
Abstract
GBA variants carriers are at increased risk of Parkinson’s disease (PD) and Lewy body dementia (LBD). The presence of pseudogene GBAP1 predisposes to structural variants, complicating genetic analysis. We present two methods to resolve recombinant alleles and other variants in GBA: Gauchian, a tool for short-read, whole-genome sequencing data analysis, and Oxford Nanopore sequencing after PCR enrichment. Both methods were concordant for 42 samples carrying a range of recombinants and GBAP1-related mutations, and Gauchian outperformed the GATK Best Practices pipeline. Applying Gauchian to sequencing of over 10,000 individuals shows that copy number variants (CNVs) spanning GBAP1 are relatively common in Africans. CNV frequencies in PD and LBD are similar to controls. Gains may coexist with other mutations in patients, and a modifying effect cannot be excluded. Gauchian detects more GBA variants in LBD than PD, especially severe ones. These findings highlight the importance of accurate GBA analysis in these patients.
Similar content being viewed by others

Introduction
The GBA gene encodes the lysosomal enzyme glucocerebrosidase, and biallelic mutations in GBA cause the autosomal recessive disorder Gaucher disease (GD [MIM: #230800, #230900 and #231000])1. Around 500 disease-causing mutations have been reported, mostly missense changes arising from single nucleotide variants (SNVs)2. Heterozygous variants in GBA [MIM: *606463] are associated with an increased risk of developing Parkinson's disease (PD)3, the second most common neurodegenerative disease, and the closely related Lewy body dementia (LBD)4. Identifying GBA mutations is difficult due to a pseudogene (GBAP1) located 6.9 kb downstream5 which has an overall homology of 96% with GBA. This rises to 98% in the region from intron 8 to the 3′-UTR, where there are five identical segments >200 bp each6. The high homology predisposes to non-allelic homologous recombination between GBA and GBAP1, leading to a wide range of structural variants (SV)7. These can be non-reciprocal, also termed gene conversion, or reciprocal, resulting in copy number variants (CNV). Throughout this paper, we use the term copy number gain (CNG) for reciprocal duplication alleles where a 20.6 kb long region of DNA between the homology segments of GBA and GBAP1 is multiplied, and copy number loss (CNL) for reciprocal fusion alleles where the same region is deleted, creating GBA-GBAP1 fusions7 (see Fig. 1). SVs that disrupt the coding sequence by gene conversion or reciprocal recombination are expected to be pathogenic for GD and risk factors for PD. Conversely, SVs not affecting the coding sequence are not pathogenic, although a modifier effect cannot be excluded7. These include CNLs outside the coding region, and all CNGs, which consist of a partial duplication of pseudogene sequence merged with a variable part of the gene, often only the 3′ UTR, with the resulting allele still containing a normal copy of the GBA coding region (Fig. 1d). The SV variability and population prevalence remain largely unknown. Pathogenic missense changes in the high homology exon 9–11 region such as the common p.L483P (NC_000001.11:g.155235252 A > G, also known as p.L444P) may arise by gene conversion, rather than simple base substitutions, with pseudogene sequence incorporated into the gene7. We refer to variants corresponding to pseudogene bases in this region as GBAP1-like. The most common GBAP1-like variants introduced by either gene conversion or reciprocal recombination in the exon 9–11 homology region include p.L483P, p.D448H (NC_000001.11:g.155235727 C > G), c.1263del55 (NC_000001.11:g.155235752_155235806del), RecNciI (which comprises three SNVs: p.L483P, p.A495P and p.Val499=), RecTL (RecNciI and p.D448H) and c.1263del+RecTL (RecNciI, p.D448H and c.1263del55).

Not to scale, corresponding roughly to g.chr1:155,210,000-155,245,000. a Wild-type allele. Only primer pair 1 will produce an amplicon. b Non-reciprocal recombination (gene conversion). Similar to non-recombinant alleles, only primer pair 1 will produce an amplicon. c Reciprocal crossover between gene and pseudogene resulting in a 20.6 kb deletion (CNL). Only primer pair 2 will produce an amplicon. d Reciprocal crossover between gene and pseudogene resulting in a 20.6 kb duplication (CNG). Both primer pair 1 and primer pair 3 will produce amplicons. Note that the normal allele is present and that amplification with primer pair 3 will produce an amplicon independently of the number of copy number gains. CNG copy number gain, CNL copy number loss.
Current sequencing approaches to characterise GBA have major pitfalls, and to date, no single approach has fully resolved recombinants6. The correct alignment of short reads when there is a highly similar pseudogene is intrinsically problematic, and GBA is challenging in exome and whole-genome sequencing (WGS)8,9,10, containing camouflaged regions11. Moreover, the reliability of the standard WGS secondary analysis pipelines such as the Genome Analysis Toolkit best practice workflow12 has not been formally assessed. Targeted short-read sequencing approaches are also possible but may require forced alignment to GBA and visual inspection and Sanger validation to detect recombinant variants, and are not likely to provide copy number information6,13. We have already performed refinement of the Illumina WGS analysis for other difficult regions due to sequence homology, demonstrating reliable resolution of SVs in such regions on Illumina WGS data in the SMN1[MIM: *600354]/SMN2[MIM: *601627] genes in spinal muscular atrophy14 and the pharmacogene CYP2D6[MIM: *124030]15. We also previously reported a method for GBA analysis using enrichment by long-range PCR, followed by sequencing on the Oxford Nanopore Technologies (ONT) MinION16, which reliably detected SNVs, including GBAP1-like variants, and could also detect non-reciprocal recombinants, but not reciprocal recombinants (Fig. 1)16.
To overcome these limitations and improve the characterisation of GBA at scale, we have developed refined pipelines based on either targeted analysis of short-read (Illumina) WGS data or targeted long-read (ONT) single-molecule sequencing. For Illumina data, we present and validate ‘Gauchian’, a novel algorithm for GBA locus analysis which can reliably resolve SVs and GBAP1-like variants. For ONT data, we have addressed the problem of reciprocal recombinants by using PCR primers designed to amplify CNGs and CNLs when they exist. We validated these methods and then applied them to large PD, LBD, and population control samples. We demonstrate that complete resolution of all variant types in GBA is possible using either Gauchian analysis of Illumina WGS data or targeted ONT sequencing. Finally, we confirm that GBA variants are more common in LBD than in PD, we report the frequency of CNVs in different populations and suggest that a possible modifier role of CNG in PD and LBD merits further study. Both methods finally enable precise characterisation of GBA at scale, thus driving the identification of causative variants forward.
Results
Cross-validation confirms both Gauchian and ONT methods
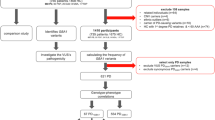
To select appropriate samples for validation of Gauchian with a broad range of mutations, we first obtained Gauchian results on 1000 Genomes Project (1kGP) samples and the Accelerating Medicines Partnership Parkinson’s Disease (AMP-PD) PD and control cohorts. We selected 37 of these for validation by ONT targeted sequencing. These included 15 samples from 1kGP with CNVs or gene conversions, and 22 samples from Parkinson’s Progression Markers Initiative (PPMI—included in AMP-PD cohort), where Gauchian showed CNVs or GBAP1-related variants (n = 7), or was discordant with available Broad Institute’s Joint Genotyping pipeline (referred to as BWA-GATK in this paper) results (n = 4), or no mutation was reported by Gauchian or BWA-GATK (n = 11). Additionally, for 5 brain DNA samples analysed first by ONT with recombinations or GBAP1-related mutations, we performed Illumina WGS and Gauchian analysis. All 42 Gauchian results were consistent with ONT. Within these validation samples, Gauchian reported five CNL, which included one in which the p.L483P was also found, one resulting in the pathogenic RecNciI, and 14 CNGs, including two samples which also carried a gene conversion, and one which carried p.L483P. Additionally, in 11 samples Gauchian called GBAP1-like variant calls within GBA, including two gene conversions. The remaining 12 samples were wild-type calls. Notably, Gauchian and ONT gave concordant results in two samples where previous BWA-GATK analysis had missed p.L483P, and two where BWA-GATK had wrongly called p.A495P (NC_000001.11:155235216:C:G). Results of cross-validation of the two methods are reported in Table 1.
To obtain further orthogonal validation, we used digital PCR (dPCR) for copy number estimation of the 20.6 kb region involved in recombination in six samples with a range of copy numbers. These included four samples where Gauchian and ONT both detected a CNG (additional copy numbers 1, 3, 5 and 6), and two where we only had ONT data, one with a CNL, and one with no CNV. The results were fully concordant (Supplementary Table 1). Finally, we applied ONT sequencing with PCR-free enrichment by adaptive sampling (UNCALLED)17 to four reciprocal recombinants, two CNG and two CNL (one pathogenic and one non-pathogenic). Inspection of the resulting alignments confirmed the presence of the SV and the breakpoints of CNL alleles (Supplementary Fig. 1).
Detection of all classes of GBA variants with targeted ONT long-read sequencing
To fully resolve the GBA gene with ONT, we used three pairs of primers, as detailed in the methods section. Primer pair 1 is designed to amplify the GBA gene, as previously described16. Primer pairs 2 and 3 are used to detect CNL and CNG alleles, respectively.
We analysed 397 samples from PD or GD patients, their relatives, and controls (Supplementary Table 2) using all three pairs of PCR primers, followed by ONT amplicon sequencing. These included 95 individuals previously sequenced with ONT using only primer pair 116. All results are shown in Supplementary Table 3. We detected two c.1263del + RecTL alleles and one RecNciI allele arising from gene conversion. Additionally, we also detected 94 coding or splice site SNVs, including the pathogenic mutations p.N409S (NC_000001.11:g.155235843 T > G, also known as p.N370S; 38% of all SNV detected) and the GBAP1-like p.L483P (20%), and the PD risk alleles p.E365K (NC_000001.11:g.155236376 C > T, also known as p.E326K, 16%) and p.T408M (NC_000001.11:g.155236246 G > A, also known as p.T369M, 4%). Notably, we also detected c.84dupG (NC_000001.11:g.155240661dup), the most common pathogenic indel in the GBA gene. As homopolymer regions are challenging for ONT18, and GBA has two coding poly-G stretches, we devised a method to detect variants within these (Supplementary Fig. 2). This additional analysis identified one single base deletion and one SNV within homopolymers that would have been missed by our old ONT pipeline (c.413delC and p.P68=, NC_000001.11:g.155239661del and NC_000001.11:g.155239989 C > T, Supplementary Fig. 2). We detected CNLs using primer pair 2 in nine samples. According to the position of the breakpoints (Supplementary Fig. 3), five of them were pathogenic, and four were not. Two non-pathogenic CNLs were in cis with p.L483P, a pattern already described7. We also detected a CNG using primer pair 3 in seven samples, four of which also carried a GBAP1-like variant (two c.1263del+RecTL and two p.L483P).
Comprehensive GBA analysis by Gauchian in short-read WGS population data
A total of 10623 samples were analysed with Gauchian, including 2504 samples from the 1kGP cohort, 2325 PD and 2598 LBD samples from the AMP-PD knowledge portal, and their respective controls. We identified 55 non-pathogenic CNLs and 146 CNGs (roughly correspond to DGV variant accessions dgv55e214 and esv3587619; Table 2). Additionally, we detected 97 GBAP1-like variants (including those generated by pathogenic CNL or gene conversion) in the exons 9–11 homology region of GBA in all three cohorts (Table 3 and Supplementary Table 4).
BWA-GATK variant calls were available for all AMP-PD samples analysed by Gauchian. For all PD and LBD case/control populations, BWA-GATK called 44 GBAP1-like variants, and Gauchian called 86, almost doubling the variant calls. Due to the sequence homology and misalignment of reads in exons 9–11, the BWA-GATK pipeline under-called all GBAP1-like variants except p.A495P and p.D448H. For p.A495P, GATK called 11 false positives (including two confirmed as false positives by ONT amplicon sequencing- see earlier) and for p.D448H BWA-GATK called two false positives. The false-positive calls by BWA-GATK are due to alignment errors caused by GBAP1 haplotypes containing GBA bases (see Fig. 2c). Gauchian also detected other coding SNVs and indels that are not GBAP1-like in the three cohorts (see Supplementary Table 5 for all variants). All these calls were concordant with BWA-GATK except in one sample where Gauchian called p.L483R, a rare pathogenic variant in the same codon as the common GBAP1-like p.L483P, but BWA-GATK did not. This variant is in the exon 9–11 homology region, and the variant reads misaligned to GBAP1, causing the false-negative by BWA-GATK (Supplementary Fig. 4).

a Median mapping quality (red line) across 2504 1kGP samples plotted for each position in the GBA/GBAP1 region (hg38). A median filter is applied in a 50 bp window. The eleven exons of GBA are shown as orange boxes. GBAP1 and MTX1 exons are shown as green and purple boxes, respectively. The 4 kb major homology region (98.1% sequence similarity, exons 9–11) between GBA and GBAP1 is shaded in pink and highlights an area of low mapping accuracy. The light blue box shows the 10 kb unique region between the two genes in which copy number calling is performed in Gauchian. b Distribution of normalised depth in the 10 kb CN calling region in 2504 1kGP samples, showing peaks at CN1 (CNL), 2 (no CNV) and 3-8 (CNG). c Recombinant haplotypes in the exons 9–11 homology region, distinguished by GBA/GBAP1 differentiating bases (x-axis). Reference genome sequences are shaded in yellow. There is an error in hg38 where the first three sites of GBAP1 show GBA bases, which could lead to alignment errors. The GBA recombinant haplotypes are shown in the white background, including those where one or a few nearby sites are mutated to the corresponding GBAP1 base, resulting from either gene conversion or CNL. Grey bases indicate that the base can be either GBA or GBAP1 depending on the breakpoint position of the CNL/conversion. Shaded in purple are two example GBAP1 haplotypes, found by Gauchian, that have been partially converted to GBA and can cause false-positive GBA variant calls by standard secondary analysis pipelines. For the first example, the reverse-p.L483P variant on GBAP1 directs aligners to align GBAP1 reads to GBA, causing the nearby p.A495P false-positive call. For the second example, the reverse-c.1263del variant inserts 55 bp to GBAP1, driving GBAP1 reads to align to GBA, causing the nearby p.D448H false-positive call. CNG copy number gain, CNL copy number loss, CNV copy number variant.
To investigate the Mendelian consistency of Gauchian calls and the prevalence of de novo variants, we analysed 602 trios in the 1kGP dataset. In eight trios, the proband carried a GBA missense variant, one in the exons 9–11 homology region and 7 outside of this region. In all cases, the variants were inherited by one parent and no de novo variants were detected (Supplementary Table 6).
Prevalence of GBA recombinant and non-recombinant variants in healthy, PD and LBD populations
Gauchian allowed us to provide a comprehensive large-scale analysis of all classes of GBA mutations in healthy, PD and LBD populations. Non-pathogenic CNVs, where the breakpoints do not alter the GBA coding region, were ten times more frequent in 'Africans' than 'Europeans' (11.3 vs 1.1%, Table 2). This was primarily driven by a striking difference in the prevalence of CNGs (10.8 vs. 0.6% for controls from both cohorts; p < 2.2e-16). Additionally, 'Africans' also had more copies gained, with a median gain of three copies compared to one for 'Europeans'.
As non-pathogenic CNVs in GBA have not previously been considered as possible PD or LBD risk factors, we compared these across the combined disease cohorts to their controls. We detected no difference in the prevalence of all non-pathogenic CNVs (1.10 vs 1.25%), CNGs (0.67 vs 0.63%) or CNLs (0.43 vs 0.63%, Table 2). Addressing SNVs next, we noticed that p.N409S was found at a very high frequency in the PD cohort of AMP-PD (in both cases and controls, 5.5 and 12.6% respectively), because of the recruitment of a large number of individuals with Ashkenazi Jewish ancestry19, where it is very common. After excluding individuals carrying it from both cohorts for consistency, GBA variants were more common in each disease cohort than in the respective controls as expected (Table 4) (PD 7.8 vs 3.9%; LBD 11.7 vs 3.5%). This was also true for severe GBA variants20 (PD 1.7 vs 0.8%; LBD 3.1 vs 0.1%). The overall OR for mutations in each disease against its controls was higher in LBD than in PD (3.68 v 2.07; p = 0.0098), and this was even more striking for the severe mutations (30.83 v 2.12; p = 0.0009).
We also noted that some individuals carried both a CNG and another GBA variant, mostly a GBAP1-like variant in the exon 9–11 homology region (Table 2), as also seen in the cohort analysed by ONT. There were no individuals with a CNG and another GBA variant in the 1kGP cohort.
Considering all samples analysed by Gauchian, seven out of 146 with a CNG also had a GBAP1-like variant, against 71 of 10,407 without a CNG (4.8 vs 0.68%; p value = 9.77e-5). Three additional individuals carried a non-GBAP1-like variant and a CNG. In the PD and LBD cohorts and their controls, nine of ten individuals carrying a CNG as well as a coding variant in GBA were patients (four PD and five LBD). One healthy control carried a CNG and p.T408M, which is a mild PD risk allele but does not cause GD. As we did not find any healthy controls with a CNG and a pathogenic variant, we considered whether the combination of both is more detrimental than a coding variant alone (excluding again p.N409S carriers, one of whom also had a CNG). We did not detect a significant added risk for disease in the combined PD and LBD AMP-PD data against their controls (OR for CNG and other variants vs other variants alone 2.31, 95% CI 0.37–45.01).
Discussion
The recent dramatic improvements in sequencing techniques have allowed a much better understanding of human genetic variation, but several regions, including some key disease-related genes, have remained challenging. One example is GBA, responsible for the autosomal recessive lysosomal storage disorder GD1, and one of the most important genetic determinants of risk for PD and the closely related LBD21. Here we present and validate Gauchian, a novel GBA caller for Illumina WGS data, capable of detecting SVs and SNVs within GBA. Using ONT targeted sequencing, we demonstrate that in the cases of discrepant calls between Gauchian and BWA-GATK analysis, the Gauchian calls are correct. We also demonstrate that a refined ONT amplicon-sequencing pipeline can detect reciprocal recombinants, and indels as well as mutations within homopolymers in coding exons. Importantly, both methods detect CNGs and CNLs arising from reciprocal recombination and allow straightforward classification of CNLs into those that do and do not affect the coding region, previously a complex task8,22. We thus provide two complementary new tools for fully resolving the GBA gene, which will be helpful to the community. Illumina WGS data can now be analysed robustly, and ONT targeted sequencing can be applied in a cost-effective way where analysis of GBA is sufficient (see cost details in Supplementary Table 7). Furthermore, the principles used in Gauchian can be applied to the analysis of other regions camouflaged due to gene duplication11.
To explore the potential of Gauchian in the population and in disease contexts, we applied it to a total of 10,623 samples from the 1000 Genomes Project and PD and LBD cohorts with their controls from the AMP-PD initiative. This allowed us to provide the first large-scale data on CNVs, and to evaluate the frequency of all classes of GBA variants in PD and LBD with greater accuracy than before. Reciprocal recombinants in particular are likely missed in PD studies16, including a recent targeted short-read study which detected none in 3402 patients8, although one study using exome data with qPCR validation reported CNGs in 1.2% of PD and 0.7% of controls22. In non-diseased individuals, we noted that CNGs were more common in those with “African” ancestry, with greater copy number variability. These results are consistent with the greater African genetic diversity, with recent evidence of “African” genomes demonstrating unexplored structural variation23 and more variability in copy numbers of SMN1, SMN2 and CYP2D614,15. They further highlight the need to study non-European genomes, which has yielded additional insights into Alzheimer’s disease24 and is being expanded in PD25.
In the PD and LBD cohorts, Gauchian analysis almost doubled the pathogenic variants detected in the homology region compared to BWA-GATK (86 vs 44) and eliminated false positives. We also performed a direct comparison of GBA variant frequencies between PD and LBD, after excluding the common p.N409S variant due to selection bias in the PD cohort19. The prevalence of GBA pathogenic or PD-risk variants was significantly higher in LBD than in PD (11.7 vs 7.8%), and this difference was even larger for severe pathogenic variants (3.1 vs 1.7%). The OR for GBA mutations in LBD compared to controls was higher in our analysis than in the original report in this cohort21 (3.68 v 2.90), and a previous study (2.55)4, due to the detection of additional mutations and the filtering of p.N409S. GBA mutations increase the risk of cognitive decline in PD26, and the odds ratio for GBA variants is higher in LBD than in PD with dementia27. Severe GBA variants in particular, which cause the neuronopathic form of GD1, have a higher risk of PD28 and a faster cognitive decline in PD than mild variants29. If PD and LBD are considered as a spectrum of phenotypes with variable cognitive involvement, our findings further suggest that GBA variants, especially severe ones, tend to predispose to a phenotype on the LBD end of the spectrum. The main limitation of this analysis is the use of LBD and PD cohorts recruited separately, with the selection process necessitating the exclusion of p.N409S, and further comparisons in unselected matched cohorts are needed. The variable penetrance and phenotypic heterogeneity of PD and LBD patients with GBA mutations is attracting a lot of attention, with lysosomal gene variants acting as genetic modifiers30. An effect of common intronic haplotypes was suggested31, but not seen by us in the AMP-PD cohort and part of the RAPSODI cohort used here32. A possible influence of CNGs has not yet been investigated. Although these do not alter the GBA coding region, they could affect expression and function, for example by acting as a competing endogenous microRNA sponge33. CNGs were not enriched in PD or LBD. There were, however, rare carriers of both a CNG and a pathogenic or PD-associated GBA variant. In the PD and LBD cohorts, nine out of ten of these were patients and the only control coding variant was the mild PD risk allele p.T408M. This raises the possibility that CNGs are modifiers, increasing the penetrance of other GBA variants. We have not, however, phased the CNGs and other variants, and did not show a statistically significant increased risk for carriers of a CNG and mutation compared to mutation alone. Therefore, further population and mechanistic work is required.
In conclusion, we have demonstrated that SNV detection and complete resolution of all classes of SVs is possible using the novel Gauchian caller with Illumina WGS, which outperforms BWA-GATK analysis, or with targeted ONT sequencing. We also demonstrate that CNVs are relatively common, and suggest that these merit investigation as possible modifiers of PD or LBD risk. Given the importance of this gene and the rapid progress to targeted clinical trials in PD34, we propose that the adoption of either workflow should be considered by research and diagnostic labs, based on local resources and data availability.
Methods
Population cohorts and samples used
We downloaded WGS CRAM files from the 1kGP. These were generated by 2 × 150 bp reads on Illumina NovaSeq 6000 instruments from PCR-free libraries sequenced to an average depth of at least 30x and aligned to the human reference, hs38DH, using BWA-MEM v0.7.15. We downloaded WGS CRAM files from PD19 and DLB21 cohorts and their controls from the AMP-PD knowledge portal. These were generated by sequencing 2 × 150 bp reads to >25x coverage and processing against hs38DH using the Broad Institute’s implementation of the Functional Equivalence Pipeline35. We also downloaded AMP-PD variant calls, generated using BWA-GATK. Where samples had been recorded as European/Caucasian/white, or African/black in the original database, we refer to them as 'European' or 'African' for consistency and simplicity, despite the lack of scientific validity, in quotation marks as suggested36.
Selected DNA samples were obtained from the PPMI37 and the NHGRI Sample Repository for Human Genetic Research at the Coriell Institute for Medical Research. DNA samples from living individuals for ONT analysis were obtained from a clinical cohort, RAPSODI38, which aims to define the risk of PD in GBA mutation carriers. Recruitment and analysis are ongoing, and the clinical results will be reported separately. DNA from saliva (Oragene DNA OG-500 kit, DNA Genotek) was extracted according to the manufacturer's protocol. Brain samples from 16 PD patients were obtained from the Queen Square Brain Bank, and DNA was extracted with phenol-chloroform39 or MagAttract HMW DNA kit (Qiagen) from the frontal cortex, cerebellum or midbrain. Ethics approval was provided by the National Research Ethics Service London—Hampstead Ethics Committee for RAPSODI, NRES Committee central—London for QSBB samples, and UCL Ethics Committee for PPMI samples. All participants provided informed consent.
Gauchian— a WGS-based GBA caller
Gauchian builds upon the strategies to solve closely related paralogs, as described in our previously developed SMN1/2 and CYP2D6 callers14,15. Gauchian calculates the total number of copies of GBA, GBAP1 and GBA/GBAP1 gene hybrids. Reciprocal recombinations across homologous regions lead to CNG and CNL of the 20.6 kb region between the homologous parts of the two genes. Since the breakpoint may vary in position, to detect CNVs, Gauchian uses the sequencing depth in the 10 kb unique region between GBA and GBAP1 (chr1:155220429-155230539; hg38) (Fig. 2a). The number of reads aligned to this region is normalised and corrected for GC content, and the copy number is called from a Gaussian mixture model (Fig. 2b). A deviation of this copy number (CN) from the diploid expectation indicates the presence of a CNV, e.g. one copy indicates a CNL, and three or more copies indicate a CNG. Thus, this number plus two gives the total copies of GBA and GBAP1 combined, i.e. CN (GBA + GBAP1). Included in this CN calculation, in addition to GBA and GBAP1 genes, are gene hybrids where part of GBA and GBAP1 are fused. CNG always leaves an intact copy of GBA, while CNL can create pathogenic GBA-GBAP1 fusions if the deletion breakpoint falls within the GBA gene coding region.
Next, Gauchian identifies the breakpoint of the CNV, following a similar approach as previously described15. To do this, we identified 82 reliable sites (Supplementary Table 8) that differ between GBA and GBAP1. Gauchian estimates the GBA CN at each GBA/GBAP1 differentiating site based on CN (GBA + GBAP1) and the numbers of reads supporting GBA- and GBAP1-specific bases. CNV breakpoints are identified when the CN of GBA changes. For example, a transition between CN1 and CN 2 indicates the breakpoint of a CNL, and a transition between CN 3 and CN 2 indicates the breakpoint of a CNG. The exact breakpoint is further refined by haplotype phasing as described in the next paragraph.
To identify recombinant variants, Gauchian analyses the 1.1 kb homology region in exons 9–11 (Fig. 2c) containing ten GBA/GBAP1 differentiating sites that are 14–315 bp away from each other, several of which are critical GBAP1-like variants. These include p.L483P, p.D448H, c.1263del55, RecNciI, RecTL and c.1263del+RecTL (Fig. 2c). The high homology and the frequent gene conversion between GBA and GBAP1 make exons 9–11 a challenging region for standard secondary analysis pipelines, which often miscall variants due to misalignments of recombinant variant reads. Additionally, three positions in the GBAP1 reference sequence in hg38 erroneously contain the GBA bases (Fig. 2c, yellow shading), so GBA p.L483P reads would likely align to GBAP1, causing false-negative calls (GBA/GBAP1 is among the regions enriched for discordant variant calls between hg19/hg3840). In addition, we found GBAP1 haplotypes that have been partially converted to GBA. Those converted bases would direct GBAP1 reads to align to GBA, causing false-positive GBA variant calls at nearby positions (Fig. 2c, purple shading). Gauchian takes a novel approach that does not rely on accurate alignments between GBA and GBAP1. Based on the linking information of reads and read pairs covering the ten differentiating sites in either GBA or GBAP1, Gauchian phases all the haplotypes at these sites originating from either GBA or GBAP1 and identifies hybrid haplotypes (i.e. a mixture of GBA and GBAP1 bases on the same haplotype). This allows us to identify CNL breakpoints as well as small and big gene conversion events.
To assess the relative abundance of the different haplotypes, Gauchian uses CN (GBA + GBAP1) and haplotype-supporting read counts at the differentiating bases to call the CN of each haplotype. Gauchian compares two scenarios: one copy of the wild-type GBA haplotype vs. two copies of the wild-type GBA haplotype. Gauchian determines which scenario is more likely given the number of supporting reads in the data. If we call only one copy of the wild-type GBA haplotype, this indicates that the individual is a carrier of the disease-causing variant. If an individual is a carrier of more than one variant haplotype and there is no haplotype that carries the GBA base at all variant sites of interest, Gauchian calls this sample compound heterozygous. Homozygous variants are called when the CN of the GBA base is called 0.
In addition to GBAP1-like variants in exons 9–11 homology region, Gauchian targets all known GBA pathogenic or likely pathogenic variants as classified by ClinVar (Supplementary Table 9), including non-GBAP1-like variants, and GBAP1-like variants outside the exons 9–11 homology region. For these, since variants don’t correspond to GBAP1, or, if they do, the region between GBA and GBAP1 is not highly similar and alignments are accurate, Gauchian parses read alignments and calls the CN of variants based on the number of variants supporting reads as described for SMN/CYP2D6 callers. Gauchian is a targeted caller for known variants and thus does not call novel variants. The analysis takes roughly 1 min per sample when run on a standard machine using a single thread and can be speeded up with multiprocessing.
ONT long-read sequencing with PCR enrichment
GBA enrichment was obtained via PCR (Supplementary Table 10), with primer pair 1, as previously described41, modified to carry the ONT barcode adaptor sequence. The product was an 8.9 kb amplicon containing the entire GBA coding region and introns (chr1:155232524-155241392; hg38). Samples were barcoded using the 96-sample barcoding kit (EXP- PBC096). Amplicons were purified with Agencourt AMPure XP magnetic beads at a ratio of 0.4x. Library preparation was carried out according to the ONT protocol (version: PBAC96_9069_v109_revO_14Aug2019 – long fragment selection) and sequencing with a MinION device on R9.4 flow cells.
Primary acquisition of sequencing data was carried out with MinION (version 20.10.3)42, and base-calling and demultiplexing with Guppy (version 4.2.2). The resulting reads were aligned to GRCh38.p13, without the alternative reference contigs, using NGMLR (version 0.2.7)43 unless otherwise stated. Clair (version 2.1.1)44 was used for SNV calling. Since the most recent Clair ONT models were trained with up to 578-fold coverage, each sample was down-sampled to 550-fold. SNV calls were filtered with the Clair genome quality (GQ) score with a threshold set at 650. We only called SNV in GBA coding exons and ten flanking bases. Intronic haplotypes for some of these samples with no coding mutations were recently reported separately32. Phasing of SNV was carried out with Whatshap (version 1.0)45 and data manipulation with Samtools (version 1.10)46, Bedtools (version 2.29.1)47 and Tabix (version 1.7-2). Optimisation from our previous method16 comprised Guppy instead of Albacore for base-calling and Clair instead of Nanopolish for SNV calling. The pipeline used here identified all previously reported coding SNVs in 95 samples which were re-analysed. Analysis was run on a machine with intel 4 cores/8 threads, 16 Gb RAM DDR4 and 1 Tb SSD memory drive. Guppy was run on the UCL computational cloud, using 2 NVIDIA A100 GPUs in parallel. The total analysis took 2 h for a maximum of 96 samples.
To detect homopolymers, we devised a method that involves analysis of.bam files with Samtools depth to obtain the depth of coverage across these A (chr1:155239990-155239995 and chr1:155239657-155239661; hg38). The depth of coverage at each position was then adjusted for the depth of coverage at the 100 flanking positions, and the result was compared with the mean of all other samples in the run. If the adjusted depth of coverage at one position was more than five median absolute deviations from the median adjusted depth of coverage of the other samples in the run at that position, this was considered evidence of a deletion (coverage lower than the mean for that position) or an SNV (coverage higher than the mean for that position). Variants detected with this method were validated with Sanger sequencing48.
To detect and amplify reciprocal recombination events, two additional sets of primers were used41. These primers were specifically designed to amplify the recombinant alleles: the set MTX1-r/GBA-nf (primer pair 2) only amplifies recombinants with GBA gene sequence at the 5′UTR end and GBAP1 sequence at the 3′-UTR end (CNL), while the set of primers ΨMTX1-r/ΨGBA-nf (primer pair 3) only amplifies recombinants with the 3′-UTR end and GBAP1 sequence at the 5′-UTR end (CNG, see Fig. 1). Samples underwent PCR using these pairs of primers. If a product was detected on agarose electrophoresis, the sample was re-amplified with the same primer pair modified to carry the ONT barcode adaptors, and the amplicons were barcoded and sequenced as described. The primer sequences and PCR conditions are given in Supplementary Table 11. To define the breakpoint of CNL, the products of primer pair 2 were aligned to GBA (to avoid alignment to GBAP1; chr1:155222384-155241249; hg38) using LAST (Version 1243)49. The resulting alignment was analysed with Clair to look for variants at positions where GBA and GBAP1 differ (Supplementary Table 8). If a sample displayed an SNV in a certain position, it meant that the breakpoint must be upstream of that but downstream of the next sentinel position where no variant is detected (Supplementary Fig. 3).
GBA enrichment with UNCALLED
To validate GBA SV without PCR enrichment, we used UNCALLED, which uses adaptive sampling to enable real-time enrichment or depletion on MinION runs via the MinKNOW API ReadUntil17. UNCALLED analyses the signal generated by the DNA molecules passing through each pore of the device in real-time and decides whether they align to a reference sequence provided. It can then prematurely eject the molecule from the pore if not of interest, freeing up sequencing capacity for new reads and ultimately achieving purely computational enrichment. The target region for enrichment was chr1:155193567-155264811, with repetitive regions masked with the UCSC RepeatMasker.
Digital PCR
dPCR was performed by QIAGEN (Hilden, Germany) on the QIAcuity instrument. Three probes were selected: DCH101-0776005A (chr1:155231010-155231209; hg38) and DCH101-0776012A (chr1:155232410-155232609; hg38) target the region affected by the recombination event, while DCH101-1260927A (chr1:155208699-155208804; hg38) is outside of this region and was used as a reference for the analysis. Each sample was tested three times, and the result is the average of the three assays.
Illumina sequencing
WGS was performed on the Illumina NovaSeq instruments using Illumina TruSeq Nano DNA Library Prep9.
Statistics and reproducibility
Analysis was carried out on R (version 4.0.5). Odds Ratios (OR) were calculated with logistic regression. To check for an additive effect on the risk of CNGs on GBA variant carriers, multivariate logistic regression was used, with disease status as the outcome variable, and GBA-carrier status and CNG-carrier status as independent variables. The sample size is reported in Tables 2, 3.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Gauchian can be downloaded from: https://github.com/Illumina/Gauchian ONT and UNCALLED scripts used can be downloaded at https://github.com/marcotoffoli Individual-level genome sequence data for the PD patients, LBD patients, and neurologically healthy controls are available at AMP-PD (https://amp-pd.org). The datasets of DNA from QSBB brain samples and NHGRI samples generated during this study (Illumina WGS and targeted ONT sequencing) will be made available on the European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/home), accession number PRJEB48317. The datasets only include read alignments to GBA/GBAP1 regions (other regions of the genome have been removed or masked) to minimise the amount of genetic information made available for public access. The datasets of DNA from PPMI samples generated during this study (targeted ONT sequencing) are incorporated into the aggregated ‘Current Biospecimen Analysis Results’ dataset in the PPMI repository. The data were clearly labelled as project 195 data (https://www.ppmi-info.org/). ONT sequencing data on living individuals are not available due to consent/IRB restrictions. 1kGP project https://www.ncbi.nlm.nih.gov/bioproject/PRJEB31736 All other data are available from the corresponding author on reasonable request, when compatible with consent/IRB restrictions. RAPSODI, https://rapsodistudy.comOMIM, http://www.omim.org/ The NCBI reference sequence for GBA on which the numbering of exons is based is NM_000157.4.
References
Hruska, K. S., LaMarca, M. E., Scott, C. R. & Sidransky, E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 29, 567–583 (2008).
Do, J., McKinney, C., Sharma, P. & Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegeneration 14, 36 (2019).
Sidransky, E. et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 361, 1651–1661 (2009).
Guerreiro, R. et al. Investigating the genetic architecture of dementia with Lewy bodies: a two-stage genome-wide association study. Lancet Neurol. 17, 64–74 (2018).
Horowitz, M. et al. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics 4, 87–96 (1989).
Zampieri, S., Cattarossi, S., Bembi, B. & Dardis, A. GBA analysis in next-generation era: pitfalls, challenges, and possible solutions. J. Mol. Diagn. 19, 733–741 (2017).
Tayebi, N. et al. Reciprocal and nonreciprocal recombination at the glucocerebrosidase gene region: implications for complexity in Gaucher disease. Am. J. Hum. Genet. 72, 519–534 (2003).
Woo, E. G., Tayebi, N. & Sidransky, E. Next-generation sequencing analysis of GBA1: the challenge of detecting complex recombinant alleles. Front. Genet. 12, 684067 (2021).
Auwera, G. A. V. der et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33 (2013).
Bodian, D. L. et al. Utility of whole-genome sequencing for detection of newborn screening disorders in a population cohort of 1,696 neonates. Genet. Med. 18, 221–230 (2016).
Ebbert, M. T. W. et al. Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome Biol. 20, 97 (2019).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv:1303.3997 [q-bio] (2013).
den Heijer, J. M. et al. A large-scale full GBA1 gene screening in Parkinson’s disease in the Netherlands. Mov. Disord. 35, 1667–1674 (2020).
Chen, X. et al. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet. Med. 22, 945–953 (2020).
Chen, X. et al. Cyrius: accurate CYP2D6 genotyping using whole-genome sequencing data. Pharmacogenomics J. 21, 251–261 (2021).
Leija‐Salazar, M. et al. Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol. Genet. Genom. Med. 7, e564 (2019).
Kovaka, S., Fan, Y., Ni, B., Timp, W. & Schatz, M. C. Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. Nat. Biotechnol. https://doi.org/10.1038/s41587-020-0731-9 (2020).
Huang, Y.-T., Liu, P.-Y. & Shih, P.-W. Homopolish: a method for the removal of systematic errors in nanopore sequencing by homologous polishing. Genome Biol. 22, 95 (2021).
Iwaki, H. et al. Accelerating medicines partnership: Parkinson’s disease. Genetic resource. Mov. Disord. 36, 1795–1804 (2021).
Beutler, E., Gelbart, T. & Scott, C. R. Hematologically important mutations: Gaucher disease. Blood Cells Mol. Dis. 35, 355–364 (2005).
Chia, R. et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat. Genet. 53, 294–303 (2021).
Spataro, N. et al. Detection of genomic rearrangements from targeted resequencing data in Parkinson’s disease patients. Mov. Disord. 32, 165–169 (2017).
Ebert, P. et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372, eabf7117 (2021).
Dehghani, N., Bras, J. & Guerreiro, R. How understudied populations have contributed to our understanding of Alzheimer’s disease genetics. Brain 144, 1067–1081 (2021).
Program, T. G. P. G. GP2: the global Parkinson’s genetics program. Mov. Disord. 36, 842–851 (2021).
Alcalay, R. N. et al. Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology 78, 1434–1440 (2012).
Nalls, M. A. et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 70, 727–735 (2013).
Gan-Or, Z. et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 84, 880–887 (2015).
Liu, G. et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann. Neurol. 80, 674–685 (2016).
Blauwendraat, C. et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 143, 234–248 (2020).
Schierding, W. et al. Common variants co-regulate expression of GBA and modifier genes to delay Parkinson’s disease onset. Mov. Disord. 35, 1346–1356 (2020).
Toffoli, M. et al. Intronic haplotypes in the GBA gene do not predict age at diagnosis of Parkinson’s disease. Mov. Disord. 36, 1456–1460 (2021).
Thomson, D. W. & Dinger, M. E. Endogenous microRNA sponges: evidence and controversy. Nat. Rev. Genet. 17, 272–283 (2016).
Mullin, S. et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations. JAMA Neurol. 77, 427 (2020).
Regier, A. A. et al. Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat. Commun. 9, 4038 (2018).
Birney, E., Inouye, M., Raff, J., Rutherford, A. & Scally, A. The language of race, ethnicity, and ancestry in human genetic research. Preprint at arXiv:2106.10041 [q-bio] (2021).
Marek, K. et al. The Parkinson progression marker initiative (PPMI). Prog. Neurobiol. 95, 629–635 (2011).
Higgins, A. L. et al. The remote assessment of parkinsonism supporting the ongoing development of interventions in Gaucher disease. Neurodegen. Dis. Man. 11 (2021).
Nacheva, E. et al. DNA isolation protocol effects on nuclear DNA analysis by microarrays, droplet digital PCR, and whole genome sequencing, and on mitochondrial DNA copy number estimation. PLoS ONE 12, e0180467 (2017).
Li, H. et al. Exome variant discrepancies due to reference-genome differences. Am. J. Hum. Genet. 108, 1239–1250 (2021).
Jeong, S.-Y. et al. Identification of a novel recombinant mutation in Korean patients with Gaucher disease using a long-range PCR approach. J. Hum. Genet. 56, 469–471 (2011).
Jain, M., Olsen, H. E., Paten, B. & Akeson, M. The Oxford nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 17, 239 (2016).
Sedlazeck, F. J. et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468 (2018).
Luo, R. et al. Exploring the limit of using a deep neural network on pileup data for germline variant calling. Nat. Mach. Intell. 2, 220–227 (2020).
Martin, M. et al. WhatsHap: fast and accurate read-based phasing. Preprint at bioRxiv https://doi.org/10.1101/085050 (2016).
Li, H. et al. The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Stone, D. L. et al. Glucocerebrosidase gene mutations in patients with type 2 Gaucher disease. Hum. Mutat. 15, 181–188 (2000).
Frith, M. last. GitLab https://gitlab.com/mcfrith/last (2022).
Acknowledgements
This study was supported in part by the Intramural Research Programme of the National Institutes of Health (National Institute of Neurological Disorders and Stroke; project numbers: 1ZIANS003154) and the JPND through the MRC grant code MR/T046007/1.
We thank the New York Genome Center and the Coriell Institute for Medical Research for generating and releasing the 1kGP WGS data. We thank the AMP-PD Knowledge Platform for hosting WGS data for patient and control cohorts.
We thank Salomé Bagayan, Diem Cao, Angela Henry, Wen Liang, Leland Mencik, Lisa Robison, Christina Toma and Sasha Treadup at Illumina for helping with the sequencing of validation samples, and Iska Steffens at QIAGEN for performing digital PCR.
Data and biospecimens used in the analyses presented in this article were obtained from the Parkinson’s Progression Markers Initiative (PPMI) (www.ppmi-info.org/specimens). As such, the investigators within Revised April 2019 PPMI contributed to the design and implementation of PPMI and/or provided data and collected biospecimens, but did not participate in the analysis or writing of this report. For up-to-date information on the study, visit www.ppmi-info.org.
PPMI—a public-private partnership—is funded by The Michael J. Fox Foundation for Parkinson’s Research and funding partners, including [list the full names of all PPMI funding partners found at www.ppmi-info.org/fundingpartners].
Data used in the preparation of this article were obtained from the AMP-PD Knowledge Platform. For up-to-date information on the study, https://www.amp-pd.org.
AMP-PD—a public-private partnership—is managed by the FNIH and funded by Celgene, GSK, the Michael J. Fox Foundation for Parkinson’s Research, the National Institute of Neurological Disorders and Stroke, Pfizer, Sanofi and Verily.
Clinical data and biosamples used in preparation of this article were obtained from the Fox Investigation for New Discovery of Biomarkers (BioFIND), the Harvard Biomarker Study (HBS), the Parkinson’s Progression Markers Initiative (PPMI) and the Parkinson’s Disease Biomarkers Programme (PDBP).
BioFIND is sponsored by The Michael J. Fox Foundation for Parkinson’s Research (MJFF) with support from the National Institute for Neurological Disorders and Stroke (NINDS). The BioFIND Investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study, visit michaeljfox.org/biofind.
Harvard NeuroDiscovery Biomarker Study (HBS) is a collaboration of HBS investigators [full list of HBS investigator found at https://www.bwhparkinsoncenter.org/biobank] and funded through philanthropy and NIH and Non-NIH funding sources. The HBS Investigators have not participated in reviewing the data analysis or content of the manuscript.
Parkinson’s Disease Biomarker Programme (PDBP) consortium is supported by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health. A full list of PDBP investigators can be found at https://pdbp.ninds.nih.gov/policy. The PDBP Investigators have not participated in reviewing the data analysis or content of the manuscript.
We are grateful to the Queen Square Brain Bank, and to individuals who donated their brains. The Queen Square Brain Bank is supported by the Reta Lila Weston Institute for Neurological Studies and the Medical Research Council UK.
Author information
Authors and Affiliations
Contributions
MT Acquisition, analysis and interpretation of data. Prepared first manuscript draft. Creation of software to detect changes in homopolymers in ONT data. XC Acquisition, analysis and interpretation of data. Prepared first manuscript draft. Creation of Gauchian software. FJS Conception, design and supervision of the work. Substantively revised the manuscript. C-YL Acquisition of data. SM Substantive contribution to conception and design of the work. Revised the manuscript. AH Acquisition of data. SK Acquisition of data. MEG-S Acquisition of data. ES Acquisition of data. SWS Substantive contribution to conception and design of the work. Revised the manuscript. AHVS Conception, design and supervision of the work. Substantively revised the manuscript. MAE Conception, design and supervision of the work. Substantively revised the manuscript. CP Conception, design and supervision of the work. Substantively revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
XC and MAE were employees of Illumina Inc. during the research and writing of this manuscript and are now employees of Pacific Biosciences Inc. SWS serve on the Scientific Advisory Council of the Lewy Body Dementia Association. SWS is an editorial board member for the Journal of Parkinson’s Disease and JAMA Neurology. AHVS is supported by the UCLH NIHR BRC. MT, FJS, C-YL, SM, AH, SK, MEG-S, ES and CP declare no competing interests.
Ethics
Ethics approval was provided by the National Research Ethics Service London—Hampstead Ethics Committee for RAPSODI, NRES Committee central—London for QSBB samples and UCL Ethics Committee for PPMI samples. All participants provided informed consent.
Peer review
Peer review information
Communications Biology thanks Yiran Guo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Manuel Breuer. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Toffoli, M., Chen, X., Sedlazeck, F.J. et al. Comprehensive short and long read sequencing analysis for the Gaucher and Parkinson’s disease-associated GBA gene. Commun Biol 5, 670 (2022). https://doi.org/10.1038/s42003-022-03610-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03610-7
This article is cited by
-
Clinical, mechanistic, biomarker, and therapeutic advances in GBA1-associated Parkinson’s disease
Translational Neurodegeneration (2024)
-
Gaucher disease provides a unique window into Parkinson disease pathogenesis
Nature Reviews Neurology (2024)
-
Association study of GBA1 variants with MSA based on comprehensive sequence analysis -Pitfalls in short-read sequence analysis depending on the human reference genome-
Journal of Human Genetics (2024)
-
GBA1 in Parkinson’s disease: variant detection and pathogenicity scoring matters
BMC Genomics (2023)
-
Accurate long-read sequencing identified GBA1 as major risk factor in the Luxembourgish Parkinson’s study
npj Parkinson's Disease (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
