissimilarity metrics that put limited weight on abundant ASVs (Fig. 4a, b and Table S6). However, Bray–Curtis, which more heavily weighs abundant ASVs, identified significantly higher multivariate dispersion for fish from the inner bay disturbed zone than for fish from the outer bay zone (Fig. 4c and Table S6). The same pattern was observed with phylogenetic dissimilarity metrics. Only the two metrics taking into account relative abundances (i.e., GUniFrac and WUniFrac) revealed significant differences in dispersion patterns among the three zones. Using GUniFrac, an index that adjusts the weight of abundant ASVs based on tree branch lengths, gut microbiomes of fish from the inner bay disturbed zone were significantly more spread in multivariate space than gut microbiomes of fish from the outer bay zone (Fig. 4e and Table S6). Gut microbial communities were significantly more variable in fish from the inner bay zone than in fish from the outer bay zone using both GUniFrac and WUniFrac (Fig. 4e, f and Table S6).

Compositional variability of the whole gut microbiome (a–f) and core gut microbiome (g–l) of Chaetodon capistratus across reefs. Compositional variability is measured as the distance to the centroid (mean ± SE) of each group (fish at each reef) in multivariate space. Multivariate analyses were computed with non-phylogenetic (Jaccard: panels a, g; Modified Gower: panels b, h; and Bray–Curtis: panels c, i) and phylogenetic (Unifrac: panels d, j; Generalized Unifrac: e, k; Weighted Unifrac f, l) metrics that differ in how much weight they give to relative abundances. On one end of the spectrum, Jaccard and Unifrac only use presence-absence data, whereas on the other end of the spectrum Bray–Curtis and Weighted Unifrac give a lot of weight to abundant ASVs in dissimilarity calculations. Significance depicts differences in multivariate dispersion between reef zones (ANOVA). Diamonds depict means.
The three PERMANOVA models explained a small portion of the variance in the composition of the whole gut microbiome using all metrics (2.29–9.22%; Fig. 5a and Table S8). Nevertheless, gut microbiome composition was significantly different between fish from all three zones (zone model), between fish collected inside and outside the bay (position model) and between fish collected on inner bay reefs that differ in coral cover (cover model) when using Jaccard, modified Gower and Bray–Curtis distances (Fig. 5a and Table S8). Whole gut microbiomes differed using phylogenetic metrics UniFrac and GUniFrac but not when emphasizing microbial relative read abundance (WUniFrac) (Fig. 5a and Table S8). Pairwise Adonis with Bonferroni corrected P values revealed significant differences among all pairs of zones using non-phylogenetic metrics (Table S10). Pairwise tests were significant using the Unifrac distance except between gut microbiomes of fish from the inner bay and inner bay disturbed zones. None of the pairwise tests using GUnifrac and WUnifrac were significantly different among zones (Table S10).

Proportion of the variance explained in Permutational Analysis of Variance (PERMANOVA) comparing the composition of the whole gut microbiome (a) and the core gut microbiome (b) of Chaetodon capistratus. Three independent PERMANOVA analyses were conducted. The “zone” model compares gut microbiomes among the three zones of the bay (inner bay, inner bay disturbed, and outer bay). The “position” model contrasts the composition of gut microbiomes of fish collected at reefs inside and outside of the bay. The “cover” model compares gut microbiomes of fish on disturbed and undisturbed reefs inside of the bay. Three non-phylogenetic (circles) and three phylogenetic (triangles) dissimilarity metrics were used. They place more (red) or less (blue) weight on relative abundances.
Whole fish gut microbiomes featured differential relative read abundances across reefs of the inner bay disturbed, inner bay, and outer bay zones (Fig. S7). Gut microbiomes of fish from the inner bay disturbed zone had a lower proportion of microbial reads assigned to Endozoicomonadaceae (Proteobacteria), but a higher proportion of Vibrionaceae and Rhodobacteraceae. In contrast, the relative contribution of Spirochetes and Firmicutes was highest in the guts of fish in the inner bay disturbed zone (Fig. S7). Within Spirochetes, the relative abundance of Brevinemataceae was highest in gut microbiomes of fish from the inner bay disturbed zone, while Clostrideaceae within Firmicutes contributed more to gut microbiomes of fish on inner bay reefs but relatively little to the gut microbiomes of fish in the outer bay zone. Shewanellaceae (phylum Proteobacteria) represented a higher proportion of the gut microbiome of fish on inner bay disturbed reefs (Fig. S7).
Beta diversity of the core gut microbiome
Patterns in multivariate dispersion were largely consistent between whole and core gut microbiomes. Differences among the three reef zones were significant for metrics that place more weight on ASV relative abundance (common ASVs) (Fig. 4h, i and Table S7). The variability of the core gut microbiome differed significantly between fish from the inner bay and inner bay disturbed zones and between fish from the inner bay disturbed and outer bay zones with highest variability levels in the inner bay disturbed zone. However, none of the phylogenetic metrics showed significant differences in dispersion among zones (Fig. 4j–l and Table S7)
As with the whole gut microbiome, the three PERMANOVA models explained a limited amount of variance in the composition of the core gut microbiome (Fig. 5b and Table S9). Yet, composition differed significantly among fish from the three zones (zone model), between fish in the inner bay and outer bay zones (position model) as well as between zones of differential coral cover within the bay (cover model) (Fig. 5b and Table S9). The core gut microbiome appeared largely similar in composition using all phylogenetic metrics but Unifrac (Table S9). Similar to the whole microbiome, pairwise Adonis tests with Bonferroni corrected P values showed significant differences among almost all pairs of zones when using taxonomic metrics (Table S11). Of the phylogenetic metrics, the only significant differences were found between the inner versus outer bay, and between the inner bay disturbed versus outer bay zone using Unifrac (Table S11). Differences in the composition of the core microbiome among reef zones was largely driven by changes in the relative abundance of ASVs assigned to the genus Endozoicomonas (class Gammaproteobacteria) (Fig. S5b). For example, the most common Endozoicomonas ASV (ASV1) was much more represented in the guts of fish from the outer bay and inner bay zones than in the guts of fish in the inner bay disturbed zone, while Endozoicomonas ASVs relative abundances appeared more evenly distributed towards the inner bay disturbed zone. In contrast, bacteria in the genus Brevinema (phylum Spirochetes) were most abundant relative to other members of the core in fish of the inner bay disturbed zone and least abundant in the outer bay zones. The giant bacterium Epulopiscium (family Lachnospiraceae, order Clostridia), which is known to aid the digestion of algae in surgeonfishes, contributed more to the core gut microbiome of fish on reefs in the inner bay disturbed zone than the inner and outer bay zones (Fig. S5b).
Prevalent ASVs in each reef zone
A machine learning-based, de-noising algorithm (PIME) was used to detect sets of ASVs in the whole gut microbiome that significantly contribute to differences between reef zones. The initial out-of-bag (OOB) error rate (i.e., the prediction error in a Random Forest model) for our unfiltered dataset was greater than 0.1 (PIME, OOB 0.27) indicating that PIME filtering would effectively remove noise. PIME identified a prevalence cut-off of 65% for the highest improved accuracy (OOB = 2.25) indicating that the model was 97.75% accurate (Table S12). The validation step showed that randomized errors (Fig. S8b) corresponded with the predicted prevalence cut-off value of 0.65 indicating the absence of false positives (Type I error).
After selecting ASVs that were present in at least 65% of the fish guts within each zone, the filtered dataset comprised 17 ASVs in eight families; i.e., Endozoicomonadaceae, Ruminococcaceae, Pirellulaceae, Lachnospiraceae, Brevinemataceae, Cyanobiaceae, Rhodobacteraceae, and Peptostreptococcaceae (Fig. 6 and Tables S12, S13,S14). Fish of the inner bay zone showed the highest richness levels with 13 ASVs, compared to eight and nine ASVs in fish of outer bay and inner bay disturbed zones, respectively (Fig. 6). An Endozoicomonas ASV (ASV1), which was also a dominant component of the core, had a much higher relative abundance in fish of the outer bay zone than in fish of the inner bay disturbed zone (Fig. 6). Communities differed most in composition between fish of the outer bay and inner bay disturbed zone, whereas fish of the inner bay zone reflected an intermediate community between these two comprising the highest richness of Endozoicomonas with six ASVs. As in the core community, the Endozoicomonas assemblage was slightly less diverse in the disturbed zone (three ASVs) and featured more similar relative read abundances than in the outer bay zone (four ASVs) where a single ASV was dominant. Two distinct ASVs of the giant bacterium Epulopiscium (family Lachnospiraceae) were prevalent in fish in both the inner and inner bay disturbed zones but were more abundant on disturbed reefs. Disturbed reefs uniquely featured anaerobic gut bacteria in the genus Romboutsia (family Peptostreptococcaceae) (Fig. 6).

Comparison of fish gut microbiomes among three reef zones. The whole fish gut microbial dataset was filtered using Prevalence Interval for Microbiome Evaluation (PIME)134 to detect which ASVs were responsible for differences among zones. Using machine learning, PIME de-noises the data by reducing within-group variability. Based on the algorithm, we selected a 65% prevalence cut-off resulting in a filtered dataset of 17 ASVs at a low error rate (OOB = 2.25) and high model accuracy (97.75%).
Discussion
Detecting how the spatial turnover of microbiomes varies within and among host populations, and in relation to habitat characteristics is essential to understanding and predicting the response of host species to environmental change. We show that whole gut microbial communities were significantly more diverse and variable in fish from inner bay disturbed reefs than from the outer bay zone in terms of some but not all measured diversity components. Conspicuously, the core microbiome, a small set of microbial strains that may form sustained relationships with the fish host, also showed higher dispersion on degraded reefs suggesting greater variability of microbial assemblages among individual fish. Significant differences in diversity and group dispersion were observed mostly in the relative abundances of the frequent and common taxa. Highly variable host-associated microbial communities have been observed in humans with immunodeficiency syndromes (reviewed in ref. 49) and in marine animals such as scleractinian corals and anemones under acute stress43,50,51,52. Zaneveld et al.43 referred to this pattern of variability as the “Anna Karenina principle” applied to host-associated microbiomes (AKP). They argued that this is a common but often overlooked response of organisms that become unable to regulate their microbiome. Our results are consistent with patterns expected under the Anna Karenina principle, which could potentially imply that fish experience some level of stress in association with habitat degradation.
Reductions in coral cover may increase foraging costs if, for example, fish spent more energy to search, capture, and handle their prey. Indeed, physiological stress imposed by environmental conditions may cause immune signals that imbalance the gut microbiome30,43,53,54. Disturbance to the microbiome, in turn, can affect the brain and further alter behaviors related to movement such as the ability to forage30,55. The low coral cover may also increase stress through intra- and interspecific competition for resources. For example, social stress in the form of aggressive interactions among conspecifics was shown to alter the behavior and microbial assemblages associated with mice by setting off immune responses critical to host health56. In Indo-Pacific obligate feeding butterflyfishes coral degradation was shown to decrease aggressive encounters among and within Chaetodon species57 as well as change the frequency of pair formation58, and the way species responded to the loss of the coral resource depended on their level of dietary specialization57,58. Foraging on degraded reefs may also increase predation risk when architectural complexity is reduced59. Anxiety-like behaviors induced by exposure to predators can lead to sustained physiological stress in vertebrates (reviewed in ref. 60).
Another possible explanation for more variable gut microbiomes on disturbed reefs could be increased behavioral heterogeneity among fish individuals (e.g., feeding behavior). Where preferred food sources are scarce, foraging behavior may become more diverse and lead to increased individual specialization on alternative food items61,62 translating into more varied gut microbiomes. In this scenario, the higher variation in gut microbial assemblages would be the result of behavioral adjustments (acclimatization) to alternative habitat conditions without necessarily causing stress. Higher alpha diversity across fish gut microbiomes in the inner bay disturbed zone supports this explanation. Although C. capistratus is able to consume a broad range of diet items39,63,64, deviations from its preferred coral prey may come with fitness consequences as shown for Indo-Pacific butterflyfishes33,65,66. For example, other authors found that obligate corallivorous Chaetodon species have reduced energy reserves at reefs where they diversify or shift their diet in response to limited coral availability33,66.
Apart from patterns of microbiome variability, the significant differences in the composition of the whole gut microbiome (as opposed to the core microbiome) in nearly all comparisons (i.e., among all three zones, between inner and outer bay, and between inner bay disturbed and undisturbed) may primarily reflect changes in diet. Specifically, in the inner bay disturbed zone where coral cover was low, the microbial assemblage suggests (i) potential changes in the invertebrate prey community and (ii) a more broad, likely omnivorous trophic profile indicated by a distinct Endozoicomonas community in codominance with anaerobic fermentative bacteria. The increased prevalence of fermentative microbes at disturbed reefs might reflect the consumption of algae and potentially sponges given their high availability in this zone. However, we lack information on the effects of sponge consumption on fish gut microbiomes. Epulopiscium, often considered a host-specific symbiont of herbivorous surgeonfishes (family: Acanthuridae)26,27,67,68, was present in the core microbiome and identified as distinct to the inner bay with predominance at disturbed reefs. This may suggest that C. capistratus can assimilate nutrients from algae and that this metabolic function is enhanced on degraded reefs by the increase in key microbial functional groups. Alternatively, the fish in our study may take up these microbes while foraging for invertebrates on the epilithic algal matrix. Overall, levels of Epulopiscium were approximately similar to those previously found in omnivores and detritivores in the Red Sea26 with the two most prevalent ASVs matching a strain previously isolated from the turf algal grazer Naso tonganus69. Additionally, the presence of Rhodobacteraceae, which are often found associated with algal biofilms70,71, may suggest detritivory but might also be related to the consumption of mucus from stressed72 and diseased corals where Rhodobacteraceae are also found73,74. The lower relative abundance of a compositionally distinct Endozoicomonas community on disturbed reefs could reflect different proportions of prey species featuring Endozoicomonas75,76 in the diet of C. capistratus.
In contrast, a single dominant Endozoicomonas ASV along with a few Firmicutes characterized the gut microbiome of C. capistratus on outer bay reefs. The presence of some Endozoicomonas ASVs shared between fish guts and potential prey (i.e., hard corals, soft corals, zoanthids, sponges), including matches to microbial sequences previously detected in two coral species (Orbicella faveolata and Poritis asteroides) in our study area at Bocas del Toro73,77, suggests the horizontal acquisition of these microbes via feeding on corals. In addition, we identified an ASV in the genus Ruegeria as indicative of the outer bay reefs, which matched a sequence previously retrieved from the soft coral species Pterogorgia anceps on the Caribbean coast of Panamá (GenBank Accession: MG099582) and which was also present across samples of potential prey taxa including hard and soft corals and sponge-infauna. Endozoicomonas originating from the food could potentially play a role in promoting the assimilation of nutrients via interactions with resident bacteria78.
The core microbiome composition significantly differed across the inner bay between fish from disturbed and undisturbed reefs where environmental conditions are generally homogeneous except for the proportion of live coral cover. Despite our models accounting for relatively little variance, this finding may suggest that bacterial communities that are most likely to have intimate metabolic interactions with C. capistratus might fail to provide important functions to hosts in severely degraded habitats. However, we cannot exclude the contribution of other factors that were not measured in this study such as microbial plasticity mediated by diet, gut colonization history79, and/or potential genetic differentiation between the inner bay and outer bay fish populations80,81,82.
Our analysis identified ten Endozoicomonas ASVs as part of the core microbiome indicating potential true resident symbionts. Members of the genus Endozoicomonas spp. are known as bacterial symbionts of marine sessile and some mobile invertebrates and fishes44,76,83,84,85,86,87. Reverter et al. (2017)86 found Endozoicomonas associated with butterflyfish gill mucus in Chaetodon lunulatus and Parris et al. (2016)87 found Endozoicomonas in the gut of damselfishes (family: Pomacentridae) and cardinalfishes (family: Apogonidae) pre- and to a lesser extent post-settlement on the reef. Corallivory in butterflyfishes has evolved in close association with coral reefs32,88 and this likely involved adaptive mechanisms to metabolize defense compounds from corals and many other sessile invertebrates (e.g., polychaetes). Adapted gut microbial communities may help butterflyfish hosts cope with toxins or facilitate the digestion of complex prey tissues as in insects89, mammalian herbivores90, and surgeonfishes26. It is likely that the gut microbial profile of C. capistratus —featuring high abundance Endozoicomonas—facilitates the digestion of complex coral prey. More detailed knowledge will be required to understand whether the potential intake of Endozoicomonas via fish browsing on sessile invertebrates plays a role in nutrient uptake in trophic strategies such as fish corallivory.
We detected increases in gut microbiome variability, diversity, and spatial community turnover. These patterns extended to the core microbiome suggesting signs of potentially altered functioning that may affect fish hosts on reefs with extremely low levels of live coral cover. Nonetheless, the density of C. capistratus was comparable across both inner bay zones indicating that the lack of live coral cover may not immediately impact the persistence of populations. Significantly lower densities at the outer bay may potentially relate to spatial patterns of larval recruitment91 and/or differences in wave exposure across reefs affecting the energy expenditure fish allocate towards swimming performance and feeding92. Additional work should focus on linking changes in the gut microbiome to direct measures of diet and host health. Our results give insight into the poorly understood spatial fluctuations in host-associated microbial communities across a natural system. This work highlights intricate links between ecosystem-scale and microbial-scale processes, which have so far been mostly overlooked. We suggest there is an urgent need to integrate measurements of the role of microbes in the response of reef fishes to the global loss of coral reefs.
Methods
Study area
Bahía Almirante, located in the Bocas del Toro Archipelago on the Caribbean coast of Panamá, is a coastal lagoonal system of ~450 km2 where numerous, relatively small, and patchy fringing coral reefs occur93. Hydrographic and environmental conditions vary across the semi-enclosed bay but are generally characterized by limited water exchange with the open ocean42. Furthermore, areas of the bay are subjected to uncontrolled sewage and dredging due to increasing coastal development and agricultural runoffs from the adjacent mainland94,95,96,97. A total of nine discontiguous reefs distributed from the mouth towards the inner bay were selected for this study based on distinct hydro-geographical zones and disturbance history, resulting in three distinct reef zones with three replicates each (total n = 9 reefs) (Fig. 1a). Throughout the manuscript, we will refer to these three distinct reef zones as “outer bay”, “inner bay”, and “inner bay disturbed”. Outer bay reefs [Salt Creek (SCR), Cayo Corales (CCR), and Popa (PPR)] are located at the mouth of the bay marking a transition zone between the inner bay and the open ocean. These reefs represent typical Caribbean reef communities featuring both massive and branching coral colonies with higher benthic cover and diversity as compared to the inner bay (Fig. 1b). Inner bay reefs [Almirante (ALR), Cayo Hermanas (SIS), and Cayo Roldan (ROL)] are largely coral and sponge dominated reefs and have lower coral diversity than the outer bay reefs (Fig. 1c). Inner bay disturbed reefs [Punta Puebla (PBL), Punta STRI (PST) and Runway (RNW)] were heavily impacted by the 2010 hypoxic event42, which resulted in the current cover of largely dead coral comprised of formally prevalent Agaricia and Porites species (Fig. 1d). Prior to this disturbance, both study zones located inside of the bay exhibited comparable benthic communities of similar health states. For example, the Punta STRI reef (PST) at the now disturbed zone featured 26.9% coral cover in 200598.
Benthic habitat and fish density
Visual surveys of benthic cover and focal fish species densitiy were conducted between May and June 2016. At each of the nine reefs, three 20 m transects were placed parallel to the shore at 2–4 m depth. Benthic community cover was estimated from 100 cm × 70 cm photographic quadrats placed every 2 m, resulting in a total of 10 quadrats per transect. Photos were analyzed on the CoralNet platform99 using a stratified random sampling design (10 rows × 10 columns with 1 point per cell for a total of 100 points per image). The first 15 of all photos were manually scored to train the algorithm. The remaining photos were then processed by the automated assignment tool, and assignments were subsequently verified for each point. Due to the difficulty involved with photo-based taxonomic identification, analyses were conducted at the level of broad benthic categories which comprised the following: live hard coral, dead hard coral, live soft coral, sponge, zoanthid, other invertebrates, seagrass, grazable substrate, macroalgae, rubble, sand, shade and “unknown”. Within the live hard coral, dead hard coral, and live soft coral categories, identification was done at the genus or species level when possible. The mean cover of each benthic category was calculated per reef. To estimate focal fish species density, C. capistratus individuals were counted along each 20 × 5 m belt transects used subsequently for the benthic surveys while swimming slowly using scuba (except at CCR).
Sample collection
The foureye butterflyfish, Chaetodon capistratus, is a common member of Caribbean coral reef fish communities (IUCN classified as least concern)100 with a distribution that extends across the subtropical Western Atlantic101,102 (Fig. 1e). The following protocol of fish capture and euthanization had been approved by the Smithsonian Tropical Research Institute’s Institutional Animal Care and Use Committee (IACUC). An average of 11 individual adult fish were collected at each of the nine reefs (min = 7; max = 16; total = 102) by spearfishing in February and March 2018 (Table S15). Captured fish were immediately brought to the boat, anesthetized with clove oil, and placed on ice in an individual and labeled sterile Whirl-Pak bag. Upon return to the research station, fish were weighed (g wet weight), and both standard length (mm SL) and total length (mm TL) were measured with a digital caliper. The intestinal tract of each fish was removed under a laminar flow hood using tools decontaminated with 10% sodium hypochlorite. The intestinal tracts were then preserved in 96% ethanol in individual 15 ml or 5 ml centrifuge tubes and stored at −20 °C until DNA extraction. To assess microbial communities present in the fish’s environment, we also obtained samples of seawater and potential prey taxa. At each of nine reefs, a total of four liters (2 × 2 L at each reef) of seawater was collected immediately above the reef substratum using sterile Whirl-Pak bags and filtered through a 0.22 µm nitrocellulose membrane (Millipore) and a total of 18 seawater samples was included into downstream analysis. Small pieces of hard coral (Siderastrea siderea, n = 2; Porites furcata, n = 2; Agaricia tenuifolia n = 2), soft coral (Antillogorgia bipinnata, n = 1; Plexauridae sp.; n = 1), sponges (Amphimedon compressa, n = 1; Chondrilla caribensis, n = 1; Demospongiae spp., n = 4), macroalgae (Amphiroa sp., n = 1), turf (n = 1), and zoantharia (Zoanthus pulchellus, n = 1; Palythoa caribaoerum, n = 1) were collected opportunistically at least at one of the three habitat zones and kept in sterile Whirl-Pak bags on ice on the boat. At the field station, samples were individually placed in 50 ml or 15 ml centrifuge tubes with 96% ethanol and stored at −20 °C until DNA extraction.
DNA analysis
The mid- and hindgut of the gastrointestinal tract of each fish was opened longitudinally to isolate the digesta and the mucosa tissue by lightly scraping the intestinal epithelium. Between 0.05 and 0.25 g of both tissue types combined was used for DNA extraction using the Qiagen Powersoil DNA isolation kit following the manufacturer's instructions with minor modifications to increase yield (see supplementary methods). Each tissue sample of potential prey organisms (invertebrates and macroalgae) was homogenized in separate vials. Additionally, infaunal communities (small worms) were isolated from two sponges, Amphimedon compressa and Dysidea sp. and the tissue homogenized for each sponge separately. DNA was extracted (0.25 g per sample) following the same protocol as described for the intestinal microbiomes. Seawater DNA was isolated from nitrocellulose membrane filters using the Qiagen Powersoil Kit following a modified protocol described in ref. 103.
A dual Polymerase Chain Reaction (PCR) approach was used to amplify the V4 hypervariable region (primers 515F104 and 806R105) of the 16 S ribosomal rRNA gene of each sample (Table S16). Subsequently, the product of all samples was sequenced by combining them into a single Illumina MiSeq sequencing run. Our protocol followed the 16 S Illumina Amplicon Protocol of the Earth Microbiome Project106 using locus-specific primers to which Illumina “overhang” sequences were appended. These overhang sequences served as a template to add dual index Illumina sequencing adapters in a second PCR reaction (see supplementary methods for detailed PCR protocols). The final product was sequenced on the Illumina MiSeq sequencer (reagent kit version 2, 500 cycles) at the Smithsonian Tropical Research Institute with a 20% PhiX spike. The absence of contaminants was confirmed with negative DNA extractions and negative PCR amplifications (see supplementary methods for detailed DNA extraction and PCR protocols).
Analysis of sequence data
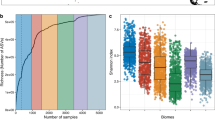
All analyses were conducted with the statistical software R version 3.6.158107. Illumina adapter and primer sequences were removed from forward and reverse reads using Cutadapt108 with a maximum error rate of 0.12 (-e 0.12). The remaining reads were filtered and trimmed based on their quality profiles and potential chimeras were removed using DADA2 version 1.12.1109. Sequences were discarded if they had more than two expected errors (maxEE = 2), at least one ambiguous nucleotide (maxN = 0), or at least one base with a high probability of erroneous assignment (truncQ = 2). Forward and reverse reads were trimmed to 220 and 180 bp respectively to remove lower quality bases while maintaining sufficient overlap between paired-end reads. Sequences were kept when both the forward and reverse reads of a pair passed the filter. Quality filtered reads were de-replicated and Amplicon Sequence Variants (ASVs) inferred. Paired-end reads were merged and pairs of reads that did not match exactly were discarded. Taxonomy was assigned to each ASV using a DADA2 implementation of the naive Bayesian RDP classifier110 against the Silva reference database version 132111. ASVs identified as chloroplast, mitochondria, Eukaryota, or those that remained unidentified at the kingdom level were removed from the dataset. Sequences of each ASV were aligned using the DECIPHER package version 2.0112. The PHANGORN package version 2.5.5113 was then used to construct a maximum likelihood phylogenetic tree (GTR + G + I model) using a neighbor-joining tree as a starting point. Fourteen samples containing few sequences (<10,000) were removed from the dataset (Fig. S9). The remaining samples were rarified without replacement to even sequencing depth (n = 10,369 sequences) for downstream analysis. Our approach followed the recommendation for the normalization of sequencing data114. Statistical analysis was conducted using phyloseq version 1.28.0115.
Delineation of the core gut microbiome
To identify the persistent bacteria associated with the fish gut (i.e., the “core microbiome”19,116) including taxa that might be potentially beneficial to the fish host, we employed a statistical approach taking into account both relative abundance and relative frequency of occurrence of ASVs as opposed to the common procedure of using an arbitrary minimum frequency threshold based on presence-absence data only116. Indicator species analysis117 (labdsv package version 2.0-1)118 was used to identify which ASVs were relatively more abundant and predominantly found in fish guts and not in their surrounding environment. We calculated an Indicator Value (IndVal) Index between each ASV and two groups of samples: (1) all fish gut samples, and (2) all seawater and sessile invertebrate samples upon which fish potentially feed. Statistical significance of the association between ASVs and groups of samples was tested using 1000 permutations. ASVs were considered indicators of fish guts (i.e., components of the core) based on a maximum probability of P value = 0.01. Sequences of ASVs identified as part of the core microbiome were compared to the non-redundant nucleotide (nr/nt) collection database of the National Centre for Biotechnology Information (NCBI) using the Basic Local Alignment Search Tool for nucleotides (BLASTn)119. We extracted metadata associated with all sequences that matched each query at 100% similarity or the first five top hits to identify within what taxa, environment, and/or habitat each core ASV and close relatives had been previously found.
Diversity analysis
The workflow of our microbial community analysis is visualized in a diagram (Fig. 7). To account for the presence of rare sequence variants caused by sequencing errors or other technical artifacts120, we used Hill numbers121 following the approach recommended by ref. 122 for sequence data to compare alpha diversity among groups of samples. Hill numbers allow scaling the weight put on rare versus abundant sequence variants while providing intuitive comparisons of diversity levels using “effective number of ASVs” as a measuring unit121,122,123. This approach allowed for balancing the over-representation of rare ASVs that might be inflated due to sequencing errors124. We calculated three metrics that put more or less weight on common species: (1) observed richness, (2) Shannon exponential that weighs ASVs by their frequency, and (3) Simpson multiplicative inverse that overweighs abundant ASVs. Alpha diversity was calculated and visualized using boxplots for the whole and core fish gut microbiomes. Because Shapiro–Wilk tests indicated that the data were not normally distributed, non-parametric Kruskal–Wallis tests were used to compare alpha diversity among reefs (n = 9) and the three reef zones (outer bay, inner bay, and inner bay disturbed) with post hoc Dunn tests.

Microbial community analysis workflow illustrating how we subsetted the whole fish gut microbiome dataset to delineate the core gut microbiome and gut microbial communities by zone, respectively. To identify the core microbiome, we used indicator analysis117 between the whole fish gut microbiome and the environmental sample fraction consisting of samples of potential fish prey taxa and the surrounding seawater. Diversity analysis was done for the whole and core fish gut microbiome, respectively. The whole fish gut microbiome was filtered for prevalence with a machine learning-based algorithm (PIME)134 to detect community differences among zones that reflect fish-microbiome responses to the habitat gradient. Created with BioRender.com. The fish icon is adapted from a color photograph of Chaetodon capistratus obtained from https://biogeodb.stri.si.edu/caribbean/en/pages with permission by D R Robertson. Icons of benthic organisms obtained from the IAN Symbol Libraries: Tracey Saxby and Joanna Woerner, Integration and Application Network (ian.umces.edu/media-library). https://creativecommons.org/licenses/by-sa/4.0/.
To test the hypothesis that fish gut microbiomes are more variable between individuals at disturbed sites, we calculated non-parametric Permutational Analysis of Multivariate Dispersion (PERMDISP2) (betadisper function, vegan package implemented in phyloseq115,125). PERMDISP2 is a measure of the homogeneity of variance among groups and compares the average within-group distance to centroid between each predefined group of samples in multidimensional space. We used a range of phylogenetic and non-phylogenetic dissimilarity metrics that differentially weigh the relative abundance of ASVs to identify the effect of abundant ASVs [Phylogenetic: Unifrac, Generalized Unifrac (package GUniFrac)126 and Weighted Unifrac127; non-phylogenetic: Jaccard128, modified Gower with log base 10129 and Bray–Curtis130]. P values were obtained by permuting model residuals of an ANOVA (Analysis of Variance) null-model 1000 times (betadisper function, vegan115,125).
Differences in microbial composition were tested using Permutational Multivariate Analysis of Variance (PERMANOVA) with the Adonis function in vegan131 computed with 10,000 permutations. Comparisons were made (1) among fish gut microbiomes of the three reef zones (“zone model”), (2) between fish gut microbiomes of outer bay reefs versus inner bay reefs (“position model”) and (3) between fish gut microbiomes of inner bay reefs and inner bay disturbed reefs which differed in coral cover (“cover model”). PERMANOVA is robust to the effect of heterogeneity of multivariate dispersions in balanced or near balanced designs as in our study132. Pairwise Adonis with Bonferroni corrected P values was computed using the pairwise Adonis function (version 0.4)133.
Finally, we used the Prevalence Interval for Microbiome Evaluation (PIME) package (version 0.1.0)134 to identify sets of ASVs that are frequently found in fish guts in each zone at the Bahía Almirante (outer bay, inner bay, inner bay disturbed). This analysis is aimed at identifying microbial ASVs that are differentially prevalent among zones. PIME uses a supervised machine learning Random Forest algorithm to reduce within-group variability by excluding low-frequency sequences potentially confounding community comparisons of microbiome data134. PIME identifies the best model to predict community differences between groups by defining a prevalence threshold that retains as many ASVs as possible in the resulting filtered communities (i.e., the random forest classifications) while minimizing prediction error (out-of-bag error, OOB). To do so, the algorithm uses bootstrap aggregating (100 iterations) of each sample group at each filtering step (prevalence interval) by 5% increments. Random Forest calculates a global prediction from a multitude of decision trees based on the bootstrap aggregations and estimates the out-of-bag error rate (OOB) from omitted subsamples during aggregating135. Validation was done by randomizing the original dataset (100 permutations) and subsequently estimating Random Forest error to determine if group differences in the filtered dataset were due to chance (pime.error.prediction function, PIME)134. A second function (pime.oob.replicate, PIME)134 repeated the Random Forest analysis using the filtered dataset for each prevalence interval without randomizing group identity. In a preliminary step, we assessed whether the OOB error for our unfiltered data was >0.1, which indicated that de-noising with PIME would improve model accuracy.
Statistics and reproducibility
Statistical analyses were carried out for the 16 S sequencing data and in-situ transect data (benthic photographic quadrats and visual censuses of fish communities). Details allowing the reproducibility of all analyses are provided in the methods section (including sampling sizes and numbers of replicates). A diagram illustrating the statistical workflow is also included (Fig. 7) and the R code is available on the project website: https://github.com/bocasbiome/web/.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Sequencing data has been submitted to the NCBI Short Read Archive (SRA) database (https://www.ncbi.nlm.nih.gov/sra) under bioproject number Accession: PRJNA718434 ID: 718434. Raw data are available on Dryad Digital Repository https://doi.org/10.5061/dryad.m905qfv28153.
Code availability
Source code is available at https://github.com/bocasbiome/web/154.
References
Kiers, E. T., Palmer, T. M., Ives, A. R., Bruno, J. F. & Bronstein, J. L. Mutualisms in a changing world: an evolutionary perspective. Ecol. Lett. 13, 1459–1474 (2010).
Idjadi, J. & Edmunds, P. Scleractinian corals as facilitators for other invertebrates on a Caribbean reef. Mar. Ecol. Prog. Ser. 319, 117–127 (2006).
Norström, A., Nyström, M., Lokrantz, J. & Folke, C. Alternative states on coral reefs: beyond coral–macroalgal phase shifts. Mar. Ecol. Prog. Ser. 376, 295–306 (2009).
Richardson, L. E., Graham, N. A. J., Pratchett, M. S., Eurich, J. G. & Hoey, A. S. Mass coral bleaching causes biotic homogenization of reef fish assemblages. Glob. Chang. Biol. 24, 3117–3129 (2018).
Wilson, S. K., Graham, N. A. J., Pratchett, M. S., Jones, G. P. & Polunin, N. V. C. Multiple disturbances and the global degradation of coral reefs: are reef fishes at risk or resilient? Glob. Chang. Biol. 12, 2220–2234 (2006).
Apprill, A. The role of symbioses in the adaptation and stress responses of marine organisms. Ann. Rev. Mar. Sci. 12, 291–314 (2020).
Alberdi, A., Aizpurua, O., Bohmann, K., Zepeda-Mendoza, M. L. & Gilbert, M. T. P. Do Vertebrate gut metagenomes confer rapid ecological adaptation? Trends Ecol. Evol. 31, 689–699 (2016).
Voolstra, C. R. & Ziegler, M. Adapting with microbial help: microbiome flexibility facilitates rapid responses to environmental change. BioEssays 42, e2000004 (2020).
Webster, N. S. & Reusch, T. B. H. Microbial contributions to the persistence of coral reefs. ISME J. 11, 2167–2174 (2017).
Wilkins, L. G. E. et al. Host-associated microbiomes drive structure and function of marine ecosystems. PLoS Biol. 17, e3000533 (2019).
Ley, R. E. et al. Evolution of mammals and their gut microbes. Science 320, 1647–1651 (2008).
Ley, R. E., Lozupone, C. A., Hamady, M., Knight, R. & Gordon, J. I. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6, 776–788 (2008).
Egerton, S., Culloty, S., Whooley, J., Stanton, C. & Ross, R. P. The gut microbiota of marine fish. Front. Microbiol. 9, 873 (2018).
Llewellyn, M. S., Boutin, S., Hoseinifar, S. H. & Derome, N. Teleost microbiomes: the state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 5, 1–1 (2014).
Tarnecki, A. M., Burgos, F. A., Ray, C. L. & Arias, C. R. Fish intestinal microbiome: diversity and symbiosis unravelled by metagenomics. J. Appl. Microbiol. 123, 2–17 (2017).
Wang, A. R., Ran, C., Ringø, E. & Zhou, Z. G. Progress in fish gastrointestinal microbiota research. Rev. Aquac. 10, 626–640 (2018).
Legrand, T. P. R. A., Wynne, J. W., Weyrich, L. S. & Oxley, A. P. A. A microbial sea of possibilities: current knowledge and prospects for an improved understanding of the fish microbiome. Rev. Aquac. 12, 1101–1134 (2019).
Rawls, J. F., Mahowald, M. A., Ley, R. E. & Gordon, J. I. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell 127, 423–433 (2006).
Shade, A. & Handelsman, J. Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14, 4–12 (2012).
Sullam, K. E. et al. Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol. Ecol. 21, 3363–3378 (2012).
Ainsworth, T. D. et al. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 9, 2261–2274 (2015).
Hernandez-Agreda, A., Leggat, W., Bongaerts, P. & Ainsworth, T. D. The microbial signature provides insight into the mechanistic basis of coral success across reef habitats. MBio. 7, e00560–16 (2016).
Roeselers, G. et al. Evidence for a core gut microbiota in the zebrafish. ISME J. 5, 1595–1608 (2011).
Clements, K. D., Angert, E. R., Montgomery, W. L. & Choat, J. H. Intestinal microbiota in fishes: what’s known and what’s not. Mol. Ecol. 23, 1891–1898 (2014).
Jones, J. et al. The microbiome of the gastrointestinal tract of a range-shifting marine herbivorous fish. Front. Microbiol. 9, 2000 (2018).
Miyake, S., Ngugi, D. K. & Stingl, U. Diet strongly influences the gut microbiota of surgeonfishes. Mol. Ecol. 24, 656–672 (2015).
Ngugi, D. K. et al. Genomic diversification of giant enteric symbionts reflects host dietary lifestyles. Proc. Natl Acad. Sci. USA 114, E7592–E7601 (2017).
Degregori, S., Casey, J. M. & Barber, P. H. Nutrient pollution alters the gut microbiome of a territorial reef fish. Mar. Pollut. Bull. 169, 112522 (2021).
Gómez, G. D. & Balcázar, J. L. A review on the interactions between gut microbiota and innate immunity of fish. FEMS Immunol. Med. Microbiol. 52, 145–154 (2008).
Butt, R. L. & Volkoff, H. Gut microbiota and energy homeostasis in fish. Front. Endocrinol. 10, 9 (2019).
Hughes, T. P. et al. Spatial and temporal patterns of mass bleaching of corals in the Anthropocene. Science 359, 80–83 (2018).
Bellwood, D. R. et al. Evolutionary history of the butterflyfishes (f: Chaetodontidae) and the rise of coral feeding fishes. J. Evol. Biol. 23, 335–349 (2010).
Berumen, M., S., M. & McCormick, M. Within-reef differences in diet and body condition of coral-feeding butterflyfishes (Chaetodontidae). Mar. Ecol. Prog. Ser. 287, 217–227 (2005).
Pratchett, M. S. Dietary overlap among coral-feeding butterflyfishes (Chaetodontidae) at Lizard Island, northern Great Barrier Reef. Mar. Biol. 148, 373–382 (2005).
Nagelkerken, I., van der Velde, G., Wartenbergh, S. L. J., Nugues, M. M. & Pratchett, M. S. Cryptic dietary components reduce dietary overlap among sympatric butterflyfishes (Chaetodontidae). J. Fish. Biol. 75, 1123–1143 (2009).
Bouchon & Harmelin-Vivien Impact of coral degradation on a chaetodontid fish assemblage, Moorea, French Polynesia. Fifth Int. Coral Tahiti 5, 427–432 (1985).
Graham, N. A. J. Ecological versatility and the decline of coral feeding fishes following climate driven coral mortality. Mar. Biol. 153, 119–127 (2007).
Pratchett, M. S., Wilson, S. K. & Baird, A. H. Declines in the abundance of Chaetodon butterflyfishes following extensive coral depletion. J. Fish. Biol. 69, 1269–1280 (2006).
Birkeland & Neudecker. Foraging behavior of two Caribbean Chaetodontids: Chaetodon capistratus and C. aculeatus. Copeia 1981, 169–178 (1981).
Gore, M. A. Factors affecting the feeding behavior of a coral reef fish, Chaetodon capistratus. Bull. Mar. Sci. 35, 211–220 (1984).
Liedke, A. M. R. et al. Resource partitioning by two syntopic sister species of butterflyfish (Chaetodontidae). J. Mar. Biol. Assoc. UK 98, 1767–1773 (2018).
Altieri, A. H. et al. Tropical dead zones and mass mortalities on coral reefs. Proc. Natl Acad. Sci. USA 114, 3660–3665 (2017).
Zaneveld, J. R., McMinds, R. & Vega Thurber, R. Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2, 17121 (2017).
Neave, M. J., Apprill, A., Ferrier-Pagès, C. & Voolstra, C. R. Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas. Appl. Microbiol. Biotechnol. 100, 8315–8324 (2016).
Ricaboni, D., Mailhe, M., Khelaifia, S., Raoult, D. & Million, M. Romboutsia timonensis, a new species isolated from human gut. N. Microbes N. Infect. 12, 6–7 (2016).
Zhang, L. et al. Characterization of the microbial community structure in intestinal segments of yak (Bos grunniens). Anaerobe 61, 102115 (2020).
Gerritsen, J. et al. A comparative and functional genomics analysis of the genus Romboutsia provides insight into adaptation to an intestinal lifestyle. Preprint at bioRxiv https://doi.org/10.1101/845511 (2019).
Fernández-Cadena, J. C. et al. Detection of sentinel bacteria in mangrove sediments contaminated with heavy metals. Mar. Pollut. Bull. 150, 110701 (2020).
Williams, B., Landay, A. & Presti, R. M. Microbiome alterations in HIV infection a review. Cell. Microbiol. 18, 645–651 (2016).
Ahmed, H. I., Herrera, M., Liew, Y. J. & Aranda, M. Long-term temperature stress in the Coral Model Aiptasia supports the ‘Anna Karenina principle’ for bacterial microbiomes. Front. Microbiol. 10, 975 (2019).
Beatty, D. S. et al. Variable effects of local management on coral defenses against a thermally regulated bleaching pathogen. Sci. Adv. 5, eaay1048 (2019).
Zaneveld, J. R. et al. Overfishing and nutrient pollution interact with temperature to disrupt coral reefs down to microbial scales. Nat. Commun. 7, 11833 (2016).
Ma, Q. et al. Impact of microbiota on central nervous system and neurological diseases: the gut-brain axis. J. Neuroinflammation 16, 53 (2019).
Pita, L., Rix, L., Slaby, B. M., Franke, A. & Hentschel, U. The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome 6, 46 (2018).
Johnson, K. V. A. & Foster, K. R. Why does the microbiome affect behaviour? Nat. Rev. Microbiol. 16, 647–655 (2018).
Werbner, M. et al. Social-stress-responsive microbiota induces stimulation of self-reactive effector T helper cells. mSystems 4, e00292-18 (2019).
Keith, S. A. et al. Synchronous behavioural shifts in reef fishes linked to mass coral bleaching. Nat. Clim. Chang. 8, 986–991 (2018).
Thompson, C. A., Matthews, S., Hoey, A. S. & Pratchett, M. S. Changes in sociality of butterflyfishes linked to population declines and coral loss. Coral Reefs 38, 527–537 (2019).
Almany, G. R. Differential effects of habitat complexity, predators and competitors on abundance of juvenile and adult coral reef fishes. Oecologia 141, 105–113 (2004).
Clinchy, M., Sheriff, M. J. & Zanette, L. Y. Predator-induced stress and the ecology of fear. Funct. Ecol. 27, 56–65 (2013).
Bolnick, D. I., Svanbäck, R., Araújo, M. S. & Persson, L. Comparative support for the niche variation hypothesis that more generalized populations also are more heterogeneous. Proc. Natl Acad. Sci. USA 104, 10075–10079 (2007).
Svanbäck, R. & Bolnick, D. I. Intraspecific competition drives increased resource use diversity within a natural population. Proc. R. Soc. B Biol. Sci. 274, 839–844 (2007).
Neudecker, S. Foraging patterns of Chaetodontid and Pomacanthis fishes at St. Croix (U.S. Virgin Islands). Proc. Fifth International Coral Reef Symposium. 415–414 (1985).
Lasker, H. Prey preferences and browsing pressure of the butterflyfish Chaetodon capistratus on Caribbean gorgonians. Mar. Ecol. Prog. Ser. 21, 213–220 (1985).
Cole, A. J., Pratchett, M. S. & Jones, G. P. Diversity and functional importance of coral-feeding fishes on tropical coral reefs. Fish Fish. 9, 286–307 (2008).
Pratchett, M. S., Wilson, S. K., Berumen, M. L. & McCormick, M. I. Sublethal effects of coral bleaching on an obligate coral feeding butterflyfish. Coral Reefs 23, 352–356 (2004).
Fishelson, L., Montgomery, W. L. & Myrberg, A. A. A unique symbiosis in the gut of tropical herbivorous surgeonfish (Acanthuridae: teleostei) from the red sea. Science 229, 49–51 (1985).
Miyake, S., Ngugi, D. K. & Stingl, U. Phylogenetic diversity, distribution, and cophylogeny of giant bacteria (Epulopiscium) with their surgeonfish hosts in the Red Sea. Front. Microbiol. 7, 285 (2016).
Choat, J. H., Robbins, W. & Clements, K. The trophic status of herbivorous fishes on coral reefs II. Mar. Biol. 145, 445–454 (2004).
Elifantz, H., Horn, G., Ayon, M., Cohen, Y. & Minz, D. Rhodobacteraceae are the key members of the microbial community of the initial biofilm formed in Eastern Mediterranean coastal seawater. FEMS Microbiol. Ecol. 85, 348–357 (2013).
Pujalte, M. J., Lucena, T., Ruvira, M. A., Arahal, D. R. & Macián, M. C. In The Prokaryotes: Alphaproteobacteria and Betaproteobacteria (Springer, 2014).
Glasl, B., Herndl, G. J. & Frade, P. R. The microbiome of coral surface mucus has a key role in mediating holobiont health and survival upon disturbance. ISME J. 10, 2280–2292 (2016).
Sunagawa, S. et al. Bacterial diversity and White Plague Disease-associated community changes in the Caribbean coral Montastraea faveolata. ISME J. 3, 512–521 (2009).
Roder, C. et al. Bacterial profiling of White Plague Disease in a comparative coral species framework. ISME J. 8, 31–39 (2014).
Morrow, K. M., Moss, A. G., Chadwick, N. E. & Liles, M. R. Bacterial associates of two caribbean coral species reveal species-specific distribution and geographic variability. Appl. Environ. Microbiol. 78, 6438–6449 (2012).
Chiarello, M. et al. Exceptional but vulnerable microbial diversity in coral reef animal surface microbiomes. Proc. R. Soc. B Biol. Sci. 287, 20200642 (2020).
Sunagawa, S., Woodley, C. M. & Medina, M. Threatened corals provide underexplored microbial habitats. PLoS ONE 5, e9554 (2010).
Zhang, C. et al. Ecological robustness of the gut microbiota in response to ingestion of transient food-borne microbes. ISME J. 10, 2235–2245 (2016).
Uren Webster, T. M. et al. Environmental plasticity and colonisation history in the Atlantic salmon microbiome: a translocation experiment. Mol. Ecol. 29, 886–898 (2020).
Fietz, K. et al. Mind the gut: genomic insights to population divergence and gut microbial composition of two marine keystone species. Microbiome 6, 82 (2018).
Smith, C. C., Snowberg, L. K., Caporaso, J. G., Knight, R. & Bolnick, D. I. Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. ISME J. 9, 2515 (2015).
Uren Webster, T. M., Consuegra, S., Hitchings, M. & Garcia de Leaniz, C. Interpopulation variation in the Atlantic salmon microbiome reflects environmental and genetic diversity. Appl. Environ. Microbiol. 84, e00691-18 (2018).
Fiore, C. L., Labrie, M., Jarett, J. K. & Lesser, M. P. Transcriptional activity of the giant barrel sponge, Xestospongia muta holobiont: molecular evidence for metabolic interchange. Front. Microbiol. 6, 364 (2015).
Neave, M. J., Michell, C. T., Apprill, A. & Voolstra, C. R. Endozoicomonas genomes reveal functional adaptation and plasticity in bacterial strains symbiotically associated with diverse marine hosts. Sci. Rep. 7, 40579 (2017).
Pogoreutz, C. et al. Dominance of Endozoicomonas bacteria throughout coral bleaching and mortality suggests structural inflexibility of the Pocillopora verrucosa microbiome. Ecol. Evol. 8, 2240–2252 (2018).
Reverter, M., Sasal, P., Tapissier-Bontemps, N., Lecchini, D. & Suzuki, M. Characterisation of the gill mucosal bacterial communities of four butterflyfish species: a reservoir of bacterial diversity in coral reef ecosystems. FEMS Microbiol. Ecol. 93 (2017).
Parris, D. J., Brooker, R. M., Morgan, M. A., Dixson, D. L. & Stewart, F. J. Whole gut microbiome composition of damselfish and cardinalfish before and after reef settlement. PeerJ 4, e2412 (2016).
Reese, E. S. Coevolution of corals and coral feeding fishes of the family Chaetodontidae. In Proc. 3rd International Coral Reef Symposium, 267–274 (Rosenstiel School of Marine and Atmospheric Science, Miami, Florida., 1977).
Hammer, T. J. & Bowers, M. D. Gut microbes may facilitate insect herbivory of chemically defended plants. Oecologia 179, 1–14 (2015).
Kohl, K. D., Weiss, R. B., Cox, J., Dale, C. & Denise Dearing, M. Gut microbes of mammalian herbivores facilitate intake of plant toxins. Ecol. Lett. 17, 1238–1246 (2014).
Emslie, M. J., Pratchett, M. S., Cheal, A. J. & Osborne, K. Great Barrier Reef butterflyfish community structure: the role of shelf position and benthic community type. Coral Reefs 29, 705–715 (2010).
Noble, M. M., Pratchett, M. S., Coker, D. J., Cvitanovic, C. & Fulton, C. J. Foraging in corallivorous butterflyfish varies with wave exposure. Coral Reefs 33, 351–361 (2014).
Greb, L. et al. Ökologie und Sedimentologie eines rezenten Rampensystems an der Karibikküste von Panamá (Inst. für Geologie und Paläontologie, Stuttgart, 1996).
Aronson, R., Hilbun, N., Bianchi, T., Filley, T. & McKee, B. Land use, water quality, and the history of coral assemblages at Bocas del Toro, Panamá. Mar. Ecol. Prog. Ser. 504, 159–170 (2014).
Collin, R., D’Croz, L., Gondola, P. & Del Rosario, J. B. Climate and hydrological factors affecting variation in chlorophyll concentration and water clarity in the Bahia Almirante, Panama. Smithson. Contrib. Mar. Sci. 323–334 (2009).
D’Croz, L., Rosario, J. B.del. & Gondola, P. The effect of fresh water runoff on the distribution of dissolved inorganic nutrients and plankton in the Bocas del Toro Archipelago, Caribbean Panamá. Caribb. J. Sci. 41, 414–429 (2005).
Seemann, J. et al. Assessing the ecological effects of human impacts on coral reefs in Bocas del Toro, Panama. Environ. Monit. Assess. 186, 1747–1763 (2014).
Guzmán, H. M., Barnes, P. A. G., Lovelock, C. E. & Feller, I. C. A site description of the CARICOMP mangrove, seagrass and coral reef sites in Bocas del Toro, Panamá. Caribb. J. Sci. 41, 430–440 (2005).
Beijbom, O. et al. Towards automated annotation of benthic survey images: variability of human experts and operational modes of automation. PLoS ONE 10, e0130312 (2015).
Rocha, L. A., Jogan, J., Király, G., Feráková, V. & Bernhardt, K.-G. Chaetodon capistratus. The IUCN Red List of Threatened Species. https://doi.org/10.2305/IUCN.UK.2010-4.RLTS.T165695A6094300.en (2010).
Froese, R. & D. P. E. FishBase. FishBase. 2019. www.fishbase.org (2020)
Smith, L. C. National Audubon Society Field Guide to Tropical Marine Fishes Caribbean, Gulf of Mexico, Florida, Bahamas, Bermuda (Alfred A. Knopf, 1997).
Nguyen, B. N. et al. Environmental DNA survey captures patterns of fish and invertebrate diversity across a tropical seascape. Sci. Rep. 10, 1–14 (2020).
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414 (2016).
Apprill, A., McNally, S., Parsons, R. & Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 75, 129–137 (2015).
Weber, L. et al. EMP 16S Illumina amplicon protocol. https://doi.org/10.17504/protocols.io.nuudeww (2018).
R Core Team. R: a language and environment for statistical computing. (R Foundation for Statistical Computing, Vienna, Austria, 2019).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10 (2011).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581 (2016).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Wright, E. S. Using DECIPHER v2.0 to analyze big biological sequence data in R. R. J. 8, 352–359 (2016).
Schliep, K., Potts, A. J., Morrison, D. A. & Grimm, G. W. Intertwining phylogenetic trees and networks. Methods Ecol. Evol. 8, 1212–1220 (2017).
Weiss, S. et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5, 27 (2017).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217 (2013).
Astudillo-García, C. et al. Evaluating the core microbiota in complex communities: a systematic investigation. Environ. Microbiol. 19, 1450–1462 (2017).
Dufrêne, M. & Legendre, P. Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecol. Monogr. 67, 345–366 (1997).
Roberts, D. W. labdsv: ordination and multivariate analysis for ecology. (2019).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Leray, M. & Knowlton, N. Random sampling causes the low reproducibility of rare eukaryotic OTUs in Illumina COI metabarcoding. PeerJ 5, e3006 (2017).
Hill, M. O. Diversity and evenness: a unifying notation and its consequences. Ecology 54, 427–432 (1973).
Alberdi, A. & Gilbert, M. T. P. A guide to the application of Hill numbers to DNA‐based diversity analyses. Mol. Ecol. Resour. 19, 1755–0998.13014 (2019).
Jost, L. Entropy and diversity. Oikos 113, 363–375 (2006).
Chiu, C. H. & Chao, A. Estimating and comparing microbial diversity in the presence of sequencing errors. PeerJ 2016, e1634 (2016).
Oksanen, J. et al. Community Ecology Package. Vienna R Found. Stat. Comput. https://doi.org/10.4135/9781412971874.n145 (2012).
Chen, J. et al. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 28, 2106–2113 (2012).
Lozupone, C. A., Hamady, M., Kelley, S. T. & Knight, R. Quantitative and qualitative diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585 (2007).
Jaccard, P. The distribution of the flora in the alpine zone.1. N. Phytol. 11, 37–50 (1912).
Anderson, M. J., Ellingsen, K. E. & McArdle, B. H. Multivariate dispersion as a measure of beta diversity. Ecol. Lett. 9, 683–693 (2006).
Bray, J. R. & Curtis, J. T. An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 27, 325–349 (1957).
Anderson, M. J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46 (2001).
Anderson, M. J. & Walsh, D. C. I. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: what null hypothesis are you testing? Ecol. Monogr. 83, 557–574 (2013).
Martinez Arbizu, P. pairwiseAdonis: pairwise multilevel comparison using adonis. R package version 0.3. https://github.com/pmartinezarbizu/pairwiseAdonis (2019).
Roesch, L. F. W. et al. Pime: a package for discovery of novel differences among microbial communities. Mol. Ecol. Resour. 20, 415–428 (2020).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Klaus, J. S., Janse, I., Heikoop, J. M., Sanford, R. A. & Fouke, B. W. Coral microbial communities, zooxanthellae and mucus along gradients of seawater depth and coastal pollution. Environ. Microbiol. 9, 1291–1305 (2007).
Ward, R. J. et al. Gastrointestinal Bacterial Symbionts: Reproductive Strategy and Community Structure. Thesis, Cornell Univ. (2009).
Séré, M. G. et al. Bacterial communities associated with Porites White Patch Syndrome (PWPS) on three Western Indian Ocean (WIO) coral reefs. PLoS ONE 8, e83746 (2013).
Moran, D., Turner, S. J. & Clements, K. D. Ontogenetic development of the gastrointestinal microbiota in the marine herbivorous fish Kyphosus sydneyanus. Microb. Ecol. 49, 590–597 (2005).
Mausz, M., Schmitz-Esser, S. & Steiner, G. Identification and comparative analysis of the endosymbionts of Loripes lacteus and Anodontia fragilis (Bivalvia: Lucinidae). (University of Vienna, 2008).
Bano, N., DeRae Smith, A., Bennett, W., Vasquez, L. & Hollibaugh, J. T. Dominance of mycoplasma in the guts of the long-jawed mudsucker, Gillichthys mirabilis, from five California salt marshes. Environ. Microbiol. 9, 2636–2641 (2007).
Frade, P. R., Roll, K., Bergauer, K. & Herndl, G. J. Archaeal and Bacterial Communities associated with the surface mucus of Caribbean corals differ in their degree of host specificity and community turnover over reefs. PLoS ONE 11, e0144702 (2016).
Turnbaugh, P. J. et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484 (2009).
Ley, R. E., Turnbaugh, P. J., Klein, S. & Gordon, J. I. Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023 (2006).
Kimes, N. E. et al. The Montastraea faveolata microbiome: ecological and temporal influences on a Caribbean reef-building coral in decline. Environ. Microbiol. 15, 2082–2094 (2013).
Smriga, S., Sandin, S. A. & Azam, F. Abundance, diversity, and activity of microbial assemblages associated with coral reef fish guts and feces. FEMS Microbiol. Ecol. 73, no–no (2010).
Zhang, X. et al. Effects of dietary supplementation of Ulva pertusa and non-starch polysaccharide enzymes on gut microbiota of Siganus canaliculatus. J. Oceanol. Limnol. 36, 438–449 (2018).
Klaus, J. S., Janse, I. & Fouke, B. W. Coral black band disease microbial communities and genotypic variability of the dominant cyanobacteria (CD1C11). Bull. Mar. Sci. 87, 795–821 (2011).
Lu, J., Santo Domingo, J. W., Hill, S. & Edge, T. A. Microbial diversity and host-specific sequences of Canada goose feces. Appl. Environ. Microbiol. 75, 5919–5926 (2009).
Ueki, A., Goto, K., Ohtaki, Y., Kaku, N. & Ueki, K. Description of Anaerotignum aminivorans gen. Nov., sp. nov., a strictly anaerobic, amino-acid-decomposing bacterium isolated from a methanogenic reactor, and reclassification of Clostridium propionicum, Clostridium neopropionicum and Clostridium lactatifermentans as species of the genus Anaerotignum. Int. J. Syst. Evol. Microbiol. 67, 4146–4153 (2017).
Bowman, K. S., Rainey, F. A. & Moe, W. M. Production of hydrogen by Clostridium species in the presence of chlorinated solvents. FEMS Microbiol. Lett. 290, 188–194 (2008).
Bueno de Mesquita, C. P., Sartwell, S. A., Schmidt, S. K. & Suding, K. N. Growing‐season length and soil microbes influence the performance of a generalist bunchgrass beyond its current range. Ecology 101, e03095 (2020).
Clever, F. et al. The gut microbiome variability of a butterflyfish increases on severely degraded Caribbean reefs. Dryad Datasets. https://doi.org/10.5061/dryad.m905qfv28 (2022).
Clever, F. & Scott, J. J. R code for reproducing the statistical analyses and figures of ‘The gut microbiome variability of a butterflyfish increases on severely degraded Caribbean reefs’. Commun. Biol. https://github.com/bocasbiome/web/ (2022).
Acknowledgements
We thank Lucia Rodriguez for field assistance, Joan Antaneda for her help in the laboratory, Ross Whippo for conducting the fish survey, and Clare Fieseler for taking photos of the benthos. The staff of the Bocas del Toro Research Station provided logistical support. We are grateful to Kristin Saltonstall and Marta Vargas for their support at the Smithsonian Tropical Research Institute’s (STRI) Ecological and Evolutionary Genomics Laboratory. Friederike Clever was supported by a Smithsonian Short Term Fellowship. This project was funded, in part, by a grant from the Gordon and Betty Moore Foundation to STRI and UC Davis (PIs: William Wcislo and Jonathan Eisen; https://doi.org/10.37807/GBMF5603) and a Ph.D. studentship by Manchester Metropolitan University to Friederike Clever. A research permit was issued by the Ministerio de Ambiente Panamá (No. SE/A-113-17).
Author information
Authors and Affiliations
Contributions
F.C., M.L., and J.J.S. conceived the study. F.C., M.L., and R.F.P. designed the study with input from A.H.A. F.C., J.J.S., and M.L. conducted the fieldwork. E.C.R.G. and F.C. dissected the fish guts. F.C. extracted the DNA. J.M.S. and M.L. prepared the DNA for sequencing and processed the sequencing data. A.H.A., J.A.E., M.L., R.F.P., and W.O.M. contributed reagents and supplies. E.C.R.G. analyzed the photographic benthic quadrats. F.C., J.M.S., and M.L. analyzed the data and wrote the first draft of the manuscript with input from L.G.E.W. and R.F.P. All authors reviewed the manuscript and contributed to the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Linn Hoffmann and Caitlin Karniski.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Clever, F., Sourisse, J.M., Preziosi, R.F. et al. The gut microbiome variability of a butterflyfish increases on severely degraded Caribbean reefs. Commun Biol 5, 770 (2022). https://doi.org/10.1038/s42003-022-03679-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03679-0
This article is cited by
-
The coral microbiome in sickness, in health and in a changing world
Nature Reviews Microbiology (2024)
-
Gut microbial communities of hybridising pygmy angelfishes reflect species boundaries
Communications Biology (2023)
-
The gut microbiome variability of a butterflyfish increases on severely degraded Caribbean reefs
Communications Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
