
Abstract
High resolution hydroxyl radical protein footprinting (HR-HRPF) is a mass spectrometry-based method that measures the solvent exposure of multiple amino acids in a single experiment, offering constraints for experimentally informed computational modeling. HR-HRPF-based modeling has previously been used to accurately model the structure of proteins of known structure, but the technique has never been used to determine the structure of a protein of unknown structure. Here, we present the use of HR-HRPF-based modeling to determine the structure of the Ig-like domain of NRG1, a protein with no close homolog of known structure. Independent determination of the protein structure by both HR-HRPF-based modeling and heteronuclear NMR was carried out, with results compared only after both processes were complete. The HR-HRPF-based model was highly similar to the lowest energy NMR model, with a backbone RMSD of 1.6 Å. To our knowledge, this is the first use of HR-HRPF-based modeling to determine a previously uncharacterized protein structure.
Similar content being viewed by others


Introduction
Mass spectrometry (MS) has rapidly gained in popularity not only in the identification and mass measurement of proteins, but in the characterization of protein higher order structure. Numerous MS-based technologies have been successfully used to characterize protein higher order structure, including hydrogen-deuterium exchange1, limited proteolysis2, chemical crosslinking3, and covalent labeling4. Covalent labeling includes a number of techniques, all of which involve reaction of some reagent with amino acid side chains usually available on the surface of the folded protein. A variety of covalent labeling reagents have been used, including acylation reagents5, diethylpyrocarbonate6, carbenes7, trifluoromethyl radicals8,9, and iodine radicals10. Here, we present an approach based on the use of hydroxyl radicals as a covalent labeling reagent. Hydroxyl radicals generate high-quality data for a variety of amino acids, providing a generalizable probe for protein topography4,11,12,13,14. We also demonstrate that this approach is capable of producing high-quality reliable protein structures that are validated in a blind test against a parallel determination by NMR methods.
The approach we use begins with data from a technique known as hydroxyl radical protein footprinting (HRPF)15. Hydroxyl radicals are useful and popular due to the wide variety of methods for in situ generation16,17,18,19,20,21,22,23, broad reactivity13,14, small size, hydrophilic nature, and well-characterized reaction pathways with various amino acids24. Work from Chance and co-workers found that apparent rates of reaction could be correlated with average solvent accessible surface area (<SASA>) once the inherent rate of reaction of the amino acid was corrected using the free amino acids as a surrogate11,25. Work from Sharp and co-workers confirmed these findings, further reporting that amino acids with lower inherent reactivity could display altered inherent reactivity based on sequence context12,26. Sharp and co-workers further used amino acid-resolution HRPF (known as HR-HRPF) coupled with computational modeling to demonstrate the ability to differentiate between accurate computational models and inaccurate computational models, opening possibilities for using HR-HRPF data to determine protein structure12.
HR-HRPF data are then used to facilitate computational predictions of structure. The Lindert group developed the first software to use covalent labeling data in automated Rosetta protein structure prediction27,28. Recently, Biehn and Lindert reported a more robust and less computationally expensive method for using HR-HRPF data to generate protein models using conical neighbor count instead of <SASA>, which successfully identified ab initio models of accurate atomic detail for three of the four benchmark proteins examined29. However, while these studies indicate the potential of HR-HRPF for the determination of protein structure, no protein of unknown structure has been determined using HR-HRPF data to inform computational modeling.
To accurately test the ability of HR-HRPF-based modeling to generate accurate novel protein structural models, we used the technology to determine the structure of the immunoglobulin-like domain (NRG1-Ig) of human neuregulin 1 (NRG1). NRG1 is a signaling glycoprotein that interacts with the ErbB/HER family of receptor tyrosine kinases via its EGF-like domain30,31,32. NRG1-mediated signaling plays an important role in neuronal and cardiac development, and regulation of synaptic plasticity31,32,33,34. Dysregulation of these signaling pathways is implicated in human disease, such as schizophrenia and certain forms of cancer35,36. Due to a combination of alternative splicing and proteolytic processing, NRG1 exhibits a high diversity of isoforms, both soluble and membrane-bound, and a number of these isoforms include the Ig-like domain32,37. In contrast to the EGF-like domain, the functional role of the 13.3 kDa NRG1-Ig domain is less well understood. It is believed to be involved in binding to heparan sulfate proteoglycans of the extracellular matrix38,39, and there are reports that it can affect ErbB receptor activation40,41,42.
In this manuscript, two teams worked independently to characterize the structure of NRG1-Ig. The first team used HR-HRPF to quantitatively measure topography of various amino acid side chains of the NRG1-Ig. Models of the protein were generated via Rosetta ab initio modeling, scored with the HRPF-guided Rosetta score term, then subjected to a Rosetta relaxation ensemble29 from which a top-scoring model was identified. Meanwhile, the second team determined the structure of NRG1 using standard heteronuclear solution NMR techniques. During structure determination, no data was shared between groups to prevent any bias. After both teams had generated their structural models, the HR-HRPF constrained structure was compared to the NMR structure, to assess the accuracy of the HR-HRPF method. The results of this study serve as a rigorous and unbiased test of the ability of HR-HRPF to facilitate a reliable determination of soluble protein structures.
Results and discussion
HR-HRPF of NRG1-Ig
NRG1-Ig was expressed in E. coli and purified as described in Supplementary Information and Fig. S1; structural homogeneity was verified by size exclusion chromatography and NMR. Proteolytic digestion of NRG1-Ig was optimized for maximum sequence coverage after complete digestion to maximize HR-HRPF data and reproducibility. GluC was found to generate considerably higher sequence coverage than trypsin (Fig. S2, Supplementary Information), with 98.3% of the NRG1-Ig sequence shown. GluC has also successfully been used in the past for HR-HRPF analysis, as the amino acids recognized by GluC are only minor oxidation targets43. Therefore, GluC was used for HR-HRPF analysis.
After purification and digestion optimization, multi-dose Fast Photochemical Oxidation of Proteins (FPOP)12,22,44 was performed on NRG1-Ig. For the purposes of this study, only FPOP data from native NRG1-Ig were used. A mixture of 10 µM NRG1-Ig, 17 mM glutamine, 1 mM adenine, 50 mM sodium phosphate, 2.2 mM Tris (pH 8.1), and hydrogen peroxide at 10 mM, 25 mM, 50 mM or 100 mM were used for FPOP labeling. Adenine dosimetry was measured for each experiment to determine delivered radical dose, in order to account for variability in radical generation or scavenging45. A control for each FPOP peroxide concentration was conducted under the same conditions without laser irradiation to measure and correct for background oxidation.
Samples were then digested using our optimized GluC protocol. LC-MS/MS using electron transfer dissociation (ETD) was performed to measure the amount of oxidation at each amino acid for each oxidized peptide. Oxidation of twenty amino acids were measured (examples in Fig. 1, with full data in Fig. S3, Supplementary Information). Under FPOP conditions, the relationship between oxidation of the dosimeter and oxidation of a target residue will approach linearity, with the slope of the linear regression of the relationship being directly proportional to the reactivity of the oxidation target; a more complete explanation is given in Fig. S4, Supplementary Information. The slope of the regression was used to determine the protection factor (PF); 95% confidence intervals for slopes were used to represent uncertainty in PF measurement. PF was converted to the natural log of PF (lnPF), which was defined as the natural log of the normalized relative intrinsic reactivity value for a particular residue14 divided by the regression slope. Values measured for lnPF for all amino acids measured are given in Fig. S5 and Table S1, Supplementary Information.

Each figure shows the calculated oxidation of each residue at four different hydrogen peroxide concentrations plotted against the changes in adenine absorbance at 260 nm. The error bars represent one standard deviation from triplicate measurements for each data point. Each point represents the oxidation of one residue at a specific radical dose. The slopes of best-fit lines are radical dose responses.
NMR analysis of NRG1-Ig
Using a suite of standard multidimensional experiments (Table S2, Supplementary Information), we obtained nearly complete resonance assigments of 1H, 13C, and 15N spins of the native polypeptide range (Table 1, Fig. S6, Supplementary Information). The only resonances we were unable to observe and assign were those of backbone 1H and 15N of Lys117. The 13Cα and 13Cβ chemical shifts of the two cysteine residues were consistent with a disulfide bond formation46. Based on extensive chemical shift assignments and NOE data we obtained a well-defined solution NMR structure of NRG1-Ig (Fig. S7, Table 1, Supplementary Information). The fold of NRG1-Ig is typical of immunoglobulin-like domains, with a sandwich of two β-sheets stabilized by a disulfide bond. The smaller anti-parallel β-sheet consists of β-strands 41–58, 94–102, 86–91, while the second β-sheet consists of β-strands 77–72, 108–115, 120–130, 45–48 in a mixed topology with the last two stands running parallel. The only helical component is a single 310 turn at 104–106.
Determining the best computational models of the Ig-like domain of NRG1
We employed our recent HRPF-guided Rosetta modeling protocol29 to predict the structure of NRG1-Ig. Sulfur-containing amino acids were excluded due to the prevelance of incompletely controlled secondary oxidation12,47,48,49. As per our published protocol, only lnPF values measured from Trp, Phe, Tyr, His, and Leu were used. Incorporation of other labeling targets increased the error observed between lnPF value and optimized conical neighbor count29. This is consistent with prior observation that the correlation between amino acid solvent accessible surface area and lnPF calculated using intrinsic reactivity values measured from free amino acids14 has much higher error as the intrinsic reactivity of the amino acid decreases, due to an increasing effect of the sequence context on the inherent reactivity of the amino acid12. Our protocol used an HRPF score term, hrf_dynamics, that rewarded models demonstrating agreement with the FPOP labeling data. The hrf_dynamics score term was previously developed based on the relationship between HRPF data and conical neighbor count, an exposure metric that is less computationally expensive to calculate than <SASA>. Based on its successful elucidiation of accurate models for three of four benchmark proteins, it was pursued for this work. Upon input of a user’s HRPF data as lnPF, the predicted neighbor count was calculated by substituting the lnPF value into the equation relating lnPF and conical neighbor count. Then, neighbor count was calculated for the input model to be scored, providing an observed neighbor count. The deviation between the observed and predicted neighbor count guides the scoring of the model, and models with predicted neighbor counts closer in value to observed neighbor counts were more rewarded. In this case, the rewarding process refers to receiving a more favorable, i.e., more negative, per-residue score. Upon scoring models with hrf_dynamics, we used Rosetta relaxation ensemble movers to sample protein flexibility. The output structures from the Rosetta mover protocol were referred to as mover models. Upon generation of 20,000 Rosetta ab initio models, we scored models with Rosetta’s score function (“Ref15”) (Fig. 2a) and hrf_dynamics to determine a total score (Fig. 2b). The 20 top-scoring models were then used as inputs for the relaxation ensemble that generated thirty mover models per top-scoring structure, leading to the addition of 600 models to be included in the model distribution (Fig. 2c).

Score versus RMSD to NMR model 1 when (a) scoring with Rosetta’s score function; (b) scoring with Rosetta and hrf_dynamics; and (c) scoring with Rosetta and hrf_dynamics including mover models (dark grey). Best scoring models are denoted by a black triangle. d RMSD histograms for top 250 scoring models when scoring with Rosetta (grey) versus Rosetta and hrf_dynamics including mover models (blue). Bin widths were maintained at 0.5 Å. e Alignment of NMR model 1 (black) with the top-scoring model identified from our HRPF-guided and mover model protocol (blue). The RMSD to the NMR model was determined to be 1.6 Å.
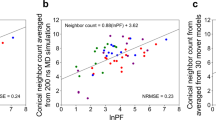
Upon examination of the 250 top-scoring models when scoring with Rosetta versus scoring with Rosetta and hrf_dynamics including mover models, we observed a decrease in the average RMSD and an increase in the percentage of models with RMSDs under 5 Å (Fig. 2d). The average RMSD of the top 250 models when scoring with Rosetta was 9.5 Å, which improved to 3.8 Å when scoring with FPOP data and including mover models. When scoring with Rosetta, 21% of the top 250 models had RMSDs below 5 Å. This improved with hrf_dynamics usage and mover model generation to 94% of models having RMSDs under 5 Å. When scoring both mover models and ab initio structures with our score term, we identified one of the generated mover models as the best scoring model. Our best scoring model exhibited an RMSD of 1.6 Å to the determined NMR structure of NRG1-Ig (Fig. 2e). The correlation between the HR-HRPF lnPF results for NRG1-Ig optimized conical neighbor count (the number of neighbors within the vicinity of a residue based on distance and angle contributions)29 from the lowest energy NMR structure was consistent with correlations previously reported for model protein structures11,12,50. The subset of amino acids considered here are robust regardless of the method of hydroxyl radical generation or amino acid-level quantitation, and no bias was introduced due to over-fitting to known structures (Fig. 3). Overall, employment of the relaxation ensemble to generate mover models resulted in a significant enrichment of accurate, high-quality, low-RMSD models in this blind prediction effort. We concluded that usage of our FPOP-guided and relaxation ensemble method increased confidence in model selection for other structure prediction efforts.

The correlation measured for NRG1 performed blinded to the NMR structure (cyan) was consistent with those reported for proteins of known structure (red: myoglobin; green: calmodulin; blue: lysozyme; violet: LMPTP).
Subsequent to our completion of the NMR structure and HR-HRPF-assisted Rosetta model of NRG1-Ig, an AlphaFold model of NRG1 was released51,52 which made a high confidence prediction for the Ig-like domain (UniProt Q02297). This is indeed a high-quality structure with a 0.85 Å RMSD of α carbons for residues 29-116 of our NMR structure, discounting the less ordered N and C termini of our his-tagged 118 residue construct (Fig. S8, Supplementary Information). We have applied our conical neighbor count scoring function to the AlphaFold model and find that it scores similarly to our NMR and HR-HRPF-assisted Rosetta model (Table S3, Supplementary Information). This underscores the utility of HR-HRFP data and our scoring procedures regardless of the method of structure prediction. It is important to note that not all proteins or regions of proteins can be predicted with confidence at this point in time; for example, AlphaFold predicts less than 30% of the 640 residue NRG1 structure with confidence (pLDDT > 70). While some of these regions are probably intrinsically disordered in free solution under most conditions53, experimental structural biology techniques including HR-HRPF-based modeling will continue to play a vital role in determining structure in the absence of confident AlphaFold predictions, as well as in testing structural models where predictions are less confident.
Conclusion
In this work, we tested the ability of HR-HRPF combined with conical neighbor count computational modeling to generate accurate, reliable structural models of a protein of truly unknown structure, NRG1-Ig. We were able to greatly increase the reliability of Rosetta modeling by application of HR-HRPF data, generating a final model with a backbone RMSD of <2 Å from the lowest energy NMR model, and with a large increase in model reliability. As the NRG1-Ig structure was unknown when HR-HRPF was performed and the NMR structure was determined independent of the HR-HRPF group, we have excluded any possibility of confirmation bias in experimental design. The consistency of our results with previous work published on proteins of known structure shown in Fig. 3 reveals a lack of confirmation bias in these previous results, and indicate no clear difference in accuracy based on the method of radical generation or amino acid-level oxidation quantification for the subset of amino acids used here (Trp, Phe, Tyr, His and Leu).
Our results as independently confirmed in a blind study by established NMR techniques demonstrate that HR-HRPF combined with conical neighbor count computational modeling is not just a tool for examining relative changes in protein topography, but is a structural biology tool that generates experimentally informed computational models of protein structure that are accurate and reliable. With the rise in computational tools for structural prediction including the recently released AlphaFold51,54, there is a need for flexible experimental methods to validate predicted structures. HR-HRPF has no theoretical limitations on the size or dynamics of measured protein structures, and can be carried out using microgram quantities of protein. Given the flexibility and low sample requirements of HR-HRPF compared with traditional high-resolution structural biology techniques, this methodology can play a significant role in the validation of computational structures, as well as the generation of accurate and reliable structural models when computational methods fail. Future work examining the ability of HR-HRPF combined with conical neighbor count to correctly identify domain-domain contacts and orientation are important for developing the application of HR-HRPF combined with conical neighbor count to address challenging problems in multi-domain protein structural biology.
Methods
Materials
Methionine amide was purchased from Bachem (Torrance, CA, USA). Adenine and L-glutamine were obtained from Acros Organics (Geel, Belgium). Catalase, trifluoroacetic acid (TFA), TCEP, and sodium phosphate were purchased from Sigma-Aldrich (St. Louis, MO). LC-MS grade formic acid, LC/MS-grade acetonitrile and water, sodium phosphate buffer, and hydrogen peroxide (30%) were obtained from Fisher Scientific (Fair Lawn, NJ, USA). Dithiothreitol (DTT) was purchased from Soltec Ventures (Beverly, MA). Sequencing-grade modified trypsin and GluC was purchased from Promega Corp (Madison, WI). Purified water (18 MΩ) was obtained from an in-house Milli- Q purification system (Millipore, Billerica, MA, USA).
Expression and purification for NMR studies
A pET-21b(+) plasmid containing a TEV-cleavable N-terminal His-tag and a 100 residue fragment comprising the NRG1-Ig domain (residues 34-133 of the UniProt Q02297 sequence) was purchased from GenScript (US distribution, Piscataway, NJ). This plasmid was transformed into BL21(DE3) E. coli cells (New England Biolabs, Ipswich, MA) using standard protocols. Transformed cells were applied onto LB agar plate with ampicillin followed by overnight incubation at 37 °C. A single colony was used to inoculate a 10 mL LB media with carbenicillin and incubated overnight at 37 °C. Cells were pelleted at 2000x g and resuspended in 3 mL of M9 media. Resuspended cells (600 µl) were used to inoculate a 50 mL M9 culture and incubated at 37 C until OD600 = 0.8. Transformed cell stock was prepared by pelleting a 5 mL aliquot, followed by resuspending in 600 µl LB and 300 ul glycerol and flash freezing with liquid nitrogen.
Glycerol stock was used to inoculate 10 mL of LB starter culture, followed by overnight incubation at 35 °C. Cells were then pelleted and resuspended in 1 L of LB medium, and incubated again at 35 °C. To produce NRG1-Ig at natural isotopic abundance expression in the 1 L culture was induced with 1 mM IPTG after reaching OD600 of ~0.6, with cells harvested 3 h after induction and frozen. For stable isotope-labeled samples, the 1 L LB culture was instead pelleted upon reaching OD600 of ~0.8, and the cell pellet was resuspended in 0.5 L of M9 minimal media containing 15NH3Cl with either 13C-glucose or 5% 13C-glucose. Incubation continued for about 1 hr at 35 °C when expression was induced with 50 µM IPTG. Cells were harvested after ~3 h by centrifugation and frozen.
Thawed cells were resuspended in lysis buffer (20 mM Tris pH 8.1, 300 mM NaCl, and 1 mM TCEP with protease inhibitors) at 4 °C and lysed using a French-press. The resulting lysate was centrifuged, and the pellet fraction containing inclusion bodies was resuspended in denaturing buffer (6 M Urea, 300 mM NaCl, 1 mM TCEP, 6 mM imidazole and 20 mM Tris pH 8.1) at 4 °C using either handheld or electric tissue homogenizer. NRG1-Ig was purified under denaturing conditions via immobilized metal affinity chromatography (IMAC) using NGC system (Bio-Rad) equipped with a 10 mL Co-NTA column. Elution of NRG-Ig1 was accomplished with a linear gradient beginning with 3% Buffer A (6 M Urea, 20 mM Tris pH 8.1 at 4 °C, 300 mM NaCl, 1 mM TCEP) and ending with 100% Buffer B (6 M Urea, 20 mM Tris, pH 8.1 at 4 °C, 200 mM imidazole, and 300 mM NaCl). The recovered U-15N,13C and natural abundance NRG1-Ig fractions were sealed in membrane tubing (Spectrapor, 6–8 kDa) and refolded by dialysis at 4 °C in four steps against a refolding buffer (20 mM Tris pH 8.1 at 4 °C, 300 mM NaCl) The refolding buffer was supplemented with 0.1 mM DDT and 50 µM ethylenediaminetetraacetic acid (EDTA) for the first dialysis stem, and only 0.1 mM DTT for the second step. U-15N,5% 13C-labeled NRG-Ig was refolded in the same manner using 0.5 mL 3.5 kDa Slide-A-Lyser cassette (Pierce). U-15N,13C- and U-15N,5%-13C-labeled NRG1-Ig were subsequently exchanged into NMR buffer (20 mM sodium phosphate pH 6.5, 100 mM NaCl) using 0.5 mL Amicon micro concentrators. U-15N,13C NMR samples (~35 µL in 1.7 mm capillary NMR tube), NRG1-Ig NC(I), and NRG1-Ig NC(II), consisted of 0.45 mM and 2.0 mM NRG1-Ig, respectively, with 0.05% sodium aside, 4 µM sodium trimethylsilylpropanesulfonate (DSS) and 7% D2O. U-15N,5% 13C NMR sample, NRG1-Ig NC5 (~40 µL in 1.7 mm capillary NMR tube), was prepared in the original TRIS refolding buffer, with 0.05% sodium aside, 5 µM sodium trimethylsilylpropanesulfonate (DSS) and 7% D2O.
NRG1-Ig purification by size exclusion chromatography for HRPF studies
A protein batch prepared by the NMR lab without isotope labels was run on Waters BEH SEC Column, 125 Å, 1.7 µm, 4.6 mm*300 mm using Thermo Fisher Dionex 3000 HPLC system. The running buffer was 20 mM Tris at pH 8.1 with 300 mM NaCl using an isocratic gradient.
Multi-dose FPOP and NRG1-Ig digestion
FPOP was performed in triplicate for three aliquots of NRG1 using a 248 nm COMPex Pro 102 high pulse energy excimer KrF laser in the presence of various hydrogen peroxide concentration (10 mM, 25 mM, 50 mM, and 100 mM)12. The experiment was done in triplicate for each hydrogen peroxide concentration. For FPOP on native NRG1, samples were prepared by mixing NRG1 to the final concentration of 10 μM in 50 mM sodium phosphate, 17 mM glutamine, and 1 mM adenine as a radical dosimeter45. Freshly prepared hydrogen peroxide at four different concentrations (10 mM, 25 mM, 50 mM, and 100 mM) was added to each sample prior laser exposure. A total volume of 20 μl of sample flowed through the excitation capillary at 17.34 µl/min. The nominal laser fluence at the plane of the excitation capillary was at 9.82 mJ/mm2 with 15% exclusion volume. After the laser irradiation, the samples were quenched in 25 ul quenching buffer containing 50 nM catalase and 20 mM methionine amide. The control sample for each hydrogen peroxide concentration was done in triplicate with the laser turned off. After laser exposure, we measured the changes in adenine UV absorbance of each oxidized sample as compared to each control at 265 nm using a nanodrop spectrophotometer. This represents the effective radical dose delivered to the protein12.
After quenching, the oxidized and control samples were denatured and reduced at 95 °C for 30 min in the presence of 5.5 mM DTT. After denaturation, the samples we put on ice for 2 min. More sodium phosphate buffer at pH 6 was added to keep its concentration at 30 mM prior to GluC addition. GluC was added in 1:20 enzyme:protein mass ratio. The samples were digested overnight for 14 h.
C18 RPLC-MS/MS C18
LC-MS/MS was done using an Acclaim PepMap 100 C18 nanocolumn (0.075 mm × 150 mm, 2 µm particle size, 100 Å pore size, Thermo Fisher Scientific) coupled to a 300 µm i.d. ×5 mm C18 PepMap 100 trap column with 5 µm particle size (Thermo Fisher Scientific) to desalt and concentrate the samples before loading onto the C18 nanocolumn for separation. The capillary pump was used to load the samples onto the C18 trap column using buffer A (water + 0.05% TFA) and buffer B (acetonitrile + 0.05% TFA). We used a nanopump for chromatographic separation using mobile phase C (water + 0.1% formic acid) and mobile phase D (acetonitrile + 0.1% formic acid). Initially, the samples were loaded onto the C18 trap column in 2% B at 5 µl/min for 6 min. The trap column was then switched inline with the nanocolumn and trapped peptides were back-eluted onto the nanocolumn using the nanopump. Elution started by increasing solvent D in a linear gradient from 2% to 40% over 22 min. The gradient then ramped up to 95% D over 5 min and held isocratic for 3 min to wash the column. Buffer D was then decreased to 2% over 1 minute and held isocratic for 6 min to re-equilibrate the column for the next run. The samples were eluted directly into a nanospray source of a Thermo Fusion Tribrid orbitrap, where the spray voltage was set at 2600 V and ion transfer tube temperature at 300 °C. A full MS scan was obtained from 150 to 2000 m/z. CID and ETD was performed every 2 s on precursor ions of +2 charge and greater for peptide identification and sequence coverage analysis. For ions with +2 charge state, ETD was performed with 20% EThcD SA collision energy to increase ETD fragmentation. The orbitrap resolution for both ETD and EThcD was 30,000 with AGC target at 5e4 and maximum injection time of 100 ms.
Peptide and amino acid level oxidation analysis
Byonic version v2.10.5 (Protein Metrics) was used to identify NRG1 peptide sequences using the NRG1-Ig protein sequence described above. For all peptides detected, the major oxidation products detected were net additions of one or more oxygen atoms. In order to calculate average oxidation events per peptide, the area under the curve for peaks of unoxidized and oxidized peptides was used according to Eq. (1). Briefly, the oxidation events per peptide were calculated by summing the intensity (I) of each peptide oxidation product multiplied by the number of oxidation events on the peptide required to generate that product and divided by the sum of I for all oxidized and unoxidized versions of that peptide, as shown in Eq. (1) P represents the average oxidation events per peptide, and I is the area under the curve for peaks of oxidized and unoxidized peptides.
The amount of oxidation at residue level quantitation in a peptide was determined by the fragment ion (z or c ion) intensities of the peptide ETD fragmentation. The oxidation fraction of a given z or c ion was calculated by dividing the oxidized sequence ion intensity to the sum of the intensity of the corresponding oxidized and unoxidized sequence ion in a particular oxidized peptide. The relative oxidation fraction of each product ion f (zi) was calculated using Eq. 3.2 where I(zi) is the intensity of the designated product ion, either summed across all spectra for RPLC, or taken at any individual point for ZIC-HILIC.
The absolute amount of oxidation of a given amino acid was determined by multiplying the average oxidation event of peptide by the absolute fractional oxidation of the corresponding sequence ions. As shown in Eq. (3), P is the average oxidation event per peptide calculated from Eq. (1), and the term in brackets is the fractional difference of two adjacent sequence ions, f(Zi) and f(Zi−1). In cases where ETD fragmentation ions are not adjacent in sequence, fractional oxidation for multiple contiguous residues within the peptide can was calculated by using non-adjacent ETD fragments in Eq. (3).
In order to take background oxidation into account, the oxidation event of each residue was calculated by subtracting the oxidation event of the same residue in control condition from its oxidation event in the oxidized sample.
Natural Protection Factor (ln(PF)) was calculated using Eq. (4) where \({R}_{i}\) represents the amino acid intrinsic reactivity for residue i while \({{Slope}}_{i}\) represents the experimentally determined radical dose response for residue i. Slopes with 95% confidence intervals were determined by linear regression analysis with the y-intercept constrained to zero.
Structural modeling
Using Rosetta’s AbInitioRelax protocol, the NRG1 Ig-like domain FASTA sequence, and fragment libraries obtained from the Robetta server, 20,000 ab initio models of NRG1 were built55,56,57,58,59. No FPOP data were included during model generation. Models were scored with the Rosetta score function named “Ref15”. Per-residue FPOP data were converted into the natural log of the protection factor (lnPF), the natural log of the normalized intrinsic reactivity divided by the FPOP labeling rate constant25,28,29. The lnPF values were supplied to the hrf_dynamics term, and models were scored based on their agreement with the labeling data29. The summed per-residue hrf_dynamics score used a weight of 9.0, as described previously. The total score was determined by adding the Rosetta and hrf_dynamics scores. Models were ranked by total score. The twenty top-scoring models were then used as input for mover model generation with the Rosetta relaxation ensemble, as described previously29. For each of the top-scoring structures, thirty mover models were obtained. The six hundred mover models were scored with Rosetta and hrf_dynamics and then included in the ab initio model distribution. The best scoring model was identified as our blind prediction for the NRG1 Ig-like domain. Upon structure determination, Cα root mean squared deviation (RMSD) values with no outlier rejection were calculated with Rosetta.
NRG1-Ig NMR
NMR data collection and processing, resonance assignment, and structure calculation followed the protocols of Northeast Structural Genomics Consortium (NESG Wiki, http://www.nmr2.buffalo.edu/nesg.wiki/Main_Page). NMR spectra (Table S2, Supplementary Information) for NRG1-Ig samples were acquired at 25 °C on AVANCE NEO 800 MHz spectrometer (Bruker BioSpin) equipped with a 1.7 mm TCI 1H(13C,15N) cryogenic probe. All spectra were Fourier-transformed using Topspin v4 (Bruker Biospin), except non-uniformly sampled 3D HBHA(CO)NH, which was reconstructed using Smile60 and Fourier-transformed with NMRPipe61. 1H chemical shifts were referenced relative to 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS), and 13C and 15N chemical shifts were referenced indirectly via gyromagnetic ratios. Visualization and analysis of NMR spectra, NOE peak picking, and integration were performed with the program CARA62. Automated assignment of backbone 1H, 15N, 13CO, 13Cα, and 13Cβ resonances was obtained with AutoAssign63 followed by interactive validation and completion. Side-chain resonances were assigned interactively using 3D (H)CCH and 3D 13C/15N-edited [1H,1H] NOESY spectra. Stereospecific assignments of Leu and Val isopropyl groups were obtained based on positive versus negative peak intensities in the 2D [13C,1H] constant-time HSQC (CT-HSQC) acquired for NRG1-Ig NC5, as described previously64. Stereospecific assignment of Asn and Gln CONH2 groups were determined from relative NOE peak intensities.
Structure calculation and automatic NOE peak assignment was performed iteratively using CYANA v 3.98.1365,66 and ASDP v1.067. Constraints for backbone φ, ψ and side-chain χ1 dihedral angles were derived using TALOS-N68, and those that were consistent with the initial structural models were used in subsequent structure calculation steps. NOE peaks with matching unambiguous assignments from CYANA and ASDP were manually checked and refined for consistency with NOE spectra and distance constraint violations, and then used to optimize NOE distance calibration function. Assignments of these peaks were kept fixed during final structure calculation with CYANA. Stereospecific assignment of CH2 groups was performed iteratively using the GLOMSA module of CYANA. Of 100 calculated conformers, 20 conformers with the lowest target function values were further refined in explicit water bath using CNS69 as previously described70 with distance constraints relaxed by 5%. The quality of NRG1-Ig structure models was analyzed with PSVS71, and the resulting statistics are summarized in Table S3, Supplementary Information. Software used for NMR data analysis and structure calculation was accessed via NMRBox72. Atomic coordinates, structural restraints, assigned NMR chemical shifts, and NOE peaklists were deposited in the Protein Data Bank (PDB ID 7SJL) and BioMagResBank (accession code 30960).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Atomic coordinates, structural restraints, assigned NMR chemical shifts and NOE peaklists were deposited in the Protein Data Bank (PDB ID 7SJL) and BioMagResBank (accession code 30960). The datasets generated during and/or analyzed during the current study, including HR-HRPF LC-MS/MS data and the final HR-HRPF-based structural model presented, are available from the corresponding author on reasonable request.
References
Engen, J. R., Botzanowski, T., Peterle, D., Georgescauld, F. & Wales, T. E. Developments in hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 93, 567–582 (2021).
Feng, Y. et al. Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol. 32, 1036–1044 (2014).
Yugandhar K., Zhao Q., Gupta S., Xiong D., Yu H. Progress in methodologies and quality-control strategies in protein cross-linking mass spectrometry. Proteomics 21, e2100145 (2021).
Liu, X. R., Zhang, M. M. & Gross, M. L. Mass spectrometry-based protein footprinting for higher-order structure analysis: fundamentals and applications. Chem. Rev. 120, 4355–4454 (2020).
Suckau, D., Mak, M. & Przybylski, M. Protein surface topology-probing by selective chemical modification and mass spectrometric peptide mapping. Proc. Natl Acad. Sci. USA 89, 5630–5634 (1992).
Pan, X., Limpikirati, P., Chen, H., Liu, T. & Vachet, R. W. Higher-order structure influences the kinetics of diethylpyrocarbonate covalent labeling of proteins. J. Am. Soc. Mass Spectrom. 31, 658–665 (2020).
Jumper, C. C., Bomgarden, R., Rogers, J., Etienne, C. & Schriemer, D. C. High-resolution mapping of carbene-based protein footprints. Anal. Chem. 84, 4411–4418 (2012).
Cheng, M., Zhang, B., Cui, W. & Gross, M. L. Laser-initiated radical trifluoromethylation of peptides and proteins: application to mass-spectrometry-based protein footprinting. Angew. Chem. Int. Ed. Eng. 56, 14007–14010 (2017).
Cheng, M. et al. Fast protein footprinting by X-ray mediated radical trifluoromethylation. J. Am. Soc. Mass Spectrom. 31, 1019–1024 (2020).
Cheng, M., Guo, C., Li, W. & Gross, M. L. Free-radical membrane protein footprinting by photolysis of perfluoroisopropyl iodide partitioned to detergent micelle by sonication. Angew. Chem. Int. Ed. Eng. 60, 8867–8873 (2021).
Kaur, P., Kiselar, J., Yang, S. & Chance, M. R. Quantitative protein topography analysis and high-resolution structure prediction using hydroxyl radical labeling and tandem-ion mass spectrometry (MS). Mol. Cell Proteom. 14, 1159–1168 (2015).
Xie, B., Sood, A., Woods, R. J. & Sharp, J. S. Quantitative protein topography measurements by high resolution hydroxyl radical protein footprinting enable accurate molecular model selection. Sci. Rep. 7, 4552 (2017).
Buxton, G. V., Greenstock, C. L., Helman, W. P. & Ross, A. B. Critical-review of rate constants for reactions of hydrated electrons, hydrogen-atoms and hydroxyl radicals (.OH/.O-) in aqueous-solution. J. Phys. Chem. Ref. Data 17, 513–886 (1988).
Xu, G. & Chance, M. R. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal. Chem. 77, 4549–4555 (2005).
McKenzie-Coe A., Montes N. S., Jones L. M. Hydroxyl radical protein footprinting: a mass spectrometry-based structural method for studying the higher order structure of proteins. Chem. Rev. 8, 7532–7561 (2021).
Sharp, J. S., Becker, J. M. & Hettich, R. L. Protein surface mapping by chemical oxidation: structural analysis by mass spectrometry. Anal. Biochem 313, 216–225 (2003).
Sharp, J. S., Becker, J. M. & Hettich, R. L. Analysis of protein solvent accessible surfaces by photochemical oxidation and mass spectrometry. Anal. Chem. 76, 672–683 (2004).
Sharp, J. S. et al. Photochemical surface mapping of C14S-Sml1p for constrained computational modeling of protein structure. Anal. Biochem 340, 201–212 (2005).
Maleknia, S. D., Chance, M. R. & Downard, K. M. Electrospray-assisted modification of proteins: a radical probe of protein structure. Rapid Commun. Mass Spectrom. 13, 2352–2358 (1999).
Maleknia, S. D. & Downard, K. M. Unfolding of apomyoglobin helices by synchrotron radiolysis and mass spectrometry. Eur. J. Biochem. 268, 5578–5588 (2001).
Aye, T. T., Low, T. Y. & Sze, S. K. Nanosecond laser-induced photochemical oxidation method for protein surface mapping with mass spectrometry. Anal. Chem. 77, 5814–5822 (2005).
Hambly, D. M. & Gross, M. L. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 16, 2057–2063 (2005).
Minkoff, B. B. et al. Plasma-generated OH radical production for analyzing three-dimensional structure in protein therapeutics. Sci. Rep. 7, 12946 (2017).
Garrison, W. M. Reaction mechanisms in the radiolysis of peptides, polypeptides, and proteins. Chem. Rev. 87, 381–398 (1987).
Huang, W., Ravikumar, K. M., Chance, M. R. & Yang, S. Quantitative mapping of protein structure by hydroxyl radical footprinting-mediated structural mass spectrometry: a protection factor analysis. Biophys. J. 108, 107–115 (2015).
Sharp, J. S. & Tomer, K. B. Effects of anion proximity in peptide primary sequence on the rate and mechanism of leucine oxidation. Anal. Chem. 78, 4885–4893 (2006).
Aprahamian, M. L. & Lindert, S. Utility of covalent labeling mass spectrometry data in protein structure prediction with Rosetta. J. Chem. Theory Comput. 15, 3410–3424 (2019).
Aprahamian, M. L., Chea, E. E., Jones, L. M. & Lindert, S. Rosetta protein structure prediction from hydroxyl radical protein footprinting mass spectrometry Data. Anal. Chem. 90, 7721–7729 (2018).
Biehn, S. E. & Lindert, S. Accurate protein structure prediction with hydroxyl radical protein footprinting data. Nat. Commun. 12, 341 (2021).
Liu, P. et al. A single ligand is sufficient to activate EGFR dimers. Proc. Natl Acad. Sci. USA 109, 10861–10866 (2012).
Mei, L. & Nave, K. A. Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 83, 27–49 (2014).
Mei, L. & Xiong, W. C. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 9, 437–452 (2008).
Xu, Y., Li, X., Liu, X. & Zhou, M. Neuregulin-1/ErbB signaling and chronic heart failure. Adv. Pharm. 59, 31–51 (2010).
Kataria, H., Alizadeh, A. & Karimi-Abdolrezaee, S. Neuregulin-1/ErbB network: an emerging modulator of nervous system injury and repair. Prog. Neurobiol. 180, 101643 (2019).
Shi, L. & Bergson, C. M. Neuregulin 1: an intriguing therapeutic target for neurodevelopmental disorders. Transl. Psychiatry 10, 190 (2020).
Laskin, J. et al. NRG1 fusion-driven tumors: biology, detection, and the therapeutic role of afatinib and other ErbB-targeting agents. Ann. Oncol. 31, 1693–1703 (2020).
Willem, M. Proteolytic processing of Neuregulin-1. Brain Res. Bull. 126, 178–182 (2016).
Pankonin, M. S., Gallagher, J. T. & Loeb, J. A. Specific structural features of heparan sulfate proteoglycans potentiate neuregulin-1 signaling. J. Biol. Chem. 280, 383–388 (2005).
Loeb, J. A. & Fischbach, G. D. ARIA can be released from extracellular matrix through cleavage of a heparin-binding domain. J. Cell Biol. 130, 127–135 (1995).
Li, Q. & Loeb, J. A. Neuregulin-heparan-sulfate proteoglycan interactions produce sustained erbB receptor activation required for the induction of acetylcholine receptors in muscle. J. Biol. Chem. 276, 38068–38075 (2001).
Centa, A., Rodriguez-Barrueco, R., Montero, J. C. & Pandiella, A. The immunoglobulin-like domain of neuregulins potentiates ErbB3/HER3 activation and cellular proliferation. Mol. Oncol. 12, 1061–1076 (2018).
Eto, K., Eda, K., Kanemoto, S. & Abe, S. The immunoglobulin-like domain is involved in interaction of Neuregulin1 with ErbB. Biochem Biophys. Res. Commun. 350, 263–271 (2006).
Li, Z. et al. High structural resolution hydroxyl radical protein footprinting reveals an extended Robo1-heparin binding interface. J. Biol. Chem. 290, 10729–10740 (2015).
Gau, B. C., Sharp, J. S., Rempel, D. L. & Gross, M. L. Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal. Chem. 81, 6563–6571 (2009).
Xie, B. & Sharp, J. S. Hydroxyl radical dosimetry for high flux hydroxyl radical protein footprinting applications using a simple optical detection method. Anal. Chem. 87, 10719–10723 (2015).
Sharma, D. & Rajarathnam, K. 13C NMR chemical shifts can predict disulfide bond formation. J. Biomol. NMR 18, 165–171 (2000).
Xu, G. & Chance, M. R. Radiolytic modification of sulfur-containing amino acid residues in model peptides: fundamental studies for protein footprinting. Anal. Chem. 77, 2437–2449 (2005).
Gau, B., Garai, K., Frieden, C. & Gross, M. L. Mass spectrometry-based protein footprinting characterizes the structures of oligomeric apolipoprotein E2, E3, and E4. Biochemistry 50, 8117–8126 (2011).
Saladino, J., Liu, M., Live, D. & Sharp, J. S. Aliphatic peptidyl hydroperoxides as a source of secondary oxidation in hydroxyl radical protein footprinting. J. Am. Soc. Mass Spectrom. 20, 1123–1126 (2009).
Stanford, S. M. et al. Diabetes reversal by inhibition of the low-molecular-weight tyrosine phosphatase. Nat. Chem. Biol. 13, 624–632 (2017).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Chukhlieb, M., Raasakka, A., Ruskamo, S. & Kursula, P. The N-terminal cytoplasmic domain of neuregulin 1 type III is intrinsically disordered. Amino Acids 47, 1567–1577 (2015).
AlQuraishi, M. AlphaFold at CASP13. Bioinforma. (Oxf., Engl.) 35, 4862–4865 (2019).
Bender, B. J. et al. Protocols for molecular modeling with Rosetta3 and RosettaScripts. Biochemistry 55, 4748–4763 (2016).
Alford, R. F. et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 13, 3031–3048 (2017).
Kim, D. E., Chivian, D. & Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531 (2004).
Leman J. K., et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 17, 665–680 (2020).
Biehn S. E., Lindert S. Protein structure prediction with mass spectrometry data. Annu. Rev. Phys. Chem. 73, 1–19 (2021).
Ying, J., Delaglio, F., Torchia, D. A. & Bax, A. Sparse multidimensional iterative lineshape-enhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data. J. Biomol. NMR 68, 101–118 (2017).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Keller R. L. J. The Computer Aided Resonance Assignment Tutorial. (CANTINA Verlag, 2004).
Zimmerman, D. E. et al. Automated analysis of protein NMR assignments using methods from artificial intelligence. J. Mol. Biol. 269, 592–610 (1997).
Neri, D., Szyperski, T., Otting, G., Senn, H. & Wuthrich, K. Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA-binding domain of the 434 repressor by biosynthetically directed fractional 13C labeling. Biochemistry 28, 7510–7516 (1989).
Guntert, P., Mumenthaler, C. & Wuthrich, K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 (1997).
Herrmann, T., Guntert, P. & Wuthrich, K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 (2002).
Huang, Y. J., Mao, B., Xu, F. & Montelione, G. T. Guiding automated NMR structure determination using a global optimization metric, the NMR DP score. J. Biomol. NMR 62, 439–451 (2015).
Shen, Y. & Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241 (2013).
Brunger, A. T. Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2, 2728–2733 (2007).
Linge, J. P., Williams, M. A., Spronk, C. A., Bonvin, A. M. & Nilges, M. Refinement of protein structures in explicit solvent. Proteins 50, 496–506 (2003).
Bhattacharya, A., Tejero, R. & Montelione, G. T. Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795 (2007).
Maciejewski, M. W. et al. NMRbox: a resource for biomolecular NMR computation. Biophys. J. 112, 1529–1534 (2017).
Acknowledgements
This work was supported by the National Institute of General Medical Sciences (R01GM127267). S.M. and R.J.D. acknowledge support from the Glycoscience Center of Research Excellence (NIH P20GM130460) and J.H.P. acknowledges support from (R01GM134335). This study made use of NMRbox: National Center for Biomolecular NMR Data Processing and Analysis, a Biomedical Technology Research Resource (BTRR), which is supported by NIH grant P41GM111135 (NIGMS). Modeling work in this publication was supported by NSF (CHE 1750666) to S.L.
Author information
Authors and Affiliations
Contributions
N.A.K. characterized recombinant NRG1-Ig by LC and mass spectrometry, collected and analyzed HR-HRPF data, assisted in the interpretation of HR-HRPF data, assisted in the comparison of HR-HRPF-based models with NMR models, and wrote portions of the manuscript. A.E. collected and analyzed NMR data, generated NMR models, assisted in the comparison of HR-HRPF-based model with NMR models, and wrote portions of the manuscript. S.E.B. developed HR-HRPF-based models and wrote portions of the manuscript. C.K.M. assisted in the analysis and interpretation of HR-HRPF data. M.J.R. and Y.K. expressed, purified, and characterized recombinant NRG1-Ig. S.K.M. and R.J.D. assisted in the interpretation of HR-HRPF data and the comparison of HR-HRPF-based models with NMR models. S.L. designed experiments and methods for the incorporation of HR-HRPF data into the modeling process, analyzed HR-HRPF-based models, compared HR-HRPF-based models with NMR models, and wrote portions of the manuscript. J.H.P. designed experiments and methods for expression, purification, and NMR analysis of recombinant NRG1-Ig, compared HR-HRPF-based models with NMR models, and wrote portions of the manuscript. J.S.S. designed experiments and methods in LC, MS, and HR-HRPF analysis of recombinant NRG1-Ig, assisted in the analysis of HR-HRPF data, assisted in the interpretation of HR-HRPF data, assisted in the selection of HR-HRPF-based models, assisted in the comparison of HR-HRPF-based models and NMR models, and wrote portions of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests: J.S.S. has a financial interest in GenNext Technologies, Inc., a company commercializing technologies for protein higher structure analysis.
Peer review
Peer review information
Communications Biology thanks Weikai Li, Vijaya Lakshmi Kanchustambham, and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editor: Gene Chong.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khaje, N.A., Eletsky, A., Biehn, S.E. et al. Validated determination of NRG1 Ig-like domain structure by mass spectrometry coupled with computational modeling. Commun Biol 5, 452 (2022). https://doi.org/10.1038/s42003-022-03411-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03411-y
This article is cited by
-
Protein complex prediction using Rosetta, AlphaFold, and mass spectrometry covalent labeling
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
