
Abstract
Non-coding RNAs (ncRNAs) form a large portion of the mammalian genome. However, their biological functions are poorly characterized in cancers. In this study, using a newly developed tool, SomaGene, we analyze de novo somatic point mutations from the International Cancer Genome Consortium (ICGC) whole-genome sequencing data of 1,855 breast cancer samples. We identify 1030 candidates of ncRNAs that are significantly and explicitly mutated in breast cancer samples. By integrating data from the ENCODE regulatory features and FANTOM5 expression atlas, we show that the candidate ncRNAs significantly enrich active chromatin histone marks (1.9 times), CTCF binding sites (2.45 times), DNase accessibility (1.76 times), HMM predicted enhancers (2.26 times) and eQTL polymorphisms (1.77 times). Importantly, we show that the 1030 ncRNAs contain a much higher level (3.64 times) of breast cancer-associated genome-wide association (GWAS) single nucleotide polymorphisms (SNPs) than genome-wide expectation. Such enrichment has not been seen with GWAS SNPs from other cancers. Using breast cell line related Hi-C data, we then show that 82% of our candidate ncRNAs (1.9 times) significantly interact with the promoter of protein-coding genes, including previously known cancer-associated genes, suggesting the critical role of candidate ncRNA genes in the activation of essential regulators of development and differentiation in breast cancer. We provide an extensive web-based resource (https://www.ihealthe.unsw.edu.au/research) to communicate our results with the research community. Our list of breast cancer-specific ncRNA genes has the potential to provide a better understanding of the underlying genetic causes of breast cancer. Lastly, the tool developed in this study can be used to analyze somatic mutations in all cancers.
Similar content being viewed by others


Introduction
Breast cancer is the most common cancer in women that has the highest frequently leading cause of cancer-related mortality amongst females worldwide1. A deeper understanding of the underlying mechanisms of breast cancer genetics and pathogenesis can be used to detect early-stage cancer to reduce morbidity and mortality due to breast cancer2. Over the last 10 years, high-throughput sequencing has comprehensively investigated the underlying genetic mechanisms that initiate or drive cancer progression and revealed many cancer-associated mutations. The International Cancer Genome Consortium (ICGC)3 has provided an extensive catalog of somatic mutations for various cancer types. This information has enabled researchers to characterize numerous protein-coding genes essential in cancer progression3,4,5,6,7.
Although most studies have mainly focused on protein-coding genes in investigating driver mutations in cancer, several lines of evidence imply that about 80% of the genome is biochemically functional8, indicating there are many mutations in non-coding regions that need to be investigated. A large portion of non-coding regions operates as regulators of oncogenes9. In addition, around 93% of disease-associated genome-wide association single nucleotide polymorphisms (GWAS SNPs) are located within these regions10,11, which can significantly influence gene expression of coding and non-coding genes.
Long non-coding RNAs (lncRNAs), an influential class of non-coding transcripts with more than 200 nucleotides, are potential cancer progression indicators and are emerging as diagnostic biomarkers in cancer and other diseases12,13,14,15,16,17. For example, GAS5 is one of the lncRNAs considerably downregulated in breast cancer18. HOTAIR is another well-known lncRNA upregulated in breast cancer, contributing to aberrant histone H3K27 methylation and cancer metastasis19,20. Furthermore, lncRNAs contribute to various regulatory activities in the cell, such as regulating gene expression via interaction with other chromatin regulatory proteins21, functioning as active enhancers22,23, and regulating chromatin structure17,24,25. Despite various studies on the impact of non-coding somatic mutations occurring in ncRNAs, the role of such ncRNAs has remained underexplored in breast cancer.
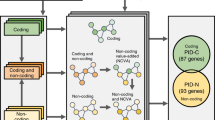
In this study, we developed a new tool, SomaGene, to identify 1030 ncRNAs that are significantly and specifically mutated in breast cancer. Using SomaGene, we show that candidate ncRNAs identified in our study are enriched considerably for regulatory features (e.g., breast-specific H3K27ac, H3K4me1, CTCF, DNase hypersensitive sites (DHS), and enhancer marks). Notably, we show that our breast cancer candidate ncRNAs have a much higher fraction of GWAS SNPs and expression of quantitative trait loci (eQTL) polymorphisms. Finally, our analyses on high-throughput chromosome conformation capture (Hi-C) data from the Human Mammary Epithelial Cell (HMEC) indicate that many of our candidate ncRNAs significantly interact with at least one protein-coding gene, which may suggest a potential enhancer role for these ncRNAs. An overview of the pipeline used in this study is shown in Fig. 1.

a ICGC cancer samples are used to identify BC-associated non-coding RNAs. Only samples with single point mutations are considered in the analysis. We also used a combined FANTOM5 robust gene list, Ensembl gene list, and the RNA Atlas28 to have a comprehensive list of non-coding RNAs. b After counting the number of mutated samples in each ncRNA, we use Fisher’s exact test, as described in the method section, to identify the mutational P value for each ncRNA. To identify significantly mutated ncRNAs, we calculate P values for 1,000,000 random permutations of breast/non-breast labels to estimate the 99% C.I. threshold of P value for each ncRNA. c We then investigate the overlapping of non-coding RNAs with breast tissue-related regulatory features (e.g., ENCODE predicted chromHMM, H3K27ac), BC-related GWAS SNPs, HMEC related eQTL polymorphisms, and HMEC related Hi-C interacting regions. d Finally, we provide a list of breast cancer-associated ncRNAs with potential enhancer activity.
We also compared enrichment of regulatory features, GWAS SNPs, and eQTL polymorphisms in the candidate set of ncRNAs with the 2nd, 3rd and last sets of ncRNAs (each set contains 1030 ncRNAs). We first sorted ncRNAs based on their mutational P value to identify 2nd, 3rd, and last sets of ncRNAs. i.e., ncRNAs that significantly mutated in breast cancer samples are positioned in the top places of the list (referring to the 1030 ncRNAs as the candidate set). We then defined the next 1030 ncRNAs after the candidate ones, in the sorted list, as the 2nd set, and the subsequent 1030 ncRNAs after the 2nd set, as the 3rd set of ncRNAs. We also defined the last 1030 ncRNAs in the sorted list as the last set of ncRNAs (i.e., those ncRNAs with the worse mutational P value in breast cancer).
We provide a list of non-coding genes with their mutational enrichment P value and annotated genomic signals at http://www.ncrna.ictic.sharif.edu and https://www.ihealthe.unsw.edu.au/research. SomaGene as an open-source R package is also available at https://github.com/bcb-sut/SomaGene.
Results
To have a comprehensive list of ncRNAs, we used a combined list of ncRNAs provided by the FANTOMCAT26, Ensembl27 consortia, and an atlas of ncRNAs28 (see method section). This includes ncRNAs from different types inclusive of pseudogene (22.9%), lncRNA intergenic (21.4%), long intergenic ncRNAs (5.6%), lncRNA divergent (13.4%), antisense (3.3%), lncRNA sense intronic (6%), miRNA (5%), misc RNA (3%), lncRNA antisense (4.8%). A full list of ncRNAs is provided in Fig. 2a. Somatic mutations from 17 cancer types were downloaded from ICGC29, including 1855 breast cancer samples containing 17,163,482 single point somatic mutations and 10,460 samples with other cancers containing 67,752,271 somatic point mutations inside the ncRNA regions.

a For the entire genome. b For the set of significantly mutated non-coding RNAs.
Background model to identify significant non-coding RNAs in breast cancer
To identify the significantly mutated ncRNAs in breast cancer samples, we counted the number of samples with somatic mutations in ncRNA from 1855 breast cancer samples and 10,460 samples with other cancers. We then calculated a P value for each ncRNA using Fisher’s exact test (see method section). We calculated P values for 1,000,000 random permutations of breast/non-breast labels for each ncRNA to identify significantly mutated ncRNAs. We estimated that an association’s probability emerges by chance (see method section). This provides a threshold for each ncRNA separately (Fig. 3). As a result, we identified 1030 ncRNAs (99% confidence interval—see method section) that significantly mutated in breast cancer samples (Supplementary Data 1). Looking into our candidate ncRNAs revealed that 27.2% of them are lncRNAs intergenic, 18.1% pseudogene, 3.8% long intergenic non-coding RNAs (lincRNA), 20.8% lncRNA divergent, 2.5% antisense, and 6.6% lncRNA sense intronic (Fig. 2b).

To determine the significance level of mutations in ncRNAs, we calculated P values for 1,000,000 random permutations of breast/non-breast labels to estimate the 99% C.I. threshold of P values. The permutation accounts for statistical significance based upon our original data sampling and avoids Type II errors that may arise from multiple testing correction approaches such as Bonferroni. We performed the permutations for each ncRNA separately. The red color indicates the 99% C.I. of observed P values in the permutations in the figure.
Breast cancer, as a heterogeneous disease, has five well-established subtypes, including LumA, LumB, Her2 + , Basal-like, and Normal-like, that show distinct molecular profiles and different underlying mechanisms. We therefore checked if 1030 ncRNAs were significantly mutated in the five well-established breast cancer subtypes. We obtained the PAM50 subtype annotation of 346 ICGC breast cancer samples from a publication by Nik-Zainal et al.30. Of 1030 significant ncRNAs, we identified 782 ncRNAs mutated in these samples. Supplementary fig. S1 shows that most ncRNAs were mutated in multiple subtypes. However, we observed that each subtype also has its unique set of ncRNAs that were not mutated in other subtypes (see more details in Supplementary Data 2).
We then checked if the mutations in the candidate set of ncRNAs are breast cancer-specific or if they are also significantly mutated in other cancers. We first identified the candidate set of ncRNAs in 17 other cancer types. We then calculated how many of the 1030 significant ncRNAs in breast cancer are also significant in other cancers. As Table 1 shows, at least 427 of 1030 ncRNAs are explicitly mutated in breast cancer samples.
To see if the 1030 BC-associated ncRNAs are more BC specific or not, we sorted the significantly mutated ncRNAs in each cancer based on their mutational P value. We took 1030 top ncRNAs to see how many of them are common with the 1030 BC-associated ncRNAs. As Table 2 shows, at least 916 (%80) of the BC-associated ncRNAs were mutated explicitly in breast cancers samples, indicating the 1030 ncRNAs identified in our study are more relevant to breast cancer than other cancers (e.g., more breast cancer-specific).
We next evaluated if 1030 significant ncRNAs were expressed ubiquitously across all breast cancer patients or if they were expressed in specific subtypes only. We used the expression dataset from Breast invasive carcinoma (BRCA) gene expression from TANRIC31. We found 504 ncRNAs of the 1030 significant ncRNAs in the TANRIC gene expression list. Of these, 106 ncRNAs were differentially expressed between the breast cancer subtypes. Supplementary fig. S2 showed that these 106 ncRNAs show some breast cancer subtype specificity. A list of differentially expressed ncRNAs is provided in Supplementary Data 3.
BC-associated GWAS SNPs are significantly enriched in the candidate non-coding RNAs
Multiple genome-wide association studies identified disease-associated genes and their respective pathways, which provided a comprehensive understanding of the disease’s etiology. It has been reported that more than 93% of disease-associated variations found by GWAS are located in the non-coding regulatory regions of genomes32, suggesting non-coding regulatory regions are relevant to disease and genetic mutations in gene regulatory regions is a significant mechanical contributor to diseases. To examine the enrichment of BC-associated GWAS SNPs in the candidate ncRNAs, we extracted the BC-associated SNPs from a pooled list of two GWAS datasets from the EBI GWAS Catalog33 and GWASdb v2 from the Wang Lab34 (for more details see the method section). As Fig. 4a shows, BC-associated GWAS SNPs are significantly enriched (P value 2.3e−27) in the candidate ncRNAs. This enrichment is much higher for those ncRNAs that contain more than 4 GWAS SNPs (>10 times enrichment). Interestingly, when we performed the enrichment analysis for ncRNAs with more than 5 GWAS SNPs, only the candidate list of ncRNAs showed enrichment for GWAS SNPs (>20 times), and there was no enrichment in the 2nd and 3rd sets of ncRNAs (Fig. 4a). Performing the same analysis on lung cancer-associated GWAS SNPs did not show such enrichment for our candidate ncRNAs (Fig. 4a), indicating that candidate ncRNAs are relevant to breast cancer. For example, candidate ncRNAs RP11-353N4.6 are known to carry breast cancer-associated GWAS SNPs35. More details on the annotated list of ncRNAs with BC-related GWAS SNPs can be found in Supplementary Data 4.

a Enrichment of breast and lung cancer-associated GWAS SNPs in the candidate non-coding RNAs. b Enrichment of breast and lung tissues associated eQTL pairs in the candidate non-coding RNAs. We repeated the enrichment analysis with many items (e.g., GWAS SNP or eQTL polymorphisms) overlapping the ncRNAs. E.g., counting the number of ncRNAs that encompass at least 2/3/4/5/6/7/8/9/ GWAS SNPs or eQTL polymorphisms. As the figure shows, the candidate set of ncRNAs significantly enriched for both BC-related GWAS SNPs and breast tissue-related eQTL polymorphisms. This enrichment is much higher than the enrichment in lung cancer-related GWAS SNPs and lung tissue-related eQTL polymorphisms.
BC-associated non-coding RNAs have a significantly higher fraction of eQTL polymorphisms
eQTL are genomic locations that explain which genetic variations may change the gene function in a relevant tissue. To assess eQTL polymorphisms in the candidate non-coding genes, we calculated the enrichment of breast mammary tissue eQTL polymorphisms downloaded from GTEx consortia36 in the candidate ncRNAs. Our analysis revealed that breast mammary tissue eQTL polymorphisms are significantly enriched (1.77 times with a P value of 4.11e−20) in the candidate ncRNAs. Interestingly, this enrichment is much higher for those ncRNAs that contain more than three eQTL polymorphisms (>2× enrichment), and such an increase has not been seen in the 2nd and 3rd sets of ncRNAs (Fig. 4b and Supplementary Data 4). We calculated the enrichment of lung-specific eQTL polymorphisms in the candidate ncRNAs as a control. As Fig. 4b shows, the enrichment of lung-specific eQTL polymorphisms in the breast-associated ncRNAs is much lower than the enrichment observed for mammary tissue eQTL polymorphisms (Fig. 4b). For example, lncRNA RP11-37B2.1 and RP11-426C22.5 are two of our candidate ncRNAs with significant P values of 7.91e−4 and 6.72e−06, respectively. These ncRNAs encompass 232 and 111 eQTL polymorphisms, respectively. lncRNA RP11-37B2.1 influences the risk of tuberculosis and the possible correlation with adverse drug reactions from tuberculosis treatment37, and RP11-426C22.5 is downregulated in SW1990/GZ Cells38. Both RP11-37B2.1 and RP11-426C22.5 overlapped with histone active mark H3K27ac, ChromHMM potent enhancer, and DHS, suggesting a potential transcription regulation role of these ncRNAs.
Enrichment of promoter and enhancer signals in the BC-associated ncRNAs
To determine if the somatic mutations are enriched in ncRNAs with enhancer activity, we first examined the enrichment of HMEC-related chromatin states provided by the ENCODE consortium within our significant ncRNAs. As Fig. 5a shows, both ENCODE promoters and enhancers have been significantly enriched within our candidate ncRNA genes (3.23 and 2.26 times with P values 4.12e−29 and 5.28e−32 for promoters and enhancers, respectively), suggesting breast cancer de novo somatic mutations are enriched in ncRNAs with enhancer and/or promoter like functions.

a Enrichment of HMEC-related promoters and enhancers identified by ENCODE (using chromatin segmentation by Hidden Markov Model (HMM). b Enrichment of promoters, enhancers, and breast tissue differentially expressed enhancers identified by FANTOM5 consortia. The enrichment is calculated by dividing the proportion of significantly mutated ncRNAs that overlap with each item by the proportion of all ncRNAs that overlap with that item. This enrichment is calculated for a significant set of ncRNAs (1030 ncRNAs) shown in red color, 2nd set (blue), 3rd set (gray) of highly mutated ncRNAs. The enrichment was also calculated for the last set of ncRNAs (brown) with a mutational P value close to 1. In other words, the last set refers to ncRNAs that had no mutation in breast cancer samples. Each set of ncRNAs contains 1030 elements.
We also investigated these enrichments in the 2nd and 3rd sets of ncRNAs (each set contains 1030 ncRNAs) and the last set of ncRNAs with worse mutational P values (see method section). As Fig. 5a shows, the same enrichment trend (but much lower) for ChromHMM predicted promoters and enhancers in the 2nd and 3rd sets of most mutated ncRNAs in breast cancer (Fig. 5a). Interestingly, there is no such trend for the last set of ncRNAs (those with no de novo mutation in breast cancer samples), supporting our hypothesis that de novo somatic mutations are enriched in enhancer-like ncRNAs. We provided an annotated list of candidate ncRNAs with ChromHMM in Supplementary Data 5.
The FANTOM5 consortium has released lists of human transcribed human promoters and tissue-specific transcribed enhancers of humans using CAGE (Cap Analysis of Gene Expression39) to study cell-type-specific enhancers. Therefore, we investigated the enrichment of FANTOM5 promoters and enhancers that overlap with the significant ncRNAs identified in this study. Figure 5b shows that both FANTOM5 promoters and enhancers are enriched in the candidate ncRNAs (1.66 and 1.76 times (P value 3.59e−34 and 3.68e−27) enrichment for FANTOM5 promoters and enhancers, respectively). In other words, 52.4% of candidate ncRNAs overlapped with FANTOM5 promoters, and 36.4% of them overlapped with FANTOM5 enhancers. However, only 34.9% and 23.5% of all ncRNAs overlapped with FANTOM5 promoters and enhancers. The same analysis on FANTOM5 mammary-specific enhancers demonstrated that the proportion of candidate ncRNAs that overlap with differentially expressed enhancers in the mammary epithelial cell is 3.84 times (P value 4.03e−05) more than the genome-wide expectation (Fig. 5b). There is also the same trend for the 2nd and 3rd sets of ncRNAs with the best mutational P values in breast cancer. An annotated list of candidate ncRNAs with FANTOM5 annotations is provided in Supplementary Data 6. We also provided a list of candidate ncRNAs that overlap with ENCODE and FANTOM5 enhancer/promoter features in Supplementary Data 7. Notably, 317 ncRNAs overlapped with ENCODE and FANTOM5 enhancer marks; 257 ncRNAs overlapped with ENCODE and FANTOM5 promoters. For example, the pseudogene NKAPP1 is differentially expressed in ABL1/ABL2 knockdown (shAA) breast cancer-associated cell lines40 and downregulated in breast cancer41. It is also a biomarker associated with breast cancer prognosis42,43. Our analysis demonstrated that NKAPP1 is one of the most significant ncRNAs, with a P value of 3.43e−06. This non-coding gene overlapped with both FANTOM5 enhancer and ChromHMM predicted enhancer and promoter. CATG00000062386 is another ncRNA gene that is significantly mutated in breast cancer samples. This FANTOMCAT specific ncRNA overlapped with FANTOM5 and ChromHMM enhancers and FANTOM5 mammary epithelial cell differentially expressed enhancers, indicating a potential enhancer role for this ncRNA in breast cancer.
Histone modifications H3K27ac and H3K4me1, CTCF binding sites, and DHS are significantly enriched for BC-associated ncRNAs
H3K27ac and H3K4me1 are histone marks present at enhancer or promoter regions. DHSs are also known as the generic markers of regulatory DNA, containing genetic variations associated with diseases44,45. In addition to these marks, CTCF binding sites also have a wide-range regulatory function in the genome, in which mutations that reside in these regions affect the binding specificity to DNA sequences and may lead to aberrant expression of cancer-related genes46. To check if these important regulatory features are enriched in the candidate ncRNAs, we investigated the enrichment of HMEC-specific chromatin histone active marks (e.g., H3K27ac and H3K4me1) CTCF binding sites and DHS in the candidate ncRNAs identified in this study. These active chromatin marks are involved in many processes, including transcriptional regulation that regulates gene expression47. Our investigation of histone active marks demonstrated that both histone marks H3K27ac and H3K4me1 are significantly enriched 2.1 times (P value 3.65e−57) and 1.6 times (1.90e−42) in the candidate ncRNAs (Fig. 6a). As these histone marks, suggesting our candidate list of ncRNAs is important for the transcriptional process in breast tissue. We performed the same analysis on histone marks H3K27me3, which is involved in the repression of transcription. Interestingly, we did not see significant enrichment (1.15 times with a P value of 5.14e−02) for histone H3K27me3 within our BC-associated ncRNAs (Fig. 6a).

a Enrichment of histone modifications (for three antibodies: H3K27ac, H3K4me1, and H3K27me3). The candidate set of ncRNAs shows significant enrichment for H3K27ac and H3K4me1 and no significant enrichment for H3K27me3, which is a repressor histone mark. b Enrichment of transcription factor binding sites (for 3 ChIP-seq experiments: HMEC + Broad+CTCF, HMEC + Broad+EZH2 and HMEC + UW + CTCF). In all experiments, the candidate set shows significant enrichment, much higher than other sets of ncRNAs. c Enrichment of DNase clusters (for three cell types: MCF − 7, MCF − 7+Hypoxia_LacAcid, and T − 47D). As the figure shows, the candidate set of ncRNAs significantly enriched for DNase clusters, much higher for most of the scores than other sets. The enrichment is calculated by dividing the proportion of significantly mutated ncRNAs that overlap with each item by the proportion of all ncRNAs that overlap with the item. This enrichment is calculated for a significant set of ncRNAs (1030 ncRNAs) as shown in red color, 2nd set (blue), and 3rd set (gray) of highly mutated ncRNAs. The enrichment was also calculated for the last set of ncRNAs (brown) that had no mutation in breast cancers. Each set of ncRNAs contains 1030 elements. Note: The score threshold is the minimum threshold for signal-value when counting the number of overlaps. See the method section for more details.
Transcription factors CTCF function as a transcriptional activator, repressor, insulator, or pausing transcription. In addition to CTCF sites, DHS also has key roles in gene regulation as regulatory element markers48. Both CTCF and DNase are functionally related to transcriptional activity and are necessary to regulate chromatin structure. Here, we choose three CTCF ChIP-seq experiments (HMEC + Broad + CTCF, HMEC + Broad + EZH2 and HMEC + UW + CTCF) and three DHS ChIP-seq experiments (MCF-7, MCF-7+Hypoxia_LacAcid, T-47D), all related to the breast cancer. We calculated the enrichment for the four sets of ncRNAs, including significant, 2nd, and 3rd sets of ncRNAs with the best mutational P values and the last set of ncRNAs with the lowest mutational P values. Our assessment of CTCF binding sites and DHS demonstrated that both CTCF (2.45 times, on average with P value 7.42e−35) and DNase (1.8 times, on average with P value 7.23e−50) are significantly enriched for our candidate ncRNA genes (Fig. 6b). Having such significant enrichment for CTCF binding sites and DNase accessible sites is strong evidence that our significant set is not chosen randomly and is related to the gene regulation process. There is also the same trend for the 2nd and 3rd sets of ncRNAs (Fig. 6b). However, the enrichments for the candidate set are much higher than the 2nd and 3rd sets. As Fig. 6b shows, there is no enrichment for the last set of ncRNAs, suggesting that these ncRNAs may not be involved in transcriptional regulation.
For example, LINC00535 is an antisense non-coding gene that is known to be associated with breast cancer49. LINC00894 is another non-coding gene that is the most downregulated lncRNA in MCF-7/TamR cells50. These ncRNAs are significantly mutated in breast cancer samples with P values 4.21e−3 and 1.53e−11. LINC00152 is another example of ncRNAs with a substantial role in enhancing breast cancer, which causes inactivation of the BRCA1/PTEN by DNA methyltransferases as tumorigenesis, mainly in triple-negative breast cancer (TNBC)51. These ncRNAs overlapped with ENCODE-predicted enhancers, histone 27 acetylation, CTCF binding sites, and DHS, suggesting a potential transcriptional regulatory role for these ncRNAs. An annotated list of candidate ncRNAs with these features is provided in Supplementary Data 8 and 9.
Chromosome conformation capture data shows a potential regulatory role for genomic loci that overlap with BC-associated ncRNAs
High-throughput chromosome conformation capture (Hi-C) based assays have been used to successfully identify regulatory regions and targets of disease-associated variations52,53. To further understand the genes in which the genomic loci encompassing candidate ncRNAs interact, we analyzed two publicly available Hi-C datasets from HMEC cell lines54. We used MHiC55 and MaxHiC56 to analyze Hi-C raw data and identify statistically significant interactions. We identified 188,982 statistically significant interactions (P value < 0.01 and read-count ≥10—see method section) in the Hi-C library 1. For 6187 of the interactions (%3.3), one side of the interaction overlapped with at least one candidate ncRNA genomic region. Another side of the interaction overlapped with protein-coding genes (promoter regions of coding genes—see method section; Supplementary Data 10). Repeating this analysis on the second Hi-C library also identified 318,034 statistically significant interactions. For 9879 of the interactions (3.1%), one end of the interactions overlapped with the genomic region of candidate ncRNAs, and another end overlapped with the promoter region of protein-coding genes (Supplementary Data 10).
We identified 1167 common significant interactions between the two libraries where one side of the interaction overlaps with genomic regions that encompassed at least one candidate ncRNAs (Supplementary Data 11) and another side with protein-coding genes. In other words, for 251 ncRNAs (the genomic regions containing 251 ncRNAs) out of a pool of 1030 candidate ncRNAs (24%), there was at least one significant interaction in both the libraries (Supplementary Data 11); this is significantly higher (1.74 times; P value 4.61e−11) than genomic regions of all ncRNAs that overlapped with one side of the interactions in both the libraries (14%). The overlapping between 251 ncRNAs and regulatory features used in this study is shown in Supplementary fig. S3.
We then repeated the enrichment analyses to see if the 251 ncRNAs supported by multiple Hi-C-based assays have better enrichment of regulatory features, GWAS, and eQTL polymorphisms than other sets of ncRNAs. As Supplementary fig. S4 shows, the 251 ncRNAs have much higher enrichment of overlapping with regulatory features, GWAS, and eQTL polymorphisms than the remaining set of ncRNAs in the candidate list (1030-251 candidate ncRNAs), as well as than the ncRNAs in the 2nd, 3rd, and last sets of ncRNAs supported by multiple Hi-C based assays (Supplementary fig. S4). This supports our hypothesis that De novo somatic point mutations are enriched in enhancer-like ncRNAs. The list of the 251 ncRNAs is provided in Supplementary Data 11.
For 757 significant ncRNAs (%82), we identified at least one interaction in either Hi-C library 1 or library 2, resulting in 21,564 interactions. For 19,674 out of 21,564 interactions (Supplementary Data 12), one end of the interaction that encompasses candidate ncRNAs (genomic regions) overlapped with either ENCODE HMM predicted enhancer or active histone mark H3K27ac (both presented in HMEC). This observation suggests a potential enhancer role for these ncRNAs; In many cases, another end of the interactions overlaps with protein-coding genes, including cancer-associated genes (Supplementary Data 12). We provided a prioritized list of candidate ncRNAs that interacted with cancer-associated protein-coding genes in the Hi-C libraries and overlapped with ENCODE HMM predicted enhancers and active histone mark H3K27ac (Table 3). For example, MYC is a BC-associated protein-coding gene acting as a transcription factor. In common with three other transcription factors (POU5F1, SOX2, and KLF4), it can induce epigenetic reprogramming of somatic cells to an embryonic pluripotent state57. In both Hi-C libraries, we found significant Hi-C interactions between MYC and two of our candidate ncRNAs, CASC8 and PVT1. PVT1 is a known enhancer for MYC58. However, CASC8 has not yet been identified as a putative enhancer for MYC. There are ~200 kb genomic distances between the CASC8 and transcription start site of MYC in which there is no protein-coding gene that overlaps with CASC8. There are strong signals of histone active marks and ENCODE predicted enhancers, and most importantly, a FANTOM5 breast differentially expressed enhancer overlapped with CASC8, suggesting a potential enhancer region in CASC8 (Fig. 7). Interestingly, there is no somatic mutation or BC-associated GWAS SNPs that overlap with MYC. However, CASC8 is significantly mutated in breast cancer samples, and most importantly, it encompasses 10 BC-associated GWAS SNPs. Altogether, this evidence may indicate a putative enhancer role of CASC8 for MYC.

Long non-coding RNA CASC8 is a breast tissue expressed lncRNA that is significantly mutated in breast cancer samples. Our analysis of HMEC-related Hi-C data shows that this lncRNA is significantly interacting with the promoter of multiple coding genes, including MYC, a known breast cancer-associated gene. There are numerous strong signals of ENCODE predicted ChromHMM potent enhancers, histone active marks H3K27ac, and H3K4me1 (all presented in HMEC) that overlap with CASC8. Notably, a FANTOM5 breast differentially expressed enhancer also overlapped with CASC8. Our analysis of GWAS SNPs and de Novo somatic point mutations revealed that CASC8 covered multiple breast cancer GWAS SNPs and many somatic point mutations related to breast cancer samples. In contrast, MYC does not encompass either GWAS SNP or BC-related somatic mutations. The figure also shows that the MYC gene interacts with PVT1, another significantly mutated lncRNA in the breast cancer sample (30 kb far from MYC). PVT1 is also overlapped with breast tissue-related regulatory features and is a previously known enhancer for the MYC gene. We used MHiC55 to analyze raw Hi-C data and MaxHiC56 to identify significant Hi-C interactions. Significant interactions are shown in blue color. ENCODE predicted chromHMM files chromatin signals were used in peak format.
Another example is ncRNA CATG00000061359, a FANTOM5-specific intergenic lncRNA significantly mutated in breast cancer samples (P value 5.32e−4). There is a significant Hi-C interaction between CATG00000061359 and gene GTPBP8 (a known breast cancer-associated gene59) in both the libraries. We also found a breast tissue-associated eQTL polymorphism in this lncRNA that influences the expression of GTPBP8 in breast tissue. Interestingly, both HMEC specific H3K27ac and HMM predicted enhancers overlap with this lncRNA. This suggests that variation in CATG00000061359 may influence the expression of GTPBP8 in breast cancer. We have provided an annotated list of candidate ncRNAs with Hi-C interactions in Supplementary Data 12.
Discussion
Somatic point mutations play a key role in tumorigenesis and the development of cancer60. Recent studies on somatic mutation evolution in cancer have identified cancer driver genes61 and mutational cancer signatures62,63,64. However, the analysis of somatic mutations has focused mainly on the protein-coding genes of the genome, and their potential impact on the non-coding RNA genes has been far less studied. ncRNAs have long been considered a non-functional part of the human genome65. However, these non-coding elements (majority lncRNAs) have recently opened a new insight into the study of breast cancer, acting as indispensable contributors to cellular activities, including the proliferation, apoptosis, survival, differentiation, and breast cancer metastasis66. In addition, ncRNAs have been used as biomarkers in many cancers, including breast cancer, through various mechanisms, including regulating the expression of protein-coding genes and functions at transcriptional, translational, and post-translational levels21,22,23,24,25. This indicates that ncRNAs may have the potential for diagnosis, prognosis, and therapeutics of cancers. This study identified ncRNAs that were mutated explicitly in breast cancer patients and then uncovered the connection between somatic point mutations in BC-associated ncRNAs and ncRNA regulatory properties in breast cancer.
As the previous study shown, tissue specificity is an important aspect of many genetic diseases, including cancers67; Our study demonstrated that the breast cancer-related ncRNAs have much higher enrichment of breast tissue/cell line-specific features; we have shown that the candidate ncRNAs with enrichment of somatic point mutations have a much higher fraction of regulatory features compared to genome-wide expectation, suggesting the potential impact of somatic mutations on the regulatory function of ncRNAs. Notably, we have shown that most of the candidate ncRNAs interact with promoters of protein-coding genes, again indicating the potential regulatory role of ncRNAs with significant enrichment of somatic point mutations in breast cancer.
Researchers have recently shown that germline mutations are associated with an increased risk of developing BC68,69 and acquired somatic mutations driving the disease, in which germline mutations may interact with somatic mutations to drive carcinogenesis or involve in tumorigenesis by contributing to critical biological and cellular processes. Our analyses revealed that the ncRNAs with enhancer-like activity are significantly overlapped with breast cancer-associated GWAS SNPs and breast tissue-related eQTL polymorphisms. This highlights the importance of somatic and germline mutations in breast cancer development and progression.
The enrichment of somatic mutations in the ncRNAs with enhancer-like activity has not been previously explored. Our Hi-C analyses also provided more evidence of enhancer activity of genomic loci encompassing BC-associated ncRNAs. Interestingly, we showed that the 251 ncRNAs supported by multiple Hi-C-based assays have much higher enrichment of regulatory features, GWAS, and eQTL polymorphisms than the remaining ncRNAs in the candidate list, indicating these ncRNAs are more likely to be involved in the gene regulation process.
However, our analyses are limited to the associations identified by pure data-driven analyses. Future experimental validation can reveal finer details about the interdependence of ncRNAs function in breast cancer.
We have developed an extensive web-based resource to communicate our results with the research community. Further works could focus on the candidate ncRNAs provided in this resource to check their complex regulatory functions and reveal the novel mechanism underlying carcinogenesis and breast cancer treatment.
This study also presented a novel computational method, SomaGene, to prioritize and predict disease-associated ncRNAs by integrating multi-level omics data, including somatic mutations, transcriptomic and epigenetic signals, and chromatin conformation data.
Methods
Tool availability
The SomaGene open-source R package, a sample dataset, and instructions on running SomaGene are provided at https://github.com/bcb-sut/SomaGene.
ICGC dataset
We used the ICGC dataset, which contains somatic point mutations from 1855 breast cancer samples and 10,419 samples from 17 different types of cancers.
A combined list of non-coding RNAs from FANTOMCAT and Ensembl consortia
We have combined two gene lists from FANTOM539 and Ensembl70 consortia (genome building hg19), enabling us to have a comprehensive list of non-coding RNA genes. We used an in-house script to combine the lists based on gene coordinates and/or gene names. If both consortiums have the same gene but different gene coordinates, we considered the FANTOM5 genes as the priority. We also added a recently published list of long non-coding RNAs from an atlas of non-coding RNAs28 into our list of ncRNAs. In total, 65,257 non-coding RNA genes were available in the combined list.
Identify significantly mutated non-coding genes
Many computational techniques have recently been used for mutational information to investigate biomarkers in human and viral genomes31,49,71,72,73. In this study, we developed SomaGene, which uses a “vcf” file from the whole genome or exome data. We included all samples from ICGC with at least one de novo mutation. We only considered single nucleotide mutations and excluded insertions or deletions from the analyses. To identify ncRNAs that significantly mutated in breast cancer samples compared to other cancers, we used Fisher’s exact test and permutation testing in the following manner:
We calculated a P value for each ncRNA using a one-sided Fisher’s exact test applied to a 2 × 2 contingency table whose elements are (1) the number of samples in breast cancer that are mutated in an ncRNA, (2) the number of samples in breast cancer that are not mutated in an ncRNA, (3) the number of samples in all cancers other than breast cancer that are mutated in an ncRNA and (4) the number of samples in all cancers other than breast cancer that are not mutated in an ncRNA (Supplementary fig. S5). To identify significant ncRNAs, we calculated P values for 1,000,000 random permutations (which can be defined by the user) of sample IDs across all cancers to estimate the probability that an association emerges by chance at a confidence interval of 99%. We identified a total of 1030 significantly mutated ncRNAs in breast cancer.
Overlap and aggregation score methods
To adequately examine the overlapping of ncRNAs with regulatory features and calculate the overlapping score with each element, each annotation’s overlapping ranges were used in our analysis. In the case of FANTOM5 promoters, enhancers, and tissue-specific enhancers, three binary variables for each ncRNA were calculated, indicating that an ncRNA has overlapped with any enhancer or promoter in the FANTOM5 dataset (Supplementary Data 6). In eQTL annotation, a set of all entries whose location overlaps with each ncRNA was extracted by concatenating the variation_id and gene_id for each location.
The chromatin segmentation annotation comprises genomic ranges, each attributed to one of 11 functional categories (segments). The percentage of overlap with a segment was calculated as the sum of proportions of nucleotides in that segment’s ranges overlapped with the ncRNA. Thus, a profile of overlapping chromatin segments was calculated with their corresponding coverage over the ncRNA (Supplementary fig. S6, Supplementary Data 5).
For histone modifications annotation, besides the proportions of nucleotides covered with the overlapping histone modification ranges in each ncRNA, the peak scores of these ranges were averaged together by the corresponding overlap percentages as their weights to obtain a single histone modification score. The coverage (overlap percentage) of each ncRNA with histone modification ranges was also calculated as the total proportion of nucleotides in the ncRNA covered with histone modification ranges.
In the case of DNase annotation, each genomic range is attributed to a group of cell types that show DNase hypersensitivity in that area. Several cell type IDs with corresponding DNase hypersensitivity scores are associated for each range. We combined the overlap annotation ranges to get an aggregated set of cell-type IDs, scores, and overlap percentages by calculating the proportion of nucleotides in the ncRNA covered with these ranges. After this step, several IDs were duplicated in many aggregated sets summed over the overlap percentages to obtain a single overlap measure. Also, it was averaged over its scores by the corresponding overlap percentages as their weights to take a single score for that ID. For each genomic range in transcription factors annotation, a group of transcription factors (from ENCODE) with their corresponding ChIP-Seq peaks are reported. While the schema of transcription factors annotation is similar to that of DNase annotation, the same procedure as described for DNase annotation was performed to obtain an aggregated set of transcription factor IDs, scores, and overlapping percentages for each ncRNA (Supplementary Data 8). The overlap of BC-related GWAS mutations loci with the overlapped ncRNA regions was identified. The total number of overlaps for each ncRNA was recorded in the output table (Supplementary Data 4).
Calculating enrichments
Every “enrichment” that is calculated throughout this study is defined as “the fraction of ncRNAs in the significant set that have the trait of interest (e.g., having overlap with or having a minimum score of a specific annotation) divided by genome-wide expectation.” It can be easily justified that the mentioned value equals the fraction of items in the whole set of ncRNAs with the trait of interest. Let’s assume that the total number of ncRNAs is N. The number of significant ncRNAs is S while A items among all and Y items within the significant set have the property of interest. Suppose a random collection of ncRNAs with size S is sampled. In that case, the probability that X items within this set have the property of interest follows a binomial distribution with parameters S andA⁄N, i.e., X ~ Binom(S,A⁄N). Thus, the expected value of X equals S×A⁄N which shows that the expected fraction of items having the property of interest in a random set of ncRNAs with size S equalsA⁄N. We then conclude that the defined enrichment can be calculated as (Y⁄S)⁄(A⁄N).
FANTOM5 promoters, enhancers, and breast differentially expressed enhancers
We used the FANTOM5 CAGE expression atlas26 to identify a set of significant ncRNAs that overlapped with FANTOM5 promoters and enhancers. The entire collection of enhancers and promoters found in the FANTOM5 data were downloaded from the FANTOM5 Phase239,74. We also downloaded FANTOM5 breast differentially expressed enhancers from ref. 75.
ENCODE chromatin state segmentation
The Chromatin State Segmentation uses a standard set of states learned by computationally integrating ChIP-Seq data for nine factors plus input using a Hidden Markov Model (HMM) across various cell types. Also, it shows a classification of chromatin, like “enhancer,” “promoter,” or “repressed.” We used this dataset to identify the set of breast-specific candidate ncRNAs that overlapped with chromatin states. The complete set of chromatin state segmentations for the models derived from HMECs, grown in vitro related to breast cancer was downloaded from the ENCODE project8.
ENCODE histone modifications by ChIP-Seq
For this study, we used HMEC-specific ChIP-Seq data in processed peak calls for histone modifications H3K27ac, H3K27me1, H3K4me3, and CTCF from the ENCODE project to identify a set of significant ncRNAs that overlapped with three types of histone modifications8.
ENCODE transcription factor ChIP-Seq data
This data shows regions of transcription factor binding derived from an extensive collection of ChIP-Seq experiments and DNA binding motifs identified within these regions by the ENCODE Factorbook repository76. The transcription factors are responsible for modulating gene transcription bind as assayed by chromatin immunoprecipitation with antibodies specific to the transcription factor followed by sequencing the precipitated DNA (ChIP-Seq). We used this dataset to identify a set of significant ncRNAs that overlapped with transcription factor ChIP-Seq. The set of transcription factor ChIP-Seq data for the HMEC cell line was downloaded from ENCODE project8.
DNase clusters
Regulatory regions tend to be DNase-sensitive which are accessible chromatin zones and functionally related to transcriptional activity. We used DHS from MCF-7 and T47D from ENCODE project to identify a set of significant ncRNAs that overlapped with DNase clusters.
Hi-C data analysis
Hi-C is an experiment for identifying the number of interactions between genomic loci in a 3D space. Our study used two replications related to the HMEC54. We used MHiC55 and Hi-C Pro77 with the default parameters for analyzing and aligning Hi-C data in 5 kb fragment size. We used MaxHiC56 as a background correction model to identify significant Hi-C interactions for true cis-interaction. Here, we included those significant interactions with P value < 0.01, read-count ≥10, with the distance between two sides of interaction more than 5 kb and <20 Mb. We then annotated Hi-C interactions with coding and non-coding genes from our combined genes list. At least 10% overlap between gene and Hi-C fragments has been considered to annotate Hi-C fragments with genes.
Genotype-tissue expression eQTLs
eQTL data were downloaded from the Genotype-Tissue Expression (GTEx) Project36. We used GTEx v7 eQTLs identified as significant in the HMEC line from the GTEx project.
Literature search for non-coding interacting genes
Our literature searches were focused on human studies and English language publications available in PubMed, Scopus, and Web of Science. Both Medical Subject Headings terms and related free words were used to increase the sensitivity of the search. We also used data and text mining techniques to extract additional associated studies78,79,80,81,82,83,84,85. A decision tree approach and a knowledge-based filtering system technique have also been used to categorize the texts from the literature search82,86,87. The search terms included “non-coding RNA” or “lncRNAs” or “genes name + cancer.” “BC” or “breast carcinoma” and “breast neoplasm.”
Genome-wide association study (GWAS) analysis
We have pooled two GWAS datasets, EBI GWAS Catalog33 and GWASdb v2, from Wang Lab34 to identify a set of significant ncRNAs that overlapped with GWAS SNPs. We first converted all GWAS SNP coordinates to UCSC hg19 using UCSC Lift Genome Annotations tools88 (www.ncbi.nlm.nih.gov/genome/tools/remap) and then used an in-house script to combine both GWAS datasets based on gene coordinates and/or gene symbols. All GWAS SNPs with P value < e−8 were included in the analyses.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
A sample dataset can be accessed at https://github.com/bcb-sut/SomaGene. The whole dataset is publicly accessible through ICGC data portal (https://dcc.icgc.org).
Code availability
The source code can be accessed at https://github.com/bcb-sut/SomaGene.
References
Torre, L. A., Siegel, R. L., Ward, E. M. & Jemal, A. Global cancer incidence and mortality rates and trends—an update. Cancer Epidemiol. Prev. Biomark. 25, 16–27 (2016).
Gerasimova, E. et al. Wavelet-based multifractal analysis of dynamic infrared thermograms to assist in early breast cancer diagnosis. Front. Physiol. 5, 176 (2014).
Consortium, I. C. G. International network of cancer genome projects. Nature 464, 993 (2010).
Futreal, P. A. et al. A census of human cancer genes. Nat. Rev. Cancer 4, 177 (2004).
Network, C. G. A. R. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061 (2008).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214 (2013).
Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 358, 1148–1159 (2008).
Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57 (2012).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Berger, S. L. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 12, 142–148 (2002).
Lee, J.-S., Smith, E. & Shilatifard, A. The language of histone crosstalk. Cell 142, 682–685 (2010).
Guttman, M. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223 (2009).
Cabili, M. N. et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927 (2011).
Alinejad-Rokny, H., Heng, J. I. & Forrest, A. R. Brain-Enriched Coding and Long Non-coding RNA Genes Are Overrepresented in Recurrent Neurodevelopmental Disorder CNVs. Cell Rep. 33, 108307 (2020).
Dashti, H. et al. Integrative analysis of mutated genes and mutational processes reveals novel mutational biomarkers in colorectal cancer. BMC bioinformatics 23, 1–24 (2022).
Ghareyazi, A. et al. Whole-genome analysis of de novo somatic point mutations reveals novel mutational biomarkers in pancreatic cancer. Cancers 13, 4376 (2021).
Heidari, R., Akbariqomi, M., Asgari, Y., Ebrahimi, D. A systematic review of long non-coding RNAs with a potential role in Breast Cancer. Mutation Res. 787, 108375 (2021).
Mourtada-Maarabouni, M., Pickard, M., Hedge, V., Farzaneh, F. & Williams, G. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene 28, 195 (2009).
Gupta, R. A. et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071 (2010).
Sørensen, K. P. et al. Long non-coding RNA HOTAIR is an independent prognostic marker of metastasis in estrogen receptor-positive primary breast cancer. Breast cancer Res. Treat. 142, 529–536 (2013).
Kopp, F. & Mendell, J. T. Functional classification and experimental dissection of long noncoding RNAs. Cell 172, 393–407 (2018).
Ørom, U. A. et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 143, 46–58 (2010).
Kim, T.-K., Hemberg, M. & Gray, J. M. Enhancer RNAs: a class of long noncoding RNAs synthesized at enhancers. Cold Spring Harb. Perspect. Biol. 7, a018622 (2015).
Quinodoz, S. & Guttman, M. Long noncoding RNAs: an emerging link between gene regulation and nuclear organization. Trends Cell Biol. 24, 651–663 (2014).
Böhmdorfer, G. & Wierzbicki, A. T. Control of chromatin structure by long noncoding RNA. Trends Cell Biol. 25, 623–632 (2015).
Lizio, M. et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 16, 1–14 (2015).
Howe, K. L. et al. Ensembl 2021. Nucleic Acids Res. 49, D884–D891 (2021).
Lorenzi, L. et al. The RNA Atlas expands the catalog of human non-coding RNAs. Nat. Biotechnol. 39, 1453–1465 (2021).
Banerjee-Basu, S. & Packer, A. SFARI Gene: an evolving database for the autism research community. Dis. Models Mechanisms 3, 133–135 (2010).
Nik-Zainal, S. et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54 (2016).
Li, J. et al. TANRIC: An Interactive Open Platform to Explore the Function of lncRNAs in Cancer. Cancer Res. 75, 3728–3737 (2015).
Albert, F. W. & Kruglyak, L. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 16, 197–212 (2015).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019).
Li, M. J. et al. GWASdb v2: an update database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res. 44, D869–D876 (2016).
Carén, H. et al. Identification of epigenetically regulated genes that predict patient outcome in neuroblastoma. BMC Cancer 11, 66 (2011).
Consortium, G. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660 (2015).
Song, J. et al. Genetic polymorphisms of long noncoding RNA RP11‐37B2. 1 associate with susceptibility of tuberculosis and adverse events of antituberculosis drugs in west China. J. Clin. Labor. Analy. 33, e22880 (2019).
Li, D. et al. Identification of lncRNAs and their functional network associated with chemoresistance in SW1990/GZ pancreatic cancer cells by RNA sequencing. DNA Cell Biol. 37, 839–849 (2018).
Hon, C.-C. et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 543, 199–204 (2017).
Dong, Y., Zhang, T., Li, X., Yu, F. & Guo, Y. Comprehensive analysis of coexpressed long noncoding RNAs and genes in breast cancer. J. Obstet. Gynaecol. Res. 45, 428–437 (2019).
Wang, J. Role of ABL Family Kinases in Breast Cancer (Duke University, 2016).
Baker, M. J., Abel, P. & Lea, R.W. inventors; University of Central Lancashire, assignee. Methods of diagnosing proliferative disorders. United States patent application US, application number: 14/443,134. (2016).
Casamassimi, A. et al. Multifaceted role of PRDM proteins in human cancer. Int J Mol Sci. 21, 2648 (2020).
Gross, D. S. & Garrard, W. T. Nuclease hypersensitive sites in chromatin. Annu. Rev. Biochem. 57, 159–197 (1988).
Gusev, A. et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet. 95, 535–552 (2014).
Barski, A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007).
Bonifer, C. & Cockerill, P. N. In Epigenetic Contributions in Autoimmune Disease 12–25 (Springer, 2011).
Mercer, T. R. et al. DNase I–hypersensitive exons colocalize with promoters and distal regulatory elements. Nat. Genet. 45, 852–859 (2013).
Zhang, Y. et al. Identification of an lncRNA‑miRNA‑mRNA interaction mechanism in breast cancer based on bioinformatic analysis. Mol. Med. Rep. 16, 5113–5120 (2017).
Zhang, X., Wang, M., Sun, H., Zhu, T. & Wang, X. Downregulation of LINC00894-002 Contributes to Tamoxifen Resistance by Enhancing the TGF-β Signaling Pathway. Biochem. (Mosc.) 83, 603–611 (2018).
Wu, J. et al. Linc00152 promotes tumorigenesis by regulating DNMTs in triple-negative breast cancer. Biomed. Pharmacother. 97, 1275–1281 (2018).
Krijger, P. H. L. & De Laat, W. Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Biol. 17, 771 (2016).
Mifsud, B. et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 47, 598 (2015).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Khakmardan, S., Rezvani, M., Pouyan, A. A., Fateh, M. & Alinejad-Rokny, H. MHiC, an integrated user-friendly tool for the identification and visualization of significant interactions in Hi-C data. BMC Genom. 21, 1–10 (2020).
Alinejad-Rokny, H. et al. MaxHiC: robust estimation of chromatin interaction frequency in Hi-C and capture Hi-C experiments. bioRxiv 2020, https://doi.org/10.1101/2020.04.23.056226 (2020).
Stosiek, P. & Kasper, M. Neo-expression of cytokeratin 7 in chronic atrophic gastritis with pernicious anemia. Der Pathol. 11, 14 (1990).
Dryden, N. H. et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res. 24, 1854–1868 (2014).
Hilakivi-Clarke, L. et al. Effects of in utero exposure to ethinyl estradiol on tamoxifen resistance and breast cancer recurrence in a preclinical model. J. Natl Cancer Institute 109, 353–365 (2017).
Kennedy, S. R., Lawrence, A. Loeb & Herr, AlanJ. Somatic mutations in aging, cancer and neurodegeneration. Mechanisms Ageing Dev. 133, 118–126 (2012).
Bailey, M. H. et al. Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385 (2018).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Bayati, M. et al. CANCERSIGN: a user-friendly and robust tool for identification and classification of mutational signatures and patterns in cancer genomes. Sci. Rep. 10, 1–10 (2020).
Hamidi, H., et al. Signatures of Mutational Processes in Human DNA Evolution. bioRxiv 2021, https://doi.org/10.1101/2021.01.09.426041 (2021).
Struhl, K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat. Struct. Mol. Biol. 14, 103–105 (2007).
Wu, Y. et al. The role of lncRNAs in the distant metastasis of breast cancer. Front. Oncol. 9, 407 (2019).
Afrasiabi, A., Keane, J. T., Heng, J. I. T., Palmer, E. E. & Lovell, N. H. Quantitative neurogenetics: applications in understanding disease. Biochem. Soc. Trans. 49, 1621–1631 (2021).
Stevens, K. N., Vachon, C. M. & Couch, F. J. Genetic susceptibility to triple-negative breast cancer. Cancer Res. 73, 2025–2030 (2013).
Wu, J., Mamidi, T. K. K., Zhang, L. & Hicks, C. Integrating germline and somatic mutation information for the discovery of biomarkers in triple-negative breast cancer. Int. J. Environ. Res. Public Health 16, 1055 (2019).
Cunningham, F. et al. Ensembl 2019. Nucleic Acids Res. 47, D745–D751 (2019).
Rajaei, P., Jahanian, K. H., Beheshti, A., Band, S. S. & Dehzangi, A. VIRMOTIF: A user-friendly tool for viral sequence analysis. Genes Dev. 12, 186 (2021).
Alinejad-Rokny, H. Proposing on optimized homolographic motif mining strategy based on parallel computing for complex biological networks. J. Med. Imaging Health Inform. 6, 416–424 (2016).
Hosseinpoor, M. et al. Proposing a novel community detection approach to identify cointeracting genomic regions. Math. Biosci. Eng. 17, 2193–2217 (2020).
Forrest, A. R. et al. A promoter-level mammalian expression atlas. Nature 507, 462–470 (2014).
Andersson, R. et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014).
Wang, J. et al. Factorbook. org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic acids research 41, D171–D176 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Javanmard, R. & JeddiSaravi, K. Proposed a new method for rules extraction using artificial neural network and artificial immune system in cancer diagnosis. J. Bionanosci. 7, 665–672 (2013).
Rad, M. P. & Pourshaikh, R. Conceptual Information Retrieval in Cross-Language Searches. Research. J. Appl. Sci. Eng. Technol. 4, 1714–1720 (2012).
Parvin, H. & Parvin, S. Divide and conquer classification. Aust. J. Basic Appl. Sci. 5, 2446–2452 (2011).
Hasanzadeh, E. et al. Text clustering on latent semantic indexing with particle swarm optimization (PSO) algorithm. Int. J. Phys. Sci. 7, 16 (2012).
Esmaeili, L., Minaei-Bidgoli, B. & Nasiri, M. Hybrid recommender system for joining virtual communities. Res. J. Appl. Sci. Eng. Technol. 4, 500–509 (2012).
Pho, K. H., Akbarzadeh, H., Parvin, H., Nejatian, S. & Alinejad-Rokny, H. A multi-level consensus function clustering ensemble. Soft Computing 25, 13147–13165 (2021).
Alinejad-Rokny, H., Anwar, F., Waters, S. A., Davenport, M. P. & Ebrahimi, D. Source of CpG depletion in the HIV-1 genome. Mol. Biol. Evol. 33, 3205–3212 (2016).
Alinejad-Rokny, H., Pourshaban, H., Orimi, A. G. & Baboli, M. M. Network motifs detection strategies and using for bioinformatic networks. J. Bionanosci. 8, 353–359 (2014).
Parvin, H. & MirnabiBaboli, M. Proposing a classifier ensemble framework based on classifier selection and decision tree. Eng. Appl. Artif. Intell. 37, 34–42 (2015).
Alinejad-Rokny, H., Sadroddiny, E. & Scaria, V. Machine learning and data mining techniques for medical complex data analysis. Neurocomputing 276, 1 (2018).
Rosenbloom, K. R. et al. The UCSC genome browser database: 2015 update. Nucleic Acids Res. 43, D670–D681 (2015).
Acknowledgements
This work was funded by the UNSW Scientia Program Fellowship and the Australian Research Council Discovery Early Career Researcher Award (DECRA) under grant DE220101210 to H.A.R. We kindly acknowledge the Government of Western Australia, Department of Health, Clinical Excellence, for their kind support on this project through the MERIT award to H.A.R. H.A.R is also supported by UNSW Scientia Program Fellowship. Analysis was made possible with computational resources provided by the BioMedical Machine Learning Bioinformatics Server with funding from the Australian Government and the UNSW SYDNEY. H.R.R is supported by IRN National Science Foundation (INSF) Grant No. 96006077.
Author information
Authors and Affiliations
Contributions
H.A.R. designed the study; H.A.R., N.R., and M.B. wrote the paper. The paper was edited by H.A.R., N.R., J.B., M.S.T., M.B., N.H.L., and H.R.R. N.R., M.B., and M.H. carried out all the analyses, including the statistical analyses, gene prioritization, annotation, permutation, and Hi-C data analysis, with the supervision of H.A.R. and H.R.R. N.R. and M.B. generated all figures and all tables with the supervision of H.A.R. and H.R.R. S.K. designed and developed the website. All authors have read and approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests. H.A-R. is an Editorial Board Member for Communications Biology but was not involved in the editorial review or decision to publish this paper.
Ethics approval and consent to participate
The ethical approval was not needed; all the data used in this study are publicly available.
Peer review
Peer review information
Communications Biology thanks Erik Knutsen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Christina Karlsson Rosenthal. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rezaie, N., Bayati, M., Hamidi, M. et al. Somatic point mutations are enriched in non-coding RNAs with possible regulatory function in breast cancer. Commun Biol 5, 556 (2022). https://doi.org/10.1038/s42003-022-03528-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03528-0
This article is cited by
-
Harnessing deep learning into hidden mutations of neurological disorders for therapeutic challenges
Archives of Pharmacal Research (2023)
-
Extensive review on breast cancer its etiology, progression, prognostic markers, and treatment
Medical Oncology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
