
Abstract
Reduced glomerular filtration rate (GFR) can progress to kidney failure. Risk factors include genetics and diabetes mellitus (DM), but little is known about their interaction. We conducted genome-wide association meta-analyses for estimated GFR based on serum creatinine (eGFR), separately for individuals with or without DM (nDM = 178,691, nnoDM = 1,296,113). Our genome-wide searches identified (i) seven eGFR loci with significant DM/noDM-difference, (ii) four additional novel loci with suggestive difference and (iii) 28 further novel loci (including CUBN) by allowing for potential difference. GWAS on eGFR among DM individuals identified 2 known and 27 potentially responsible loci for diabetic kidney disease. Gene prioritization highlighted 18 genes that may inform reno-protective drug development. We highlight the existence of DM-only and noDM-only effects, which can inform about the target group, if respective genes are advanced as drug targets. Largely shared effects suggest that most drug interventions to alter eGFR should be effective in DM and noDM.
Similar content being viewed by others

Introduction
Impaired kidney function can progress to kidney failure requiring dialysis or transplantation, which implicates high early mortality1 and public health burden2. The most common kidney function measure is the glomerular filtration rate (GFR). Chronic kidney disease (CKD) can be defined as GFR < 60 ml/min per 1.73 m2, and has a prevalence of ~10% in the population3. In large-scale epidemiological studies, GFR can be estimated from serum creatinine (eGFR) using the CKD-Epi formula4.
Diabetes mellitus (DM) is a major risk factor for impaired kidney function5 and is present in 30% of individuals starting dialysis in Europe6. Most current pharmacotherapy in DM has substantial side effects7,8,9,10. Recent work showed that SGLT2 inhibitors and GLP1R are reno-protective for individuals with DM11,12 and early evidence indicates that this might be also true for individuals without DM (noDM)13. The biological processes related to lower kidney function might differ between individuals with DM and individuals without DM. Understanding the mechanisms of kidney function variability within and between these groups is pivotal for understanding pathogenesis. Genome-wide association studies (GWAS) have pinpointed hundreds of loci for eGFR14,15,16. These loci help understand kidney function variability in the overall population. However, commonalities and differences in genetic kidney function effects between DM and noDM individuals are not well understood.
Genes underneath GWAS loci have been shown to double the success rate in drug development pipelines17,18. This renders each gene in eGFR loci a drug target to alter kidney function, particularly when the gene maps to association-driving variants that are relevant for the protein or for the gene’s expression in kidney tissue19. This opens up a clinically important question in the context of differential or shared genetic kidney function effects between DM and noDM individuals: when a locus identified for eGFR in the overall population turns out to have no effect in DM individuals, the underlying causal gene might not be informative for reno-protection in DM individuals. This is not an unlikely scenario, since only 10% of individuals in previous population-based GWAS on eGFR overall had DM16,20.
Previous DM-/noDM-stratified searches for genetic eGFR-effects have been limited21 and without identification of a significant DM/noDM-difference. It is unclear whether this was due to limited power or lack of such differential effects. GWAS for eGFR-based kidney function outcomes in DM, used as one approach to identify the genetics of Diabetic Kidney Disease (DKD), identified four loci in ~40,000 type 1 or type 2 DM patients22 and four other loci in ~19,500 type 1 DM patients23. When genetic effects on eGFR in DM versus noDM have little overlap, such focused searches will remain the best option. For shared genetics, GWAS on eGFR overall will have much better power.
The quest for understanding commonalities and differences of genetic eGFR-effects between DM and noDM has two aspects: a search for eGFR-associated loci with DM/noDM-difference will help understand whether such differential effects exist and a search for SNP-association on eGFR allowing for DM/noDM-difference may detect novel eGFR loci. Statistically, DM/noDM-difference in eGFR association and genetics-by-DM-status interaction on eGFR association are equivalent. There are various approaches to do so24. These approaches have been applied successfully for other phenotypes25,26,27,28,29, but not yet for GWAS interaction analyses for kidney function.
We thus set out to search for eGFR loci with DM/noDM-difference and for novel eGFR loci allowing for difference. For this, we gathered GWAS data on eGFR separately for 178,691 DM and 1,296,113 noDM individuals. We prioritized genes underneath identified loci using in-silico functional evidence. We also evaluated the impact of DM-/noDM-specific weights on the genetic risk score (GRS) for eGFR in data independent from the variant- and weight-identifying step30.
Results
Overview of the GWAS meta-analyses
We first analyzed GWAS data on 7,046,926 single nucleotide polymorphisms (SNPs) and their association with logarithm-transformed eGFR separately for 109,993 DM (type 2 or type 1) and 1,070,999 noDM individuals (stage 1, including 72 studies from CKDGen Consortium31 and UK Biobank, UKB32; mostly European-ancestry; Supplementary Data 1, Methods, Fig. 1a). These DM-/noDM-stratified GWAS summary statistics allowed us to apply the difference test, the joint test and the stratified tests (Methods). Based on these tests, we searched for eGFR-associated loci with DM/noDM-difference and for novel eGFR-associated loci allowing for DM/noDM-difference.

a Overview on datasets and meta-analyses. b Approaches to identify DM/noDM-differences. c Approaches to identify novel eGFR loci and suggestive DM/noDM-differences. d Genetic risk score (GRS) analyses in HUNT.
We sought replication of identified loci in independent DM-/noDM-stratified data (stage 2, nDM = 68,698, nnoDM = 225,114; Million Veterans Program, MVP33, Michigan Genomics Initiative, MGI34, Trøndelag health study, HUNT35; all European-ancestry; Supplementary Data 1, Methods). While the discovery+replication design augments confidence in identified loci, this comes at the cost of lower power36. We also conducted GWAS searches in a combined stage design (stage 1 + 2: nDM = 178,691, nnoDM = 1,296,113), to yield significant loci exploiting the full sample size, yet without independent replication.
The two stages of data were also used to separate the variant identification and weight quantification for the GRS (stage 1 data) from the GRS association analyses (stage 2 data). The analysis workflow is shown in Fig. 1.
Seven eGFR loci identified with DM-/noDM-differential effects
To search for eGFR loci with DM/noDM-difference, we applied two approaches24,37 (Fig. 1b): (i) a genome-wide difference test (PDiff < 5 × 10−8, difference test approach), and (ii) a search for genome-wide significant association with overall eGFR followed by a difference test in the same data (POverall < 5 × 10−8 and PDiff < 0.05/k, k = number of followed SNPs, overall+difference test approach, Methods).
In the discovery search (stage 1, nDM = 109,993; nnoDM = 1,070,999), we identified four eGFR loci with significant DM/noDM-difference (Supplementary Fig. 1): (i) two by the difference test approach (rs77924615 near UMOD-PDILT, rs12233328 near PDE9A; PDiff < 5 × 10−8, Supplementary Data 2), and (ii) two further loci by the overall+difference test approach (near TPPP and MED1-NEUROD2; POverall < 5 × 10−8 and PDiff < 0.05/610 = 8.2 × 10−5; corrected for 610 followed variants16, Supplementary Data 2). Details of the overall+difference test results are provided in Supplementary Note 1 and Supplementary Data 3, conditional difference analyses on known POverall < 5 × 10−8 variants in UMOD-PDILT in Supplementary Fig. 2. In stage 2 data (nDM = 68,698, nnoDM = 225,114), three of the four loci replicated (near UMOD-PDILT, MED1-NEUROD2, and TPPP; one-sided PDiff < 0.05/4 = 0.0125; Table 1, Supplementary Data 2), while the PDE9A locus variant did not (PDiff = 0.30).
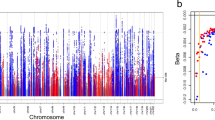
In the combined stage design (stage 1 + 2: nDM = 178,691; nnoDM = 1,296,113), we identified seven eGFR loci with significant DM/noDM-difference (Table 1, Fig. 2a, Supplementary Fig. 3): (i) six loci by the difference test approach (PDiff < 5 × 10−8, Supplementary Data 2) including the three loci already found by the discovery+replication approach now all with PDiff < 5 × 10−8 (near UMOD-PDILT, TPPP, and MED1-NEUROD2;), and three additional loci (near CSRNP, DCDC5, and NRIP1), (ii) one locus from the overall+difference test approach (near SLC22A2; POverall < 5 × 10−8 and PDiff < 0.05/610 = 8.2 × 10−5; 610 followed variants as described above, Supplementary Data 2).

We searched for DM/noDM-differential genetic associations on eGFR using the difference test approach and the overall+difference approach in combined stage 1 + 2 (CKDGen, UKB, MVP, MGI, and HUNT; nDM = 178,691; total nnoDM = 1,296,113). Seven difference loci were identified. a Shown are difference test P-values over chromosomal base position (Manhattan plot) highlighting the six loci identified by the difference test approach (red, PDiff < 5 × 10−8) and the one locus identified by the overall+difference test approach (orange, 610 variants16 with stage 1 POverall < 5 × 10−8, PDiff < 0.05/610 = 8.2 × 10−5). Loci are annotated by the name(s) of the nearest gene(s); asterix indicates loci that were also identified by the discovery+replication design (Table 1, Supplementary Fig. 1). b Shown is a comparison of DM-/noDM-specific eGFR-effect sizes for the seven identified difference lead variants based on combined stage 1 + 2 data. Effect sizes are aligned to the eGFR-decreasing alleles in noDM except for CSRNP1 (aligned to eGFR-decreasing allele in DM). Error bars reflect 95% confidence intervals of the estimated genetic effect.
Approximate conditional analyses using the difference test (combined stage 1 + 2, European only, Methods) did not identify any further independent variants within the 7 identified difference loci (regional difference plots in Supplementary Fig. 4). Interestingly, there was also no further independent variant with DM/noDM-difference in the UMOD-PDILT locus besides rs77924715, despite the multiple independent variants known for eGFR association overall16,20 (Supplementary Fig. 4a, b). Sensitivity analyses confirmed that DM/noDM-difference was also observed without log-transformation of eGFR and also after adjusting for interaction with age or other eGFR risk factors (Supplementary Note 2, Supplementary Data 4, Supplementary Table 1).
With respect to DM-/noDM specificity and magnitude of effects, we found the following (Fig. 2b): (i) six of the seven variants identified with differential eGFR-effects showed more pronounced effects in DM versus noDM (near UMOD-PDILT, TPPP, DCDC5, NRIP1, and SLC22A2) or a DM-only effect on eGFR (near CSRNP1) and (ii) one variant showed a noDM-only effect (near MED1-NEUROD2). These patterns were consistent in stage 1 and stage 2 separately (Supplementary Fig. 5). Of note, with the combined stage 1 + 2 sample size, the difference detectable for a common variant of 30% minor allele frequency at 80% power was 0.0050 or 0.0037 log(ml/min/1.73 m²) for a DM-only or noDM-only effect, respectively (Supplementary Note 3, Supplementary Fig. 6).
In summary, we established seven eGFR loci with DM/noDM-difference (near UMOD-PDILT, TPPP, MED1-NEUROD2, CSRNP1, DCDC5, NRIP1, and SLC22A2) including one with DM-only (CSRNP1) and one with noDM-only effect (MED1-NEUROD2). While all seven difference loci were known loci for eGFR16, these are the first eGFR loci reported with significant DM/noDM-difference from a genome-wide search for the difference to the best of our knowledge.
Allowing for difference identifies 32 novel eGFR-associated loci including CUBN
To search for novel eGFR-associated loci allowing for the difference between DM and noDM, we applied two approaches (Fig. 1c, Methods): (i) a genome-wide joint test38, which considers the main SNP effect on eGFR and the SNP-by-DM-status interaction effect jointly (joint-test approach) and (ii) GWAS separately in DM or noDM (stratified tests approach).
In the discovery (stage 1; nDM = 109,993, nnoDM = 1,070,999), we identified 25 novel eGFR loci after excluding previously identified GWAS eGFR loci16,20 (PJoint or PnoDM < 5 × 10−8, none by PDM < 5 × 10−8, Supplementary Data 5, Supplementary Fig. 7a). In stage 2 data (nDM = 68,698, nnoDM = 225,114;), three of the 25 loci replicated (PJoint or PnoDM < 0.05/25; near ATP12A, SERTAD2, and ABCC2, Table 2).
In the combined stage design (stage 1 + 2: nDM = 178,691, nnoDM = 1,296,113), we identified 32 novel eGFR loci (PJoint or PDM or PnoDM < 5 × 10−8, Fig. 3a, Table 2, Supplementary Data 5, Supplementary Fig. 7b): (i) 30 novel loci by the joint test; (ii) two additional loci by the noDM-only search (near FAT4, SLC2A4). While no locus was additionally identified by the DM-only search, two of the 32 novel loci were genome-wide significant in DM (PDM < 5 × 10−8; near SH3BP4, LOXL4; also identified by joint test).

We searched for novel loci associated with eGFR allowing for DM-/noDM-difference using the joint test or DM-/noDM-stratified tests approaches in combined stage 1 + 2 (nDM = 178,691; total nnoDM = 1,296,113). We found 32 novel genome-wide significant eGFR loci (P < 5 × 10−8, >500 kB distant of known eGFR loci compared to previous work16,20): 30 by joint, 17 by noDM-only and 2 by DM-only test. a Shown are P-values for eGFR based on joint, noDM-only, and DM-only test over chromosomal position. Highlighted in red are loci with suggestive DM/noDM-difference (PDiff < 0.05/34; corrected for 34 independent variants across 32 loci), blue for loci identified by joint and noDM-only test (15 loci), and purple for loci that were only identified by joint test (upper panel) or noDM-only test (middle panel). Loci were annotated by nearest genes if PDiff < 0.10 or if they were also identified by the discovery+replication design (the latter also indicated by asterix, Table 2). b Shown is a comparison of DM-/noDM-specific eGFR-effect sizes for the 32 novel eGFR locus lead variants. Highlighted in red are the locus names of loci with suggestive DM/noDM-difference (PDiff < 0.05/34; corrected for 34 independent variants across 32 loci). Effect sizes are aligned to the eGFR-decreasing alleles in noDM. Error bars reflect 95% confidence intervals of the estimated genetic effect.
These 32 novel loci included the three replicated novel loci from the discovery+replication approach (near ATP12A, SERTAD2, and ABCC2) (Table 2). The 29 additional novel loci included the CUBN locus, which is well-known for urinary albumin-to-creatinine ratio (UACR)39 and microalbuminuria40 (Fig. 4). The identified CUBN locus eGFR lead variant, rs11254238, showed (i) no correlation with the known signals for UACR (r² < 0.001, D’ > 0.24 to any of the known independent signal variants for UACR, rs141493439, rs45551835, rs557338857, rs562661763, rs74375025), (ii) no effect on UACR41 (P = 0.57, Supplementary Data 6), and (iii) a twice as large effect on eGFR in DM compared to noDM, but the difference was not significant (bDM = 0.0048, bnoDM = 0.0021, PDiff = 0.07).

Shown are P-values for associations at the wider (top) and more narrow (bottom) CUBN locus region for a eGFR (joint test P-values, nDM = 178,691 and nnoDM = 1,296,113) and b urinary albumin-to-creatinine ratio (UACR; P-values from ref. 39, n = 564,257). Lead variant for eGFR is rs11254238; color codes variants’ correlation r2 to rs11254238 in all panels.
Approximate conditional analyses (combined stage, European only, Methods) identified two additional variants independently associated with eGFR at two joint-test loci (near SERTAD2 and PIK3CG, PJoint_cond < 5 × 10−8, Supplementary Table 2). This raised the total number of newly identified eGFR-associated variants to 34.
Among the 34 novel eGFR variants, four showed a suggestive DM/noDM-difference for eGFR (PDiff < 0.0015 = 0.05/34, Table 2, Fig. 3b): one was a DM-only effect (near SH3BP4) and three showed more pronounced effects in DM (near LOXL4, ALPL, and PIK3CG). These patterns were consistent also in stage 1 and stage 2 separately (Supplementary Fig. 5). Of note, the difference test is mathematically dependent on the joint test and the stratified tests, but not on the overall test24. Therefore, the observed difference in the overall+difference test approach applied to the same data is an established significant difference, while the observed difference in loci established by the joint or stratified tests is termed here as suggestive difference.
In summary, the joint test and stratified tests identified 34 genome-wide significant independent eGFR variants across 32 novel eGFR loci. These included four loci with suggestive DM/noDM-difference, which yielded a total of 11 eGFR loci with established or suggestive DM/noDM-difference.
Interaction with Hba1C and overlap with associations for DM risk, glycemic and other traits
We explored the lead variants of the 11 eGFR loci identified with DM/noDM-difference (i.e., established/suggestive SNP-by-DM interaction) for SNP-by-HbA1c interaction in UK Biobank (n = 368,005), making full use of the continuous variable HbA1c instead of binary DM status. For all 11 variants, the SNP main effects and SNP-by-HbA1c interaction effect sizes were directionally consistent with main and interaction effects sizes in the SNP-by-DM interaction analysis (Supplementary Data 7, Methods). This underscored again the negative interaction effect for the 10 of the 11 variants with larger effects in DM or DM-only effects, while the one variant (near MED1/NEUROD2) showed a negative interaction effect for the SNP-by-DM as well as the SNP-by-HbA1c interaction.
We were also interested in whether any of the 11 loci with DM/noDM-difference overlapped with known genome-wide significant loci for type 1 DM42, type 2 DM19, or glycemic traits43. None of the 11 loci overlapped with type 1 DM, type 2 DM, or glycemic traits loci (all 11 lead variants P > 5.0 × 10−8 in published GWAS for DM risk, glucose, or insulin levels, Supplementary Data 8). None of the 11 variants was associated with type 1 DM judged at Bonferroni-corrected significance (P > 0.05/11 = 4.5 × 10−3). Three of the 11 variants were associated with type 2 DM (P < 0.05/11 = 4.5 × 10−3, rs77924615 near UMOD-PDILT, rs55722796 near MED1-NEUROD2) or fasting glucose (rs963837 near DCDC5). For all three variants, the eGFR-lowering allele was associated with decreased type 2 DM risk or fasting glucose. Thus, the SNP effect on eGFR cannot be fully explained by DM status, which would have yielded a DM risk increasing or glucose-increasing effect by the eGFR-lowering allele. The observation is in line with a pleiotropic effect on DM and eGFR, but with adverse effects for one of the two (eGFR or DM/glucose) and a beneficial effect for the other, which should be considered in drug design when applicable. When taking-into-account the DM/noDM-stratum where the eGFR effect was more pronounced, the three variants consisted of (i) two variants with stronger eGFR-effects in DM (still significant eGFR effect in noDM, near UMOD/PDILT and DCDC5) and (ii) one variant with a noDM-only effect on eGFR (near MED1-NEUROD2).
We also queried the Open Targets Genetics database44 for associations of the 11 variants with other traits. We found 126 genome-wide significant associations (P < 5 × 10−8, Supplementary Data 9), particularly for hypertension and blood counts: for 3 of the 11 variants, the eGFR-decreasing alleles were associated with increased risk of hypertension (near UMOD/PDILT, DCDC5, and PIK3CG). The variant rs55722796 near MED1/NEUROD2, solely associated with eGFR in noDM individuals, was not associated with hypertension, but the eGFR-decreasing allele (in noDM) was associated with decreased blood counts (i.e., decreased red blood cells, hemoglobin, and hematocrit).
GWAS for eGFR in individuals with DM identified 29 genome-wide significant loci
Previous searches for SNP-effects on eGFR-based kidney function outcome in DM patients had identified eight genome-wide significant loci (i.e., eGFR-based DKD loci; four identified in type 1 DM patients23, four other in type 1 or type 2 DM patients22). We wanted to understand the overlap of these with the genome-wide significant loci in our DM-only GWAS on eGFR (nDM = 178,691, combined stage, Methods): we observed 29 genome-wide significant eGFR loci in DM (PDM < 5 × 10−8, Fig. 5, Supplementary Data 10). These included: (i) 27 novel eGFR loci in DM, and (ii) two of the four previously identified DKD loci among type 1 or type 2 DM patients22 (near UMOD/PDILT and PRKAG2; r2 = 0.60 and 0.94, respectively, between eGFR and DKD lead variants). None of the previously identified DKD loci among type 1 DM patients23 were identified. Two of the 27 novel eGFR loci in DM were detected here as completely novel eGFR loci (near SH3BP4 and LOXL4; also detected by the joint test). The 29 identified eGFR loci in DM included 6 of our 11 difference loci (near UMOD/PDILT, TPPP, DCDC5, SH3BP4, NRIP1, and LOXL4).

Shown are eGFR association P-values in individuals with DM over chromosomal position in combined stage (nDM = 178,691). This DM-specific analysis identified 29 independent eGFR-associated loci in DM. Compared to known DKD loci22,23 (i.e., association with eGFR or CKD in type 1 and/or type 2 DM individuals) and known overall eGFR loci16,20, 2 loci are novel for eGFR overall and novel for DKD (red), 24 are novel for DKD but known for eGFR (orange), and 3 are known DKD and known eGFR loci (purple).
In silico evidence prioritized 18 genes by protein- or expression-altering variants or as human kidney disease monogene
Genes at GWAS loci for eGFR might pinpoint relevant drug targets for kidney function18. Dissecting eGFR loci as DM-only, noDM-only, or shared will help define the target population for potential therapy. Gene PrioritiSation (GPS) was conducted previously16 for all genes underneath the 424 eGFR-associated loci in a meta-analysis of CKDGen and UKB (i.e., our stage 1 studies) focused on European-ancestry. Since all 7 difference loci were among these 424 loci, we used this established GPS tool to extract in-silico functional evidence for the 159 genes at the 7 difference loci: here, we prioritized a gene when it mapped to an association-driving variant (i.e., 99% credible set variant with posterior probability of association ≥5%, Methods) that was relevant (i) for the protein (with high predicted deleteriousness45, i.e., CADD score ≥15) or (ii) for expression in kidney tissue (eQTL, false-discovery rate ≥5%)46,47 or (iii) when the gene was a known kidney disease monogene in human (Methods). We found 6 prioritized genes (Fig. 6, Supplementary Fig. 8): (i) PGAP3 mapping to the locus with noDM-only effect on eGFR (near MED1/NEUROD2) and (ii) five genes in loci with more pronounced eGFR-effects in DM but direction-consistent effects in noDM (TPPP, UMOD, SLC6A19, NRIP1, and SLC22A2).

Shown are gene prioritization results for the seven loci with established difference (Table 1, Supplementary Fig. 8). We highlighted six genes based on association-driving variants (PPA > 5%) that were deleteriously protein-relevant or expression-modulating, genes that were known as human kidney monogenes (OMIM or ref. 78 with subsequent manual curation) and in addition SLC22A2 due to its known link to metformin response.
We also used the same approach as in the previous GPS tool16 to prioritize the genes underneath the 32 novel eGFR loci (34 independent signals, Supplementary Data 11–15, Supplementary Table 3, Supplementary Fig. 9): (i) among the 40 genes in the four novel loci with suggestive DM/noDM-difference, 2 genes were human kidney disease monogenes (HPS1 and HPSE2, Fig. 7a); (ii) for the 341 genes in the other 28 novel eGFR loci (i.e., noDM/noDM-difference), 10 genes were prioritized (Fig. 7b): 6 genes contained an association-driving variant that was protein-deleterious (AUTS2, CUBN, DVL2, RASSF6, SLC2A4, and ZFP36L1), one gene mapped to an eQTL in glomerular tissue (TNIK), and 3 additional genes were human kidney disease monogenes (SLC2A2, SLC30A9, and SLC7A7). Particularly interesting was an association-driving variant, rs1801232, in CUBN that was a missense variant (r² = 0.73 to eGFR lead variant rs11254238). The missense variant rs17804499 in RASSF6 had a particularly high probability of being the association-driving variant (posterior probability of association = 86%), which rendered this variant and gene a compelling candidate for functional follow-up. Two genes, SLC2A2 and SLC2A4, encode the glucose transporters GLUT2 and GLUT4 and reside in two novel eGFR loci.

Shown are gene prioritization results for the 32 novel eGFR loci (Table 2, Supplementary Fig. 9): a for the four novel eGFR loci with suggestive difference, and b for the 28 other novel eGFR loci. We highlighted 12 genes based on association-driving variants (PPA > 5%) that were deleteriously protein-relevant or expression-modulating or genes that were known as human kidney monogenes (OMIM or ref. 78 with subsequent manual curation).
In summary, across the identified seven loci identified with established or suggestive DM-/noDM-difference in eGFR-effects and the 32 novel eGFR loci, we prioritized 18 genes with evidence of protein-deleterious or expression-modulating variants or being human kidney disease monogenes. These genes are compelling candidates for functional follow-up and drug development pipelines.
DM/noDM-stratified weights for GRS show similar explained variance as overall weights
We assessed whether the GRS for eGFR, computed as an effect size weighted sum of eGFR-lowering alleles, could be improved by using DM/noDM-specific eGFR-effects as weights rather than overall eGFR-effects. We built the GRS based on the previously identified 634 eGFR variants16 (i.e., identified in stage 1 studies) or based on the seven variants with established DM/noDM-difference using different weighting schemes (overall effects, DM-/noDM-specific effects, or a combination; all weights from stage 1 data). We computed the GRS for each individual in the stage 2 study HUNT (unrelated, European-ancestry; nDM = 3023, nnoDM = 25,484, Fig. 1d, Methods).
Across all weighting schemes, we found no significant DM/noDM-difference in the 634-variant GRS association with eGFR (PDiff > 0.05, Table 3). The 634-variant GRS explained more of the eGFR variance in noDM compared to DM (e.g., R2 = 6.0% vs. 4.0% using overall-effect weights). Since the absolute 634-variant GRS effect was similar in DM and noDM (e.g., beta per sdGRS = −2.54 versus −2.84 ml/min/1.73 m2 using overall-effect weights), this larger relative GRS effect in noDM can be attributed—at least in part—to the smaller eGFR variance in noDM versus DM (HUNT standard deviation = 11.8 versus 13.4 ml/min/1.73 m2, respectively). Yet, the 7-variant GRS explained more of the eGFR variance in DM compared to noDM (e.g., R2 = 0.98% vs. 0.62% using overall-effect weights). The reason for this is that six of the seven variants had larger eGFR-effects in DM than in noDM, which accumulated to a larger absolute GRS effect in DM (e.g., beta per sdGRS = −1.02 in DM compared to −0.68 in noDM using overall-effect weights).
When comparing different weighting schemes, we found no notable improvement in R2 values by using DM-/noDM-specific weights compared to overall weights (Table 3). In the 634-variant GRS, using DM-specific weights even reduced R2 in DM individuals (R² = 3.1% versus 4.0%). This may be attributable to the larger uncertainty in DM-specific weights estimated from the meta-analysis restricted to DM individuals. For the 7-variant GRS, DM-specific weights slightly increased R2 in DM individuals, but not markedly (R2 = 1.0% versus 0.98%). These findings underscore a similar performance of overall weights and DM-/noDM-specific weights when building a GRS for eGFR. The comparison of the 634-variant with the 7-variant GRS suggests that more differential effect loci and higher precision in DM-specific weights might substantially improve the genetically explained variance also in DM individuals.
Discussion
In this GWAS on eGFR in ~180,000 individuals with DM and ~1.3 million individuals without DM, we established seven loci with significant DM/noDM-differential effects on eGFR near UMOD/PDILT, TPPP, MED1/NEUROD2, CSRNP1, DCDC5, NRIP1, and SLC22A2. We also identified 32 novel eGFR-associated loci when allowing for potential difference, including four loci with a suggestive difference near SH3BP4, LOXL4, ALPL, and PIK3CG. The 11 loci with established or suggestive difference included two loci with DM-only effects on eGFR (near CSRNP1, SH3BP4), one with noDM-only effect (near MED1/NEUROD2), and all others showed more pronounced effects in DM compared to noDM. Our GWAS focused on DM individuals confirmed two known DKD loci, but also identified 27 novel loci for eGFR in DM, which make these potential new DKD loci. The DM-/noDM-stratified GRS analyses showed no improvement in explained trait variance when using DM-/noDM-stratified weights, but the seven identified DM/noDM-differential eGFR-effects explained more of the eGFR variance in DM individuals than in noDM individuals.
Our gene prioritization at the 11 loci with DM/noDM-difference and at the 28 other novel eGFR loci highlighted 18 genes: 10 genes mapped to eGFR-association-driving variants that were deleteriously protein-relevant or eQTLs in kidney tissue, which suggests them as potential drug targets to alter eGFR. Further eight genes were prioritized because they were known as human kidney disease monogenes, which made them plausible causal genes for these common variant findings. Particularly interesting was CUBN in a novel eGFR locus. The CUBN locus is the GWAS locus with the strongest effect on UACR39 and microalbuminuria40 and identified here for eGFR at genome-wide significance, but respective lead variants for eGFR and UACR are uncorrelated. The lead variant for eGFR, rs11254238, is highly correlated with a variant that alters the encoded protein, cubilin (rs1801232, r2 = 0.91). We observed a twice as large effect on eGFR in DM compared to noDM in both discovery and replication data, but not statistically significantly different. This is in line with previously observed larger CUBN variants’ effects on microalbuminuria40 and UACR41,48 in DM, which provides further evidence for an interaction of CUBN with DM status on kidney function. In fact, our previous sequencing of the CUBN gene found rare variants in CUBN that were associated with higher UACR and with better eGFR49. Our study now reports such a parallel association between UACR and eGFR now also for a common variant at the genome-wide significance levels. Together, our results provide further support for the importance to assess the physiological role of CUBN by functional studies not only for microalbuminuria, but also for finding pathophysiological explanations related to impaired filtration rate.
The RASSF6 gene has been studied in relation to the kidney as well: RASSF6 mediates apoptosis in various cells50. It is shown to be expressed in the slit diaphragm in glomeruli and the apical membranes in proximal renal tubular epithelial cells of rat kidney. The same animal study suggests the involvement of the RASSF6 pathway in contrast-induced nephropathy. Thus, the novel detection of the RASSF6 locus for eGFR here and the lead variant, rs17804499, being protein-altering and with 86% probability causal render RASSF6 a compelling candidate for the functional follow-up to help understand its role in kidney function that is elusive so far.
Another interesting gene was PGAP3 in the MED1-NEUROD2 locus, which mapped to an expression-modulating variant that was associated with eGFR in noDM, but not in DM individuals. PGAP3 is a known gene for CKD and eGFR: eQTL analyses showed colocalization of the association signal with PGAP3 expression51. PGAP3 knockout mice developed larger glomeruli with deposition of immunoglobulins52, although we acknowledge that this does not provide a mechanistic explanation for its association with eGFR. Eventually, if causal genes at eGFR loci with significant differences by DM status will be advanced as drug targets for reno-protective therapies, the knowledge about DM- or noDM specificity of the eGFR association can help define the target group for potential subsequent therapy, as DM-only, noDM-only, or both.
The difference in SNP effects between DM and noDM can be interpreted as SNP-by-DM-status interaction effect. To our knowledge, our study is the first genome-wide search for SNP-by-DM-status interaction effects for eGFR. Sensitivity analyses indicated that the observed differences were neither biased from the stratified modeling, nor from log-transformed eGFR, and the SNP-by-DM-status interaction was not explained by SNP-interaction with age, sex, hypertension, or body-mass-index53. The seven loci with an established difference are known loci for eGFR16,20. To our knowledge, the significant DM/noDM-differential effects for eGFR or eGFR-based CKD were identified here for the first time, except for the UMOD-PDILT locus21. At UMOD-PDILT, a well-known kidney function locus16,20, the two-fold higher eGFR effect in DM compared to noDM confirmed previous observations for eGFR21,54 and CKD55. Very interesting was the TPPP locus, where the three-fold higher eGFR effect in DM now draws attention to a locus that was just one of the hundreds of small effects eGFR loci. The lead variant for the difference, rs434215, modulates TPPP expression in tubulo-interstitial tissue46, but the role of TPPP in diabetes and kidney disease is yet unknown. Previous studies on TPPP focused on the nervous system: TPPP is highly expressed in the brain and shown to affect neural microtubules56.
Our GWAS on eGFR in DM provides a link to the genetics of DKD. Previous GWAS for DKD analyzed eGFR or eGFR-based CKD in type 1 and/or type 2 DM individuals22,23. Our GWAS in >178,000 DM individuals was 4- to 7-fold larger. These DM individuals were mostly from population-based studies of adult individuals. As such, the proportion of type 1 DM among DM individuals analyzed here reflects the proportion of type 1 DM of 5–10% among adult DM individuals57. Due to the substantially larger number of type 2 DM individuals, the identified DM/noDM-differential effects on eGFR may mostly reflect differences between type 2 DM versus noDM. A better distinction by type of DM would require more granular data focusing on the distinction between these two major diabetes groups and substantially larger data on type 1 DM. While the use of self-reports or one-time measurements of glucose or HbA1c to define DM here is typical also for GWAS on DM19,58, this implies heterogeneity in the DM definition and may include some individuals without clinically manifest DM.
Yet, our 29 loci with genome-wide significant association with eGFR in DM confirmed two of eight previously identified DKD loci22,23 and the 27 other are compelling new candidate loci for further analyses in studies specialized on DKD patients. A large sample size eGFR GWAS with a more heterogeneous spectrum of DM can be a powerful complementary approach to focused searches in DKD patients22,23. Several prioritized genes mapped to these novel eGFR loci in DM: TPPP already mentioned above and SLC6A19, NRIP1, HPS1 as well as HPSE2 with strong monogenic impact on kidney. All these genes reside in loci with more pronounced eGFR-effects in DM compared to noDM, but none of these effects was DM-only.
Our GWAS allowing for DM/noDM-differential effects identified 32 novel eGFR-associated loci compared to previous GWAS16,20. Some of our novel loci might be identified due to increased power by ~20% increased sample size or due to chance by using alternative statistical tests. However, four of the novel loci showed suggestive DM/noDM-difference, suggesting that their identification was facilitated by using tests allowing for differential effects59. The four suggestive difference loci included one locus, near SH3BP4, with eGFR effect only in DM.
In contrast to hundreds of loci found in GWAS for eGFR, the eGFR loci with significant DM/noDM-difference were few. This suggests a largely shared genetics of kidney function between DM and noDM individuals, which has an important implication for drug development: most drug interventions aimed at altering eGFR should thus be effective among persons with and without DM. This is mirroring what is observed for SGLT2 inhibitors—a medication originally developed for individuals with DM that is now also being tested for reno-protection among individuals without DM13. While our GWAS here is, to date, the largest for eGFR in DM individuals, we still might have missed loci with effects in DM-only. Power for interaction effects is generally smaller than for overall effects60, and particularly reduced when one subgroup, like DM, is substantially smaller (~10%) than the other28. Future work with an increased sample size might detect more eGFR loci with differences by DM status, especially those with effects in DM-only. This GWAS included mostly European individuals and our findings require replication in non-European ancestries61, particularly because of the large differences in DM prevalence across ancestries62.
In summary, our results highlight the existence of DM- and noDM-specific genetic effects on kidney function, but emphasize that the majority of eGFR locus associations do not differ between individuals with and without DM. Larger DM-/noDM-stratified data on eGFR in the future will improve the detectability of differential effect loci and the precision of DM-specific weights. This might also improve DM-/noDM-stratified GRS prediction of eGFR. The identified eGFR loci with difference between DM- and noDM individuals include loci with effects only in DM as well as loci with effects in noDM. This has highly relevant implications, if the respective genes are advanced as a drug target: the specificity of the association might help sharpen the target group for such potentially arising drug therapies.
Methods
Definition of the outcome eGFR and study-specific participant information
GFR was estimated in all study participants in all studies using serum creatinine measurements via the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula20. Study-specific information on the utilized assay and year of measurement is given in Supplementary Data 1. We used the R package nephro63, winsorized at 15 and 200 ml min–1per 1.73 m2, and logarithmized using a natural logarithm. For a better interpretation of effect size, we there used eGFR on the original scale in GRS analyses. Study-specific information on study design, sample size, sex and age, utilized serum creatinine assay, year of measurement, and average eGFR is given in Supplementary Data 1.
Each study is conducted according to the declaration of Helsinki; local ethics committees approved research protocols and participants provided written informed consent.
Definition of DM status
DM of each study participant at the time point of the serum creatinine measurement was defined either (i) as fasting plasma glucose ≥126 mg/dl (7.0 mmol/L) or diabetes therapy, or (ii) (fasting glucose unavailable) as non-fasting plasma glucose ≥200 mg/dl (11.0 mmol/L) or diabetes therapy, or (iii) (glucose unavailable) as self-reported diabetes. For UKB, DM was defined as HbA1c≥48 mmol/mol (≥6.5%) or diabetes therapy (i.e., A10 ATC codes obtained from64).
Study-specific GWAS analyses stratified by DM status
We distributed an analysis plan to the 72 participating studies of the CKDGen consortium. Each study conducted analyses separately for individuals with DM and individuals without DM. All studies imputed genotypes to the Haplotype Reference Consortium v1.1 (HRC) or 1000 Genomes Project phase 3 v5 (1000Gp3v5) ALL or phase 1 v3 (1000Gp1v3) ALL panel. Each study conducted linear regression GWAS on log(eGFR) using natural logarithm, an additive genotype model as well as adjusted for sex, age, and other study-specific covariates. Details on study-specific genotyping, imputation, and GWAS were described previously for CKDGen20. For UKB, we utilized the fastGWA tool65 to conduct GWAS for log(eGFR) based on linear mixed models while accounting for sex, age, age × sex, age2, age2 × sex, and 20 genetic principal components and assuming an additive genetic model, which allowed to include related individuals in the GWAS65. For stage 2 studies, MVP, MGI, and HUNT, the same analysis plan was distributed. Details on genotyping, imputation, and GWAS in UKB as well as for stage 2 studies are shown in Supplementary Data 1. For quality control, we excluded variants with low imputation quality, Info < 0.6, or rare variants with minor allele frequency, MAF < 0.1%. We utilized the software packages GWAtoolbox66 and EasyQC67 for the quality control of study-specific GWAS results. We conducted a correction for genomic control lambda of the results stratified by diabetes status.
DM/noDM-stratified GWAS meta-analyses
In stage 1 of our analysis, separately in DM (nDM = 88,829) and noDM (nnoDM = 620,665) strata, we conducted fixed-effect inverse-variance weighted meta-analyses of 72 GWAS of log(eGFR) using metal68, and then meta-analyzed these results with DM/noDM-stratified GWAS from UKB (nDM = 21,040; nnoDM = 414,628; European only). To adjust for population stratification within studies, we applied genomic control (GC) correction69 to each study prior to the meta-analysis. We applied a second GC correction to the DM- and noDM stage 1 meta-analysis results (GC lambda = 1.02 and 1.20, respectively). We excluded variants that were present only in ≤36 stage 1 studies (≤50%) and variants with a cumulative minor allele count of <400 in the stage 1 meta-analyses. In summary, 109,869 individuals with DM and 1,035,190 with noDM were included in stage 1. We followed variants identified at stage 1 in independent stage 2 meta-analyses. For stage 2, we included DM/noDM-stratified GWAS on log(eGFR) from MVP (nDM = 57,430, nnoDM = 122,966, hospital-based), MGI (nDM = 7469, nnoDM = 36,558, hospital-based) and HUNT (nDM = 3799, nnoDM = 65,590, population-based), totalling 68,698 individuals with DM and 225,114 with noDM, all of European-ancestry. Again, we applied study-specific GC correction prior to the meta-analysis and a second GC correction69 to the stage 2 meta-analysis results (GC lambdas = 1.00 and 1.02 for DM- and noDM, respectively). To maximize power for locus identification, we combined the double GC-corrected stage 1 and 2 meta-analysis separately by DM status via fixed-effect inverse-variance weighted meta-analyses of the two sources using metal68. The GC lambda in this final meta-analysis was comparable to previous GWAS16,70 (GC lambdas = 1.03 and 1.15, respectively). The DM-/noDM-specific summary statistics (stratified GWAS) for each variant genome-wide served to investigate potential DM-/noDM-differential genetic effects on log(eGFR) without making any assumptions on the DM association with any other covariate71. These DM-/noDM-specific summary statistics on genetic variants associated with log(eGFR) enabled the implementation of all the following statistical tests to search for DM/noDM-difference loci or novel loci allowing for difference as described below.
Approaches to search for DM/noDM-difference in genetic effects on eGFR
We used the meta-analyzed SNP-specific summary statistics, to test for difference in eGFR-effects between DM and noDM. For this, we applied a difference test for each variant using
where \({\hat{\beta }}_{{{{{\mathrm{DM}}}}}}\) and \({\hat{\beta }}_{{{{{\mathrm{noDM}}}}}}\) are the genetic effect estimates for eGFR in DM or noDM from the stratified GWAS meta-analysis, respectively, and corresponding standard errors \({{se}}_{{{{{\mathrm{DM}}}}}}\) and \({{se}}_{{{{{\mathrm{noDM}}}}}}\). The term \({r}_{{{{{\mathrm{diab}}}}}}\) reflects the correlation between \({\hat{\beta }}_{{{{{\mathrm{DM}}}}}}\) and \({\hat{\beta }}_{{{{{\mathrm{noDM}}}}}}\) across all variants (Spearman correlation coefficient, \({r}_{{{{{\mathrm{diab}}}}}}=0.14\)).
To search genome-wide for eGFR loci with DM-/noDM-differential effects, we applied two approaches24,37: (i) a genome-wide difference test (PDiff < 5 × 10−8, difference test approach), and (ii) a search for genome-wide significant association with overall eGFR followed by a difference test in the same data (POverall < 5 × 10−8 and PDiff < 0.05/k, k = number of followed SNPs, overall+difference test approach). The two approaches complement each other in terms of power to detect difference loci that depend on the magnitude and DM-/noDM specificity24,37. We implemented the approaches in two designs: first, we applied a discovery+replication design, where we searched for significant differences in the stage 1 meta-analysis and moved the selected SNPs to a replication stage using the stage 2 meta-analysis (applying a Bonferroni-corrected alpha-level accounting for the SNPs tested). Second, to make full use of stage 1 and stage 2 data, we searched for differences in stage 1 + 2 meta-analysis combined (combined stage design). We utilized the R package EasyStrata72 to apply the difference test approaches to the DM/noDM-stratified meta-analysis results.
Approaches to search for eGFR loci allowing for DM/noDM-differences
By allowing for differences between DM and noDM in SNP-effects on eGFR, one can possibly detect novel loci that have been masked in analyses on overall eGFR (without DM-status stratification). Again, we used the meta-analyzed SNP-specific summary statistics to apply two approaches to search for novel eGFR loci allowing for difference: (i) a screen using a two degrees of freedom joint test (Pjoint < 5 × 10−8, joint-test approach)38 derived from a χ2-test using38
where \({\hat{\beta }}_{{{{{\mathrm{DM}}}}}}\) and \({\hat{\beta }}_{{{{{\mathrm{noDM}}}}}}\) are the genetic effect estimates for eGFR in DM or noDM from the stratified GWAS meta-analysis, respectively, and corresponding standard errors \({{se}}_{{{{{\mathrm{DM}}}}}}\) and \({{se}}_{{{{{\mathrm{noDM}}}}}}\); and (ii) a on eGFR association in individuals with or without DM separately (stratified tests approach, PDM < 5 × 10−8 or PnoDM < 5 × 10−8). Analogously to the search for difference loci, we applied two-stage designs, the discovery+replication, and the combined stage designs. We utilized the R package EasyStrata72 to apply the joint and stratified test approaches to the DM/noDM-stratified meta-analysis results.
Variant selection and region definition
In order to derive non-overlapping locus regions and locus lead variants, we clumped genome-wide significant variants from the respective test results (PDiff < 5 × 10−8, PJoint < 5 × 10−8, PDM < 5 × 10−8, or PnoDM < 5 × 10−8) as done previously16: the most significant variant was selected genome-wide (first locus lead variant) and the corresponding locus was defined as the smallest physical interval on the corresponding chromosome containing this variant such that there were no genome-wide significant variants within 500 kb outside the two borders. Omitting the identified locus, we repeated the procedure until no further genome-wide significant variants were detected. By this, a locus region is defined by adding ±250 kb to the first and last genome-wide significant variant of an identified locus. This procedure also ensured non-overlapping loci. A locus was considered to be known for eGFR from previous GWAS, if it overlapped with one of the 427 known eGFR loci (424 from ref. 16, 3 additional from ref. 20). If a locus is not known, we call it a novel eGFR locus. For the identified variants, we assessed between-study heterogeneity based on the CKDGen meta-analysis using a Chi-Squared test and an I2 statistic73 and verified the association statistics with regards to abnormal or unusually large effect sizes.
GCTA analyses to identify independent secondary signals within loci
To evaluate whether there were multiple independent signals within locus, we conducted approximate conditional analyses with GCTA74 for each identified locus. These analyses were based on European-ancestry individuals, since appropriate trans-ethnic linkage disequilibrium (LD) reference panels were limited. As LD reference panel, we used a random subset of 20,000 unrelated individuals of European-ancestry from UKB, as done previously16. To obtain independent signals for associations derived by the difference test, the joint test, or DM- or noDM-specific association tests, we applied a stepwise approach: we conditioned via GCTA analysis on the, respectively, observed lead variant in DM and noDM separately and, second, derived DM/noDM-specific conditioned results for the locus. Separately for DM and noDM, we ensured whether any of the conditioned DM- or noDM-specific associations in the locus showed genome-wide significant association at PDM_Cond < 5 × 10−8 or PnoDM_Cond < 5 × 10−8. For loci derived by the difference test or the joint test, we applied the difference test or joint test, respectively, to the conditioned DM/noDM estimates to infer whether additional signals showed significant differences or joint effects (PDiff_Cond < 5 × 10−8 or PJoint_Cond < 5 × 10−8).
Prioritization of variants and genes
For each variant within each identified signal, we derived the variants that were the most likely to drive the association. For this, we calculated approximate Bayes factor and posterior probabilities of association (PPA) based on Z-scores using the Kichaev method75. We calculated PPAs based on unconditioned or conditioned summary statistics depending on whether the locus showed only one or multiple independent signals, respectively. Then, we obtained 99% credible sets of variants by sorting the variants within each signal by descending PPA and then summing up PPAs until a cumulative PPA of 99% was reached. To prioritize genes, we used the results from the Gene PrioritiSation (GPS) published previously16 for known eGFR loci and generated the GPS de novo for novel eGFR loci accordingly: we queried each gene underneath identified loci and prioritized the gene when it mapped to a 99% credible set variant that was protein-relevant with high predicted deleteriousness (CADD45 PHRED-Score ≥ 15) or expression-relevant in kidney tissue (eQTL, NEPTUNE46, or GTEx v776, false-discovery rate, FDR < 5% for the eQTL) and for splice quantitative trait loci in kidney tissue (sQTL)76 (FDR < 5% for the sQTL). We also prioritized genes that were known as human kidney disease monogene: for this, we queried each gene at identified loci for a documented kidney phenotype in human as done previously16 (Online Mendelian Inheritance in Man® database, OMIM77 or Groopman et al.78) with additional manual curation by expert review to focus on kidney disease monogenes.
GRS analyses
GRS analyses based on the previously identified 634 eGFR variants16 and based on the variants identified for DM-/noDM-difference on eGFR were conducted in unrelated European-ancestry individuals from HUNT (nDM = 3023, nnoDM = 25,484, stage 2 study). For each individual, the GRS was computed as the weighted sum of eGFR-lowering alleles across the 634 variants applying three different weighting schemes: (i) weighted by the respective overall per-variant effect as derived previously (i.e., estimated in CKDGen and UK Biobank, our stage 1 data16) (ii) weighted by the variant’s DM-/noDM-specific effects as derived here in stage 1 depending on whether the individual in HUNT had DM or noDM, respectively (and vice versa), (iii) weighted by the variant’s DM-/noDM-specific effect from stage 1 for HUNT individuals with DM/noDM, respectively, when the variant was among the seven with an identified significant difference, and weighted by the overall-effect size otherwise. By this, the GRS association analyses conducted in a stage 2 study were independent of the variant identification and weight estimation, which were based on stage 1 studies. Separately for individuals with and without DM, we estimated the association of the GRS on eGFR by linear regression (original scale; adjusted by sex, age, and genetic principal components). We judged the GRS effect per standard deviation of the GRS and the eGFR variance explained by the GRS separately in DM and noDM separately.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Summary genetic association results for the DM-status-specific meta-analyses for log(eGFRcrea) can be downloaded from https://ckdgen.imbi.uni-freiburg.de/. All other data are available from the corresponding author on reasonable request.
Code availability
The analysis plan can be downloaded from https://ckdgen.eurac.edu/mediawiki/index.php/CKDGen_Round_4_EPACTS_analysis_plan and the phenotype command line script from https://github.com/genepi-freiburg/ckdgen-pheno. All other code is available from the corresponding author on reasonable request.
References
Chapter 1: Definition and classification of CKD. Kidney Int. Suppl. https://doi.org/10.1038/kisup.2012.64 (2013).
El Nahas, A. M. & Bello, A. K. Chronic kidney disease: the global challenge. Lancet 365, 331–340 (2005).
Bikbov, B. et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 395, 709–733 (2020).
Levey, A. S. et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 150, 604–612 (2009).
Baumeister, S. E. et al. Effect of chronic kidney disease and comorbid conditions on health care costs: a 10-year observational study in a general population. Am. J. Nephrol. 31, 222–229 (2010).
Pippias, M. et al. Renal replacement therapy in Europe: a summary of the 2012 ERA-EDTA Registry Annual Report. Clin. Kidney J. 8, 248 (2015).
Parving, H.-H. et al. Cardiorenal end points in a trial of Aliskiren for type 2 diabetes. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa1208799 (2012).
Haller, H. et al. Olmesartan for the delay or prevention of microalbuminuria in type 2 diabetes. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa1007994 (2011).
Mann, J. F. et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 372, 547–553 (2008).
Group, T. A. to C. C. R. D. S. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 358, 2545–2559 (2008).
Neuen, B. L. et al. SGLT2 inhibitors for the prevention of kidney failure in patients with type 2 diabetes: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. 7, 845–854 (2019).
Hocher, B. & Tsuprykov, O. Renoprotective effects of GLP1R agonists and SGLT2 inhibitors. Nat. Rev. Nephrol. 2017 1312 13, 728–730 (2017).
Heerspink, H. J. L. et al. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2024816 (2020).
Tin, A. et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 51, 1459–1474 (2019).
Hellwege, J. N. Mapping eGFR loci to the renal transcriptome and phenome in the VA Million Veteran Program. Nat. Commun. https://doi.org/10.1038/s41467-019-11704-w (2019).
Stanzick, K. J. et al. Discovery and prioritization of variants and genes for kidney function in >1.2 million individuals. Nat. Commun. 12, 1–17 (2021).
Nelson, M. R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856 (2015).
King, E. A., Wade Davis, J. & Degner, J. F. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 15, e1008489 (2019).
Mahajan, A. et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 50, 1505–1513 (2018).
Wuttke, M. et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet. 51, 957–972 (2019).
Pattaro, C. et al. Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat. Commun. 7, 1–19 (2016).
van Zuydam, N. R. et al. A genome-wide association study of diabetic kidney disease in subjects with type 2 diabetes. Diabetes 67, 1414–1427 (2018).
Salem, R. M. et al. Genome-Wide association study of diabetic kidney disease highlights biology involved in glomerular basement membrane collagen. J. Am. Soc. Nephrol. 30, 2000–2016 (2019).
Winkler, T. W. et al. Approaches to detect genetic effects that differ between two strata in genome-wide meta-analyses: Recommendations based on a systematic evaluation. PLoS ONE 12, e0181038 (2017).
Bentley, A. R. et al. Multi-ancestry genome-wide gene–smoking interaction study of 387,272 individuals identifies new loci associated with serum lipids. Nat. Genet. 51, 636–648 (2019).
Winkler, T. W. et al. The influence of age and sex on genetic associations with adult body size and shape: a large-scale genome-wide interaction study. PLoS Genet. 11, e1005378 (2015).
Kilpeläinen, T. O. et al. Multi-ancestry study of blood lipid levels identifies four loci interacting with physical activity. Nat. Commun. 10, 376 (2019).
Justice, A. E. et al. Genome-wide meta-analysis of 241,258 adults accounting for smoking behaviour identifies novel loci for obesity traits. Nat. Commun. 8, 1–19 (2017).
Sung, Y. J. et al. A large-scale multi-ancestry genome-wide study accounting for smoking behavior identifies multiple significant loci for blood pressure. Am. J. Hum. Genet. 102, 375–400 (2018).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Köttgen, A. & Pattaro, C. The CKDGen Consortium: ten years of insights into the genetic basis of kidney function. Kidney Int. https://doi.org/10.1016/j.kint.2019.10.027 (2020).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Gaziano, J. M. et al. Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 70, 214–223 (2016).
Zawistowski M. et al. The Michigan Genomics Initiative: a biobank linking genotypes and electronic clinical records in Michigan Medicine patients. medRxiv. https://doi.org/10.1101/2021.12.15.21267864.
Brumpton, B. M. et al. The HUNT Study: a population-based cohort for genetic research. medRxiv https://doi.org/10.1101/2021.12.23.21268305. (2021).
Skol, A. D., Scott, L. J., Abecasis, G. R. & Boehnke, M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet. 38, 209–213 (2006).
Kooperberg, C. & LeBlanc, M. Increasing the power of identifying gene × gene interactions in genome-wide association studies. Genet. Epidemiol. 32, 255–263 (2008).
Aschard, H., Hancock, D. B., London, S. J. & Kraft, P. Genome-wide meta-analysis of joint tests for genetic and gene-environment interaction effects. Hum. Hered. 70, 292–300 (2011).
Teumer, A. et al. Genome-wide association meta-analyses and fine-mapping elucidate pathways influencing albuminuria. Nat. Commun. 10, 1–19 (2019).
Böger, C. A. et al. CUBN is a gene locus for albuminuria. J. Am. Soc. Nephrol. 22, 555–570 (2011).
Teumer, A. et al. Genome-wide association studies identify genetic loci associated with albuminuria in Diabetes. Diabetes 65, 803–817 (2016).
Chiou, J. et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 594, 398–402 (2021).
Chen, J. et al. The trans-ancestral genomic architecture of glycemic traits. Nat. Genet. 53, 840–860 (2021).
Mountjoy, E. et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat. Genet. https://doi.org/10.1038/s41588-021-00945-5 (2021).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. https://doi.org/10.1093/nar/gky1016 (2019).
Gillies, C. E. et al. An eQTL landscape of kidney tissue in human nephrotic syndrome. Am. J. Hum. Genet. https://doi.org/10.1016/j.ajhg.2018.07.004 (2018)
GTEx consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science https://doi.org/10.1126/science.aaz1776 (2020).
Ahluwalia, T. S. et al. A novel rare CUBN variant and three additional genes identified in Europeans with and without diabetes: results from an exome-wide association study of albuminuria. Diabetologia 62, 292–305 (2019).
Bedin, M. et al. Human C-terminal CUBN variants associate with chronic proteinuria and normal renal function. J. Clin. Invest. 130, 335–344 (2020).
Withanage, K. et al. Expression of RASSF6 in kidney and the implication of RASSF6 and the Hippo pathway in the sorbitol-induced apoptosis in renal proximal tubular epithelial cells. J BioChem. https://doi.org/10.1093/jb/mvs056 (2012).
Ko, Y. A. et al. Genetic-variation-driven gene-expression changes highlight genes with important functions for kidney disease. Am. J. Hum. Genet. 100, 940–953 (2017).
Wang, Y. et al. Significance of glycosylphosphatidylinositol-anchored protein enrichment in lipid rafts for the control of autoimmunity. J. Biol. Chem. https://doi.org/10.1074/jbc.M113.492611 (2013).
Keller, M. C. Gene × environment interaction studies have not properly controlled for potential confounders: the problem and the (simple) solution. Biol. Psychiatry 75, 18–24 (2014).
Köttgen, A. et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat. Genet. 41, 712–717 (2009).
Vujkovic, M. et al. Discovery of 318 novel loci for type-2 diabetes and related micro- and macrovascular outcomes among 1.4 million participants in a multi-ethnic meta-analysis. medRxiv https://doi.org/10.1101/19012690 (2019).
Hlavanda, E. et al. Brain-specific p25 protein binds to tubulin and microtubules and induces aberrant microtubule assemblies at substoichiometric concentrations†. Biochemistry 41, 8657–8664 (2002).
CDC. National Diabetes Statistics Report 2020. Estimates of diabetes and its burden in the United States. (CDC, 2020).
Xue, A. et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 9, 2941 (2018).
Aschard, H., Spiegelman, D., Laville, V., Kraft, P. & Wang, M. A test for gene–environment interaction in the presence of measurement error in the environmental variable. Genet. Epidemiol. 42, 250–264 (2018).
Randall, J. C. et al. Sex-stratified genome-wide association studies including 270,000 individuals show sexual dimorphism in genetic loci for anthropometric traits. PLoS Genet. 9, e1003500 (2013).
Hindorff, L. A. et al. Prioritizing diversity in human genomics research. Nat. Rev. Genet. https://doi.org/10.1038/nrg.2017.89 (2018).
Cheng, Y. J. et al. Prevalence of diabetes by race and ethnicity in the United States, 2011-2016. JAMA 322, 2389–2398 (2019).
Pattaro, C. et al. Estimating the glomerular filtration rate in the general population using different equations: effects on classification and association. Nephron Clin. Pract. 123, 102–111 (2013).
Wu, Y. et al. Genome-wide association study of medication-use and associated disease in the UK Biobank. Nat. Commun. https://doi.org/10.1038/s41467-019-09572-5 (2019).
Jiang, L. et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nat. Genet. https://doi.org/10.1038/s41588-019-0530-8 (2019).
Fuchsberger, C. et al. GWAtoolbox: an R package for fast quality control and handling of genome-wide association studies meta-analysis data. Bioinformatics 28, 444–445 (2012).
Winkler, T. W. et al. Quality control and conduct of genome-wide association meta-analyses. Nat. Protoc. 9, 1192–1212 (2014).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Devlin, A. B., Roeder, K. & Devlin, B. Genomic control for association. Biometrics. 55, 997–1004 (2013).
Pulit, S. L. et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. 28, 166–174 (2019).
Sung, Y. J. et al. An empirical comparison of joint and stratified frameworks for studying G × E interactions: systolic blood pressure and smoking in the CHARGE gene-lifestyle interactions working group. Genet. Epidemiol. 40, 404–415 (2016).
Winkler, T. W. et al. EasyStrata: evaluation and visualization of stratified genome-wide association meta-Analysis data. Bioinformatics 31, 259–261 (2015).
Huedo-Medina, T. B., Sánchez-Meca, J., Marín-Martínez, F. & Botella, J. Assessing heterogeneity in meta-analysis: Q statistic or I 2 Index? Psychol. Methods 11, 193–206 (2006).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012).
Kichaev, G. et al. Integrating functional data to prioritize causal variants in statistical fine-mapping sStudies. PLoS Genet. 10, e1004722 (2014).
Aguet, F. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Hamosh, A., Scott, A. F., Amberger, J., Valle, D. & McKusick, V. A. Online Mendelian inheritance in man (OMIM). Hum. Mutat. 15, 57–61 (2000).
Groopman, E. E. et al. Diagnostic Utility of exome sequencing for kidney disease. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa1806891 (2019).
Acknowledgements
The Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) supported the meta-analysis—Project-ID 387509280—SFB1350 (Subproject C6 to I.M.H.). A.M.H., B.R., and R.T. were supported by VACSR&D MVP grant CX001897. This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration, and was supported by VACSR&D MVP grant CX001897 (A.M.H.). This publication does not represent the views of the Department of Veteran Affairs or the United States Government. We conducted this research using the UK Biobank resource under the application number 20272. We thank Paola Bilani for collecting author information. Extended acknowledgements are provided in Supplementary Note 4 for all studies, in Supplementary Note 5 for MVP and in Supplementary Note 6 for LifeLines.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Consortia
Contributions
T.W., H.R., A.Teu., M.Go., B.R., K.Stan., R.T., U.Th., C.A.B., A.Hu., F.K., A.Köt., C.P., and I.M.H. wrote the manuscript. T.W., H.R., A.Teu., M.Go., C.A.B., A.Köt., C.P., and I.M.H. designed the study. B.T., D.Cu., K.B.S., M.Sch., Q.Y., A.B.Z., A.Ca., A.Co., A.d.G., A.Fer., A.J.O., A.Kör., A.Ma., A.Me., A.P.J.d.V., A.Pa., A.Pe., A.R.R., A.Tö., B.B., B.Br., B.I.F., B.J., B.K.K., B.M.P., B.P., B.Sch., B.Sp., B.W.J.H.P., C.G., C.H., C.K., C.M.K., C.N.S., C.R., C.Sa., C.W., Ch.F., D.F.R., D.I.C., D.M., D.T., E.B., E.S.T., F.Gi., G.E., G.G., G.N.N., G.S., G.We., H.B., H.G., H.H., H.Kr., H.L., H.M.S., H.Sc., I.M.N., I.R., I.T., J.Ä., J.B.J., J.C.C., J.Co., J.F.F., J.F.W., J.G., J.Ha., J.J.M.L., J.O., J.P.C., J.S.K., J.Th., J.X., K.Ch., K.D., K.Ec., K.Ho., K.Hv., K.L.M., K.Ste., K.Str., L.J.L., L.L., L.M.Y.A., L.Wal., M.A.I., M.A.L., M.A.P., M.Boc., M.C.i., M.E.K., M.Gö., M.H.d.B., M.K.E., M.Käh., M.Kub., M.Lo., M.N., M.O.M., M.Per., M.Y., N.G.M., N.Pi., N.S.J., N.V., O.D., O.M., O.P., O.T.R., P.E., P.G., P.H., P.K., P.K.E.M., P.M.R., P.Ma., P.O.S., P.P.P., P.R., P.Vo., R.d.M., R.Do., R.J.C., R.J.F.L., R.S., S.C.L., S.F.W., S.Ha., S.J.L.B., S.L., S.Va., T.B., T.E., T.L., T.S.A., T.Wo., U.V., V.Gi., V.Gu., V.H.X.F., V.S., V.W.V.J., v.D.R.M., W.B.W., W.Ki., W.Ko., W.L., W.Mä., W.Zh., X.S., Y.Fri., Y.M., Y.X.W., K.Star., U.Th., C.A.B., A.Hu., F.K., A.Köt., and C.P. managed an individual contributing study. T.W., H.R., A.Teu., M.Go., B.R., K.Stan., L.T., A.Ch., A.Ti., B.T., C.H.L.T., J.B.W., J.F.C., K.Hor., M.Co., M.Li., M.Sch., M.Wu., P.J.v.d.M., Q.Y., S.Gh., T.N., Y.L., F.Gue., A.D., A.P.J.d.V., A.P.M., A.V.S., B.I.F., B.J., B.N., B.V., C.D.L., C.L., C.M.L., C.N.S., C.Sc., C.Y.C., Ch.F., Ch.Wa., D.Cz., D.F.G., D.I.C., D.O.M.K., E.H., E.S., F.Riv., F.Riz., G.D., G.G., G.S., H.D.W., H.K.i., H.Sn., I.M.N., J.Ä., J.B.W., J.D., J.F.F., J.J., J.Ku., J.L., J.Marte., J.Marti., J.Tr., K.A.R., K.M.R., K.Mi., L.A.L., L.L., L.Ly., L.M.R., L.Wan., M.A., M.A.N., M.Boe., M.E.K., M.F., M.F.F., M.Ga., M.H.P., M.K.W., M.Kan., M.Kuo., M.L.Big., M.L.O., M.P.C., M.Stu., M.LiC., N.M., P.H., P.K.J., P.P.M., R.J.C., R.Ru., R.T., R.T.G., R.Z.H.S., S.A.P., S.C.F., S.D.G., S.Gr., S.Pat., S.Se., S.Va., S.Vo., Sh.J.H., T.B., T.C., T.H., T.S.A., Th.S.B., V.V., W.Mä., W.Zh., X.S., Y.Ch.T., Y.K., Y.M., Y.O., K.Star., C.A.B., A.Köt., C.P., and I.M.H. performed statistical methods and analysis. T.W., H.R., M.Go., B.R., K.Stan., L.T., A.Ch., A.Ho., J.B.W., J.F.C., K.Hor., M.Co., M.Li., M.Sch., M.Wu., P.J.v.d.M., S.Gh., T.N., Y.L., G.P., A.D., A.I.P., A.P.M., A.V.S., B.V., C.A.H., C.D.L., C.N.S., C.Sc., C.Y.C., Ch.Wa., D.Cz., E.C., E.H., E.S., E.W.A., F.Riz., G.E., G.Pi., H.C., H.G., H.Ki., J.B.W., J.D., J.J., J.L., J.Marte., J.Marti., J.Tr., K.L.M., L.C., L.Ly., M.E.K., M.F., M.H.P., M.P.C., M.Stu., M.Li.C., M.Ling.C., N.S.J., P.H., P.P.M., R.M., R.Ru., R.T., R.T.G., S.Ber., S.Gr., S.Pat., S.Se., T.C., T.S.A., V.H.X.F., W.Mä., Zh.Y., C.A.B., and I.M.H. performed bioinformatics. T.W., H.R., A.Teu., M.Go., B.R., K.Stan., J.F.C., K.Hor., M.Sch., M.Wu., S.Gh., Y.L., A.Ma., A.Tö., B.A., B.I.F., B.J., B.Sch., C.D.L., C.G., C.L., C.M.L., C.N.S., C.Y.C., D.F.R., D.I.C., F.Guo., H.B., H.H., H.Ki., I.T., J.Ä., J.B.W., J.D., J.F.F., J.Ha., J.Marte., J.Tr., K.D., K.En., K.L.M., K.Mi., L.L., M.F.F., M.Ga., M.Stu., N.D.P., N.V., N.Y.Q.T., P.H., P.v.d.H., R.T., R.T.G., R.Z.H.S., S.Se., S.Va., S.Vo., Sh.J.H., T.B., T.S.A., V.V., W.H., W.Ko., X.G., K.Star., U.Th., C.A.B., A.Hu., A.Köt., C.P., and I.M.H. interpreted results. A.Teu., A.A.H., A.B.Z., A.Ca., A.Co., A.d.G., A.Fra., A.Kör., A.Mo., B.H.S., B.W.J.H.P., C.H., C.N.S., C.W., Ch.Wa., D.F.R., D.I.C., D.J.P., D.T., E.B., E.S., E.W.A., F.Gi., F.Guo., F.Riv., F.Riz., G.E., H.B., H.C., H.G., J.Ä., J.B.W., J.C.C., J.C.M., J.I.R., J.P.C., J.S.K., J.Tr., K.D.T., K.Hv., K.L.M., L.A.L., L.C., L.L., L.Ly., L.Wan., M.L.B., M.A.N., M.Boe., M.E.K., M.F.F., M.H.P., M.K.E., M.K.W., M.Käh., M.Kub., M.La., M.N., M.O.M., M.Per., M.Wa., M.Ling.C., N.D.P., N.M., N.Pi., N.S.J., N.V., O.M., O.T.R., P.H., P.K.E.M., P.M.R., P.O.S., P.S., P.v.d.H., R.B., R.J.C., R.N., R.Z.H.S., S.Pad., S.Va., T.J.R., T.L., T.M., U.V., V.H.X.F., V.S., v.D.R.M., W.H., W.Ko., W.Mä., Y.Fri., Y.X.W., K.Star., C.A.B., A.Hu., and F.K. performed genotyping. D.Cu., A.B.Z., A.Ca., A.Co., A.d.G., A.Ma., A.Me., A.R.R., A.Ter., A.Tö., B.H.S., B.I.F., B.J., B.P., B.W.J.H.P., C.L., C.M.K., C.M.S., C.Sa., Ch.K.H., D.J.P., D.M., D.M.W., D.R., E.P.B., E.S.T., G.B., G.W.M., G.We., H.B., H.H., I.O., I.R., I.T., J.B.J., J.C.C., J.Co., J.Ha., J.J.M.L., J.P.C., J.S.K., K.Ch., K.D., K.Ec., K.Ho., K.Hv., K.Ma., K.N., L.J.L., L.K., M.A.P., M.Ci., M.Gö., M.I., M.K.E., M.Käh., M.Kas., M.Kub., M.Per., N.H.K., N.Pi., O.P., O.T.R., P.H., P.M.R., P.Ma., P.O.S., P.P.P., P.R., P.S., P.Vo., R.N., R.S., S.C.L., S.F.W., S.Pad., S.Vo., T.B., T.J.R., T.L., T.Wo., V.Gu., V.S., V.W.V.J., v.D.R.M., W.H., W.Mä., K.Star., U.Th., C.A.B., A.Köt., and C.P. recruited subjecs. T.W., H.R., A.Teu., M.Go., B.R., K.Stan., L.T., A.Ch., A.Ho., A.Ti., B.T., C.H.L.T., D.Cu., J.B.W., J.F.C., K.B.S., K.Hor., M.Co., M.Li., M.Sch., M.Wu., P.J.v.d.M., Q.Y., S.Gh., T.N., Y.L., G.P., F.Gue., A.A.H., A.B.Z., A.Ca., A.Co., A.D., A.d.G., A.Fer., A.Fra., A.I.P., A.J.O., A.Kör., A.Ma., A.Me., A.Mo., A.P.J.d.V., A.P.M., A.Pa., A.Pe., A.R.R., A.Ter., A.Tö., A.V.S., B.A., B.B., B.Br., B.H.S., B.I.F., B.J., B.K.K., B.M.P., B.N., B.P., B.Sch., B.Sp., B.V., B.W.J.H.P., C.A.H., C.D.L., C.G., C.H., C.K., C.L., C.M.K., C.M.L., C.M.S., C.N.S., C.R., C.Sa., C.Sc., C.W., C.Y.C., Ch.F., Ch.K.H., Ch.Wa., D.Cz., D.F.G., D.F.R., D.I.C., D.J.P., D.M., D.M.W., D.O.M.K., D.R., D.T., E.B., E.C., E.H., E.P.B., E.S., E.S.T., E.W.A., F.Gi., F.Guo., F.Riv., F.Riz., G.B., G.D., G.E., G.G., G.N.N., G.Pi., G.S., G.W.M., G.We., H.B., H.C., H.D.W., H.G., H.H., H.Ki., H.Kr., H.L., H.M.S., H.Sc., H.Sn., I.M.N., I.O., I.R., I.T., J.Ä., J.B.J., J.B.W., J.C.C., J.C.M., J.Co., J.D., J.F.F., J.F.W., J.G., J.Ha., J.I.R., J.J., J.J.M.L., J.Ku., J.L., J.Marte., J.Marti., J.O., J.P.C., J.S.K., J.Th., J.Tr., J.X., K.A.R., K.Ch., K.D., K.D.T., K.Ec., K.En., K.Ho., K.Hv., K.L.M., K.M.R., K.Ma., K.Mi., K.N., K.Ste., K.Str., L.A.L., L.C., L.J.L., L.K., L.L., L.L.y., L.M.R., L.M.Y.A., L.P., L.Wal., L.Wan., M.L.B., M.A., M.A.I., M.A.L., M.A.N., M.A.P., M.Boc., M.Boe., M.Ci., M.E.K., M.F., M.F.F., M.Ga., M.Gö., M.H.d.B., M.H.P., M.I., M.K., M.K.E., M.K.W., M.Käh., M.Kan., M.Kas., M.Kub., M.Kuo., M.L.Big., M.L.O., M.La., M.Lo., M.N., M.O.M., M.P.C., M.Per., M.Pir., M.Stu., M.Wa., M.Y., M.LiC., M.Ling.C., N.B., N.D.P., N.G.M., N.H.K., N.M., N.Pi., N.S.J., N.Sch., N.V., N.Y.Q.T., O.D., O.M., O.P., O.T.R., P.E., P.G, P.H., P.K., P.K.E.M., P.K.J., P.M.R., P.Ma., P.O.S., P.P.M., P.P.P., P.R., P.S., P.v.d.H., P.Vo., R.B., R.d.M., R.Do., R.J.C., R.J.F.L., R.M., R.N., R.Ru., R.S., R.T., R.T.G., R.Z.H.S., S.A.P., S.Ber., S.C.F., S.C.L., S.D.G., S.F.W., S.Gr., S.Ha., S.J.L.B., S.L., S.Pad., S.Pat., S.Se., S.Va., S.Vo., Sh.J.H., Sh.M.K., T.B., T.C., T.E., T.H., T.J.R., T.L., T.M., T.S.A., T.Wo., Th.S.B., U.V., V.Gi., V.Gu., V.H.X.F., V.S., V.V., V.W.V.J., v.D.R.M., W.B.W., W.H., W.Ki., W.Ko., W.L., W.Mä., W.Zh., X.G., X.S., Y.Ch.T., Y.Fri., Y.K., Y.M., Y.O., Y.X.W., Zh.Y., K.Star., U.Th., C.A.B., A.Hu., F.K., A.Köt., C.P., and I.M.H. critically reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
GlaxoSmithKline and Merck & Co employed A.Y.C. Janssen Pharmaceuticals and GlaxoSmithKline employed D.M.W. K.B.S., L.M.Y.-A. and M.A.L. are full-time employees of GlaxoSmithKline. M.S. receives funding from Pfizer Inc. for a project not related to this research. J.Ä. reports personal fees from AstraZeneca, Boehringer Ingelheim and Novartis, outside of the submitted work. D.F.G., H.H., K.S., P.S., G.S. and U.T. are employees of deCODE/Amgen Inc. Kevin Ho received support by Fresenius Medical Care North America. M.K. is employed with Synlab Holding Deutschland GmbH. W.K. reports consulting fees from AstraZeneca, Novartis, Pfizer, The Medicines Company, DalCor, Kowa, Amgen, Corvidia, Daiichi-Sankyo, Genentech, Novo Nordisk, Esperion, OMEICOS, LIB Therapeutics, speaker honoraria from Amgen, AstraZeneca, Novartis, Berlin-Chemie, Sanofi, and Bristol-Myers Squibb, and grants and non-financial support from Abbott, Roche Diagnostics, Beckmann, and Singulex, outside the submitted work. C.L. received Grants/ Research Support from Bayer Ag/ Novo Nordisk, Husband works for Vertex. As of January 2020, A.M. is an employee of Genentech, and a holder of Roche stock. W.M. is employed with Synlab Holding Deutschland GmbH. D.O.M.-K. is a partime research physician at Metabolon, Inc. M.A.N. was supported by a consulting contract between Data Tecnica International LLC and the National Institute on Aging (NIA), National Institutes of Health (NIH), Bethesda, MD, USA and consults for a number of small biotech and pharma. M.L.O. received grant support from GlaxoSmithKline during conduct of the study and received support from Novartis, Merck, Amgen, and AstraZeneca. L.S.P. has served on Scientific Advisory Boards for Janssen, and has or had research support from Merck, Pfizer, Eli Lilly, Novo Nordisk, Sanofi, PhaseBio, Roche, Abbvie, Vascular Pharmaceuticals, Janssen, Glaxo SmithKline, and the Cystic Fibrosis Foundation. He is also a cofounder, Officer and Board member and stockholder for a company, Diasyst, Inc., which markets software aimed to help improve diabetes management. A.I.P. and D.F.R. are employees of Merck Sharp Dohme Corp. Bruce.M.P. serves on the steering committee of the Yale Open Data Access Project funded by Johnson & Johnson. P.R. received fees to his institution for research support from AstraZeneca and Novo Nordisk; for steering group participation from AstraZeneca, Gilead, Novo Nordisk, and Bayer; for lectures from Bayer, Eli Lilly and Novo Nordisk; and for advisory boards from Sanofi and Boehringer Ingelheim outside of this work. V.S. has received a modest honorarium from Sanofi for consulting. He also has ongoing research collaboration with Bayer Ltd. (all outside of the present study). L.W. received institutional grants from GlaxoSmithKline, AstraZeneca, BMS, Boehringer-Ingelheim, Pfizer, MSD and Roche Diagnostics. H.W. has received grant support paid to the institution and fees for serving on Steering Committees of the ODYSSEY trial from Sanofi and Regeneron Pharmaceuticals, the ISCHEMIA and the MINT studies from the National Institutes of Health, the STRENGTH trial from Omthera Pharmaceuticals, the HEART-FID study from American Regent, the DAL-GENE study from DalCor Pharma UK Inc., the AEGIS-II study from CSL Behring, the SCORED and SOLOIST-WHF from Sanofi Aventis Australia Pty. Ltd., and the CLEAR OUTCOMES study from Esperion Therapeutics. M.P. is partly funded by the study FinnGen (www.finngen.fi), which is jointly funded by a Finnish Governmental agency Business Finland and thirteen international pharmaceutical companies: Abbvie, AstraZeneca, Biogen, Boehringer Ingelheim, Bristol-Myers Squibb, Genentech, a member of the Roche Group, GlaxoSmithKline (GSK), Janssen, Maze Therapeutics, MSD (the tradename of Merck & Co., Inc, Kenilworth, NJ USA), Novartis, Pfizer and Sanofi. C.C.K. is an Editorial Board Member for Communications Biology, but was not involved in the editorial review of, nor the decision to publish this article. The remaining authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Despoina Manousaki and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: George Inglis. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Winkler, T.W., Rasheed, H., Teumer, A. et al. Differential and shared genetic effects on kidney function between diabetic and non-diabetic individuals. Commun Biol 5, 580 (2022). https://doi.org/10.1038/s42003-022-03448-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03448-z
This article is cited by
-
X-chromosome and kidney function: evidence from a multi-trait genetic analysis of 908,697 individuals reveals sex-specific and sex-differential findings in genes regulated by androgen response elements
Nature Communications (2024)
-
Polygenic scores for estimated glomerular filtration rate in a population of general adults and elderly – comparative results from the KORA and AugUR study
BMC Genomic Data (2023)
-
KidneyGPS: a user-friendly web application to help prioritize kidney function genes and variants based on evidence from genome-wide association studies
BMC Bioinformatics (2023)
-
Genetics of diabetes-associated microvascular complications
Diabetologia (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
