
Abstract
Actinic keratosis (AK) is a common precancerous cutaneous neoplasm that arises on chronically sun-exposed skin. AK susceptibility has a moderate genetic component, and although a few susceptibility loci have been identified, including IRF4, TYR, and MC1R, additional loci have yet to be discovered. We conducted a genome-wide association study of AK in non-Hispanic white participants of the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort (n = 63,110, discovery cohort), with validation in the Mass-General Brigham (MGB) Biobank cohort (n = 29,130). We identified eleven loci (P < 5 × 10−8), including seven novel loci, of which four novel loci were validated. In a meta-analysis (GERA + MGB), one additional novel locus, TRPS1, was identified. Genes within the identified loci are implicated in pigmentation (SLC45A2, IRF4, BNC2, TYR, DEF8, RALY, HERC2, and TRPS1), immune regulation (FOXP1 and HLA-DQA1), and cell signaling and tissue remodeling (MMP24) pathways. Our findings provide novel insight into the genetics and pathogenesis of AK susceptibility.
Similar content being viewed by others
Introduction
Actinic keratoses (AKs) are keratinocyte-derived neoplasms that arise on skin exposed to chronic ultraviolet (UV) radiation1. They are highly prevalent among older individuals with light pigmentation, with prevalence estimates ranging from 11 to 60% in non-Hispanic whites (NHW) over 40 years of age2,3. Importantly, AKs can progress to develop into keratinocyte carcinoma (KC), particularly cutaneous squamous cell carcinoma (cSCC), which is among the most common and costly malignancies among NHWs4,5,6. The underlying pathogenesis of AK includes alterations in the pathways regulating cell growth and differentiation, inflammation, and immunosuppression caused by UV radiation, tissue remodeling, oxidative stress, and impaired apoptosis7.
Characterizing the genetic factors influencing AK susceptibility is an essential step toward understanding the pathogenesis of keratinocyte neoplasia. Genetic susceptibility to AK has been implicated by a genome-wide association study (GWAS) performed in a European cohort, which identified three pigmentation-related loci (i.e., IRF4, TYR, and MC1R), explaining 2.6% of the variance in the risk of AK8. A subsequently published genome-wide compound heterozygote scan reported 15 AK-associated loci, three of which (KCNK5/KCNK17, PAQR8/GSTA2, and KCNQ5/KHDC1) were replicated that were not related to pigmentation pathways9. AK has a moderate genetic component with an array-heritability estimate of ~17.0%8. However, many of the reported genetic risk loci have not been validated, and the genetic etiology of AK remains largely unknown.
To address these knowledge gaps, we performed a GWAS of AK among 63,110 non-Hispanic white individuals (16,352 AK cases and 46,758 controls) in the Kaiser Permanente Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort (discovery cohort). We then validated our findings in an independent cohort of 29,130 NHW individuals in the Mass-General Brigham (MGB) Biobank cohort (5110 AK cases and 24,020 controls). Our findings validate the previously reported susceptibility loci and identify novel loci with functional roles in pigmentation and immune system pathways, highlighting biological pathways through which human genetic variation impacts keratinocyte carcinogenesis.
Results
GERA cohort
We conducted the primary discovery analysis of AK in 16,352 AK cases and 46,758 controls from the NHW GERA sample. As compared to controls (subjects without AK diagnosis), subjects with AK were more likely to be older (65.6 vs. 58.7 years) and male (49.6% vs. 38.2%)(Table 1), consistent with previous studies2,10.
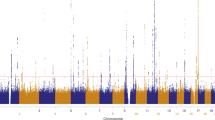
We identified eleven genome-wide significant (P < 5 × 10−8) loci associated with AK (Table 2 and Fig. 1), of which seven loci have not been previously reported. These included FOXP1 (lead SNP rs62247035), SLC45A2 (lead SNP rs16891982), HLA-DQA1 (lead SNP rs9271377), BNC2 (lead SNP rs12350739), RALY (lead SNP rs6059655), and MMP24 (lead SNP rs2425025). We confirmed genome-wide association with AK at the previously reported IRF4, TYR, and MC1R loci8. The QQ plot is presented in Fig. 2a. SNPs rs6059655 in RALY and rs2425025 in MMP24 on 20q11 are in linkage disequilibrium (r2 = 0.85).

The y axis represents log-scale P-values, and loci with the smallest P obtained from the logistic regression are labeled with the nearest or corresponding gene’s name. Known loci are in green triangles and newly identified loci are in blue triangles.

a GWAS of GERA discovery cohort (n = 63,110). b GWAS of meta-analysis combining GERA and MGB Biobank cohorts (n = 92,240). Lambda indicates inflation factor.
Conditional analyses identified an additional locus
A conditional analysis in the GERA cohort identified one additional SNP (rs35063026), which resides in the SPATA33 gene within the previously reported MC1R/DEF8 locus (OR 1.43, P = 3.7 × 10−50). This SNP was not a proxy variant of the previously reported SNP rs1805008. These two SNPs (rs35063026 and rs1805008) are ~0.25 Mb apart and in linkage equilibrium (r2 = 0.005), suggesting that they are independent signals in the same locus. We generated a regional association plot at the MC1R/DEF8 locus to illustrate the multiple independent signals at this genomic region (Supplementary Fig. 1).
Sensitivity analysis
To examine whether identified AK-associated loci were driven by cSCC risk, we performed a sensitivity analysis limiting the cohort to participants in GERA without previously diagnosed cSCC. Eight out of the eleven AK-associated loci (SLC45A2, IRF4, BNC2, HERC2, DEF8, SPATA33, RALY, and MMP24) were confirmed to be associated with AK at a genome-wide level of significance (P < 5 × 10−8). The remaining three AK-associated loci (FOXP1, HLA-DQA1, and TYR) reached Bonferroni-level of significance (P = 4.5 × 10−3 = 0.05/11; 11 SNPs tested) (Supplementary Table 1). The direction of the effect of the risk allele of AK-susceptibility loci was consistent with those of the full study cohort.
Validation in the MGB Biobank cohort
The validation cohort consisted of 5110 AK cases and 24,020 controls from the MGB Biobank cohort. Similar to our findings in the GERA cohort, MGB Biobank AK cases were more likely to be older (72.3 vs. 58.7 years) and male (53.7% vs. 46.1%) when compared to controls (Table 1). We assessed the 11 lead SNPs identified in the GERA cohort for validation in the MGB Biobank cohort. We found five SNPs validated at a genome-wide significant level, including rs16891982 at SLC45A2, rs12203592 at IRF4, rs1126809 at TYR, rs4268748 at DEF8, and rs35063026 at SPATA33 (Table 2). Three additional SNPs were validated at Bonferroni significance (P < 4.5 × 10−3 = 0.05/11; 11 SNPs tested) including rs12350739 in BNC2, rs6059655 in RALY, and rs2425025 in MMP24. The remaining loci (rs62247035 in FOXP1, rs9271377 in HLA-DQA1, rs12916300 in HERC2) did not reach statistical significance, although their direction of effect was consistent with those of the discovery cohort.
Confirmation of previously reported AK-associated loci
We investigated the three previously reported AK-associated SNPs in both the GERA and MGB Biobank cohorts (Table 3). All 3 SNPs were replicated at a genome-wide level of significance in both cohorts. Of note, the most significant SNP in the 16q24 locus in the current study was rs4268748 in the DEF8 gene, whereas in the previous study, it was rs139810560 in the MC1R gene. These genes are ~14.8 kb apart and in low linkage disequilibrium (r2 = 0.18), suggesting that this region may be subject to adaptive selection11,12,13.
A meta-analysis of GERA and MGB Biobank
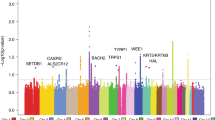
We conducted a meta-analysis combining the GERA and MGB Biobank cohorts to increase the power to detect additional novel loci (QQ plot in Fig. 2b). In addition to the 12 independent loci identified in the GERA discovery or conditional analysis, we identified one locus, TRPS1 (lead SNP rs7832568, OR = 1.07, P = 2.84 × 10−8), that was not previously reported (Table 4 and Fig. 3). Regional association plots at the novel AK-susceptibility loci are presented in Supplementary Fig. 2a–g.

The y axis represents log-scale P-values, and loci with the smallest P obtained from the logistic regression are labeled with the nearest or corresponding gene’s name. Known loci are in green triangles, newly identified loci in discovery analysis are in blue triangles, and newly identified loci in meta-analysis are in orange triangles.
Gene and pathway prioritization
We conducted gene-based and biological pathway prioritization analyses using Versatile Gene-based Association Study 2 (VEGAS2) software implemented in a command-line tool (https://vegas2.qimrberghofer.edu.au). VEGAS2 integrative tool aggregates association strength of individual markers into pre-specified biological pathways14,15. Using a mapping threshold of 10 kb upstream and downstream of gene boundaries, we found 55 genes that reached a significant threshold after correcting for multiple testing of 23,051 genes (P < 2.17 × 10−6) (Supplementary Table 2). We found a significant association with AK for the ANXA9 gene on chromosome 1, which was not identified in the discovery GWAS or meta-analysis. The pathway-based analysis identified five pathways/gene-sets that were significantly enriched after correcting for multiple testing of 9,736 pathways/gene-sets (P < 5.14 × 10−6) (Supplementary Table 3). These included melanin or secondary-metabolite biosynthesis pathways. Significant results (Benjamini–Hochberg false discovery rate [FDR] control approach with FDR of 0.1) of gene-based analysis and pathways/gene-sets analysis are presented in Supplementary Tables 2 and 3.
Heritability estimate for AK
We estimated SNP-based heritability in the GERA NHW sample using a linkage disequilibrium score regression (LDSC), and we found an SNP-based heritability estimate of 0.077 (h2SNP; 95% CI 0.05–0.10).
Discussion
Our large GWAS of AK confirmed the previously reported association between SNPs in genes related to the pigmentation pathway (IRF4 on chromosome 6p25, TYR on chromosome 11q14, and MC1R on chromosome 16q24)8,9, and also has identified previously unreported AK-associated SNPs at novel loci (FOXP1 on 3p13, SLC45A2 on 5p13, HLA-DQA1 on 6p21, TRPS1 on 8q23, BNC2 on 9p22, HERC2 on 15q13, and RALY and MMP24 on 20q11). These novel loci harbor genes implicated in pigmentation, immune regulation, and cell signaling. Several loci are in or near genes in the pigmentation pathways that are likely to be relevant to AK and associated with skin tanning ability and KC risk16,17,18,19. IRF4 activates the melanogenic enzyme TYR expression, while RALY-ASIP antagonizes the pathway. SLC45A2 and HERC2/OCA2 regulate melanin production, and BNC2 may regulate the expression of pigmentation genes20. Further, we identified AK-associated pathways, including the melanin synthesis process. Melanin pigment molecules form a coat around the nucleus of epidermal keratinocytes that may protect the keratinocytes from UV-induced DNA damage, which can lead to AK development.
The predominance of AK susceptibility loci associated with pigmentation genes suggests the critical role of components of the pigmentation pathway for AK risk. Also, it confirms the well-known heritable risk factors of AK, such as hair and eye colors or fair skin5. Previous GWAS of the Rotterdam population reported that IRF4, MC1R, and TYR genes might increase AK risk by affecting pigmentation and oncogenic functions8. A subsequent compound heterozygote scan on candidate pigmentation genes reported a suggestive association with HERC2 loci (P = 5.5 × 10−6), while the association with SLC45A2 and BNC2 did not reach statistical significance9. Our strongest signal was SNP rs12203592 in IRF4 (P = 1.97 × 10−155), consistent with the previous study. SNP rs12203592 lies within an intronic regulatory region of the interferon regulatory factor 4 (IRF4) gene that impacts skin pigmentation by modulating enhancer-mediated transcriptional regulation and physically interacting with the IRF4 gene promoter in an allele-specific manner21. This SNP has been associated with pigmentation, hair color, eye color, freckles, skin sensitivity to sun exposure, and skin cancers, including cSCC, basal cell carcinoma (BCC), and melanoma18,22,23,24,25,26,27,28. IRF4 cooperates with the melanocyte master regulator, microphthalmia-associated transcription factor (MITF), to activate the tyrosinase (TYR) expression that catalyzes melanin production and other pigments from tyrosine by oxidation29,30. In the previously published GWAS8, AK-susceptibility SNP in TYR locus (rs1393550) did not reach a genome-wide level of significance, likely due to limited sample size. The association of rs1393350 and AK risk reached a genome-wide level of significance in our study, and we identified an additional lead SNP rs1126809 at the TYR locus. SNP rs1126809 has been associated with a low tan response and increased risk of keratinocyte carcinomas and melanoma16,18,24,25,31, and it may cause changes at the post-translational modification site, leading to dysregulation of melanin synthesis within the melanosomes32. Similarly, SNP rs16891982 lies in SLC45A2, which encodes a transporter protein that mediates melanin synthesis, correlates with reduced melanin content in cultured human melanocytes33. The variant has been associated with pigmentation and melanoma23,31. In addition, SNP rs12350739, an intergenic SNP of basonuclin 2 (BNC2), and the highly conserved surrounding region function as enhancers regulating BNC2 transcription in human melanocytes34. Variants in the BNC2 locus have been associated with skin color, freckling, and KC16,24,25,34,35. SNP rs12916300 is an intronic variant in HERC2 (HECT and RLD domain containing E3 ubiquitin-protein ligase 2). Individual SNPs at this locus and the nearby gene OCA2 have been associated with pigmentation variability as well as cSCC risk24,36.
Interestingly, most AK-associated loci have been previously reported as cSCC-associated loci suggesting common biological pathways in keratinocyte carcinogenesis24. While cSCC can arise either de novo or from a preexisting AK, the annual risk of cSCC for subjects with multiple AKs ranged from 0.15 to 80%37. A previous study on gene expression patterns demonstrated that AK and cSCC were genetically related38. The direction of the effect of the risk allele of AK-susceptibility loci is similar to what was found in the previous GWAS of cSCC risk24,25. In addition to those loci related to the pigmentation pathway, variants in HLA-DQA1 were associated with AK risk. Class II HLA genes encode major histocompatibility complex (MHC) molecules that bind antigenic peptides presented by antigen-presenting cells and deliver them to the T-cell receptors on T cells. Increased AK incidence among immunosuppressed subjects may suggest a role for HLA antigens and immune response in AK pathogenesis39,40,41,42. Interestingly, rs4455710 at HLA-DQA1 locus that was identified in our meta-analysis is the same lead SNP that was identified as a cSCC risk locus in the previous GWAS24. We identified rs9271377 at the HLA-DQA1 locus in our discovery cohort, and this SNP was in strong linkage disequilibrium with rs4455710 (r2 = 0.6483). This may suggest shared immune effects involving AK and SCC, given that AKs can be a precursor to cSCC among individuals with both AKs and SCC. In addition, SNP rs62247035 at 3p13 is intronic in forkhead box P1 (FOXP1), a gene that encodes a transcriptional factor that regulates lymphocyte development and whose abnormal expression has been demonstrated in various human cancers. FOXP1 has been reported as a negative regulator of anti-tumor immune responses via its regulation on chemokine expression and MHC class II expression43,44. Notably, common variants in FOXP1 have been associated with cSCC and BCC in previous GWAS16,18,24. The SNP rs62246017 in FOXP1 was previously associated with cSCC, and it was in linkage disequilibrium with rs7638354 identified in the current study (r2 = 0.882). Interestingly, recent studies showed that immunotherapy of AK reduced the risk of SCC development by inducing T-cell immunity45,46. The finding of shared immune-related genomic loci associated with both AK and cSCC risk suggests that immune-related pathways may hold promise for novel therapeutic options in keratinocyte carcinogenesis.
Multiple SNPs were identified in 16q24 (DEF8 and SPATA33 locus) that are associated with pigmentation traits, tanning response, and skin cancer risk16,18,22,23,24,25,28,47. While the previously published GWAS identified rs139810560 in MC1R at 16q24 to be associated with AK8, and rs1805007 in MC1R was the most significant SNP at this locus in our validation cohort. Common DNA variants at the MC1R gene encoding melanocortin one receptor on the melanocytes that produce a melanin pigment have been associated with tanning ability, hair color, and KC and melanoma risk16,18,23,25,28,31. Previous studies found multiple SNPs independently related to hair color near the MC1R locus23,48. We found rs4268748 in DEF8 at the 16q24 locus to be most significantly associated with AK risk and rs35063026 in SPATA33 to be independently associated with AK risk on conditional analyses. Variants in both DEF8 and SPATA33 have been associated with pigmentation and cSCC24,47. Additionally, a previous phenome-wide association study identified a variant (rs258322) encoding cyclin-dependent kinase 10 (CDK10) that was associated with AK49. Of note, imputed expression levels of CDK10 were negatively correlated with risk allele dosage of SNP rs426874850. A previous study proposed that variants at the MC1R locus regulate the SPATA33 gene (in sun-exposed skin tissue) and the CDK10 gene (in all tissues)16. Consistent with previous studies, our findings may provide evidence that the correlation of various genes with complicated linkage disequilibrium structures in the 16q24 region may collectively play a role in keratinocyte neoplasia. Future functional studies may help elucidate the role of AK-associated SNPs in this genomic region.
In our meta-analysis, an additional novel AK-susceptibility locus, TRPS1 at 8q23, was identified. SNP rs7832568 lies inside an intronic region of TRPS1, which encodes a transcription factor that binds to a dynein light chain protein and suppresses the transcriptional activity of GATA regulated genes essential for bone and hair follicles, and is involved in sun sensitivity16,51. Interestingly, a recent genome-wide meta-analysis of cSCC identified a susceptibility SNP in the TRPS1 locus51. The TRPS1 (lead SNP rs7832568) locus will need to be validated in an external sample to confirm its implication in AK susceptibility.
Our gene-based analysis also identified the annexin A9 (ANXA9) gene on chromosome 1q21.3 as an AK risk locus. ANXA9, also known as pemphaxin, is targeted by pemphigus vulgaris antibodies in keratinocytes and may contribute to immune response and the acantholytic process52,53. In addition, a previous melanoma GWAS and a recent transcriptome-wide association study of cSCC identified ANXA9 as a susceptibility locus50,54. The role of ANXA9 in cutaneous carcinogenesis has been implicated by multi-omic methods and warrants further investigation.
This study’s strengths include the robust sample sizes of both the discovery and validation cohorts and the associated comprehensive electronic health records derived from large, independent healthcare delivery systems. There are several limitations to be considered when interpreting the results. Our findings are limited to non-Hispanic white individuals, in which AKs almost exclusively arise, and results may not be extrapolated to individuals of non-European ancestry. This study defined AKs based on clinician-rendered diagnosis using the International Classification of Disease (ICD) diagnosis codes captured in the electronic healthcare systems. As such, we cannot exclude the possibility of undiagnosed AK arising in the controls. However, given the high reliability of AK codes55, it is likely that the case definition has high validity.
In summary, this study provides independent replication of three previously reported AK susceptibility loci and identified seven novel loci contributing to AK pathophysiology. Our findings help elucidate pathways involved in AK pathophysiology and keratinocyte carcinogenesis, especially for immune regulation pathways, that we may benefit from targeted therapy. Identified loci could also serve as a framework for future functional investigations and as a basis for developing risk prediction models to identify individuals at high risk for keratinocyte neoplasia for future behavioral modification or chemoprevention trials.
Materials and methods
Case and control definition
Potentially eligible cases in both cohorts were defined as participants who had a clinician-rendered AK diagnosis (International Classification of Disease (ICD) diagnosis code version 9 of 702.0 and version 10 of L57.0) in the electronic health record. The control group included all participants without a relevant AK-ICD code. Our analysis included only self-reported NHW participants to minimize confounding risk due to ancestry differences. The Institutional Review Boards at the Kaiser Foundation Research Institute and the MGB Human Research Committee approved all study procedures.
GERA cohort: genotyping, quality control, and imputation
We report a GWAS of AK in 16,352 cases and 46,758 controls from the GERA cohort, NHW samples. The GERA cohort consists of 110,266 adults who consented to the Research Program on Genes, Environment, and Health at Kaiser Permanente Northern California (KPNC). GERA participants were genotyped at over 665,000 genetic markers on Affymetrix Axiom arrays optimized for individuals of European56. Samples with a sample call rate <0.97 have been filtered out. Standard quality control (QC) procedures were applied57, with an additional step in which SNPs with a call rate <0.90 were removed. Detailed reports of genotyping and SNP quality control have been previously described58. Data then were pre-phased with SHAPE-IT v2.559. SNPs were imputed from 1000 Genomes Project reference panel (phase I release, http://100genomes.org) using IMPUTE2 v2.3.160,61. Additional QC procedures on genotyped data were applied before conducting GWAS62. The Genome Reference Consortium Human genome build 37 (GRCh37) was used in annotating variants. SNPs in the genotyped dataset were included in the imputed dataset that passed QC. We used the information R2 from IMPUTE2 as a QC parameter, which estimates the imputed genotype’s correlation to the actual genotype. Genetic markers with an imputation R2 > 0.7 and minor allele frequency (MAF) > 0.01 were included in this study.
GWAS analysis and covariate adjustment
Logistic regression of AK for each SNP was performed using PLINK v1.9 (www.cog-genomics.org/plink/1.9/). We adjusted for age at cohort entry, sex, and top ten ancestry principal components (PCs). Principal component analysis (PCA) was performed using the smartpca program, part of the EIGENSOFT4.2 software package63. Details of the ancestry analyses are previously described58. We modeled data from each genetic marker using additive dosages accounting for the uncertainty of imputation. We defined the lead SNP as the most significant SNP within a 2 Mb (±1 Mb) window at each locus. Novel loci were defined as those located over 1 Mb apart from any previously reported locus.
Conditional models
We performed a stepwise procedure to explore independent signals within the loci identified in the GERA cohort16. Specifically, we fitted a new regression model in a 2 Mb (±1 Mb) window at each locus, including the top genome-wide significant SNP (smallest P < 5 × 10−8) at each locus identified in the association analysis step as a covariate (conditional model). We considered the top genome-wide significant SNP (smallest P < 5 × 10−8) at each locus identified from the conditional model as an independent signal and added it to the covariate list for the next iteration. A joint association of all the selected SNPs is iterated until no new genome-wide significant SNP at each locus remained associated. Conditional models were conducted using PLINK v1.9.
Sensitivity analysis
Given that AK and cSCC are reported to be genetically related38, we performed a sensitivity analysis to explore the identified AK-associated signals among those without cSCC in the GERA discovery cohort. Details on cSCC case verification in GERA are described previously24. We excluded 7,121 subjects with at least one validated cSCC case (invasive or in situ), remaining 55,989 subjects in the sensitivity analysis (11,029 AK cases and 44,960 controls). Logistic regression of AK risk for each SNP was performed using PLINK v1.9 adjusting for age, sex, and top ten PCs.
MGB Biobank cohort: genotyping, quality control, imputation, and GWAS analysis
To validate the significant GERA-identified SNPs, we evaluated associations in the NHW subjects from the MGB Biobank, consisting of 5,110 AK cases and 24,020 controls. The MGB Biobank is an extensive integrated database containing clinical data from MGB HealthCare for ~100,000 consented patients and genomic data for over 35,000 participants64. MGB samples were genotyped using three versions of SNP array offered by Illumina (Illumina, Inc., San Diego, CA), including (1) Multi-Ethnic Genotyping Array (MEGA) array including 1,416,020 SNPs, (2) Expanded Multi-Ethnic Genotyping Array (MEGA Ex) array including 1,741,376 SNPs, and (3) Multi-Ethnic Global (MEG) array including 1,778,953 SNPs. GRCh37 has been used in the annotation of the variants. Imputation was performed using the Michigan Imputation Server that uses Minimac365. MGB Biobank uses the HRC (Version r1.1 2016) reference panel for imputation. This HRC panel consists of 64,940 haplotypes of predominantly NHW ancestry. Haplotype phasing was performed using SHAPE-IT59.
We included only NHW subjects, which were self-reported by patients, to minimize the risk for confounding due to ancestry differences and to be consistent with the discovery cohort. PCA was applied to characterize the population structure and exclude racial outliers. For the PCA, QC steps of genotyped data were conducted. Briefly, any variants with an SNP call rate <0.98 or MAF < 0.01, as well as any subjects with call rate <0.98, a discrepancy between the reported and predicted sex, evidence of an excess of homozygosity, or related or duplicated subjects (identity-by-descent [IBD] > 0.2) were excluded from the PCA.
For the genome-wide association analyses, imputed SNPs were used. Only common variants of three arrays (MEG, MEGA, MEGA EX) were included in all analyses after QCs. Specifically, an info score >0.8 (high-quality imputed SNPs), SNP call rates >0.95, and MAF > 0.01 were retained in the association analyses. PLINK 1.90 was used to conduct the genome-wide association analysis, adjusted for age, sex, and the top ten PCs. All phenotyping analyses were conducted using R (version 3.6.2, http://www.R-project.org/) and STATA 15.0 (StataCorp. 2017. Stata Statistical Software: Release 15. College Station, TX: StataCorp LLC).
Replication of previously reported SNPs in GERA and MGB Biobank
To assess whether the previously described AK-associated loci replicated in the GERA cohort, we tested three susceptibility SNPs identified in the previous GWAS with a genome-wide level of significance or after multiple testing corrections28. We reported the replication analysis results in the discovery and validation cohorts.
Meta-analysis
We conducted a meta-analysis of AK combining the discovery and validation cohorts using the PLINK software package (-meta-analysis). The analysis of each contributing GWAS had been performed independently, and quality control included assessments for population stratification in each data set. Variants that are commonly observed in both discovery and validation cohorts were included in the meta-analysis. A combined (discovery-validation) fixed-effect meta-analysis was performed using a Mantel–Haenzel method with the genome-wide P-value significance threshold set at 5 × 10−8.
Genes and biological pathway prioritization
We calculated gene- and pathway-based p-values using the VEGAS2 software to prioritize genes and biological pathways14,15. A gene-based association analysis on the GWAS AK results of the GERA cohort was conducted using all variants assigned to a gene to compute gene-based P-value. Gene-based results were carried forward to run a pathway-based test. We analyzed the enrichment of the genes in 9,736 pathways or gene-sets (with 23,051 unique genes) derived from the Biosystem’s database (https://vegas2.qimrberghofer.edu.au/biosystems20160324.vegas2pathSYM). All significant results using the Benjamini–Hochberg FDR control procedure with FDR 0.1 were presented66.
SNP-based heritability
We used the LDSC software implemented in the LD Hub Web interface (http://ldsc.broadinstitute.org/ldhub/) for estimating array-heritability67. GWAS summary statistics of the GERA cohort were used to calculate heritability.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The GERA genotype data are available upon application to the KP Research Bank (https://researchbank.kaiserpermanente.org/). A subset of the GERA cohort consented for public use can be found at NIH/dbGaP: phs000674.v3.p3. The combined (GERA + MGB) meta-analysis GWAS summary statistics are available from the NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/downloads/summary-statistics), study accession number GCST90095184.
References
Goldenberg, G. & Perl, M. Actinic keratosis: update on field therapy. J. Clin. Aesthet. Dermatol. 7, 28–31 (2014).
Chetty, P., Choi, F. & Mitchell, T. Primary care review of actinic keratosis and its therapeutic options: a global perspective. Dermatol. Ther. (Heidelb.) 5, 19–35 (2015).
Dodds, A., Chia, A. & Shumack, S. Actinic keratosis: rationale and management. Dermatol. Ther. (Heidelb.) 4, 11–31 (2014).
Housman, T. S. et al. Skin cancer is among the most costly of all cancers to treat for the Medicare population. J. Am. Acad. Dermatol. 48, 425–429 (2003).
Siegel, J. A., Korgavkar, K. & Weinstock, M. A. Current perspective on actinic keratosis: a review. Br. J. Dermatol. 177, 350–358 (2017).
Roewert-Huber, J., Stockfleth, E. & Kerl, H. Pathology and pathobiology of actinic (solar) keratosis - an update. Br. J. Dermatol. 157, 18–20 (2007).
Berman, B. & Cockerell, C. J. Pathobiology of actinic keratosis: ultraviolet-dependent keratinocyte proliferation. J. Am. Acad. Dermatol. 68, S10–S19 (2013).
Jacobs, L. C. et al. IRF4, MC1R and TYR genes are risk factors for actinic keratosis independent of skin color. Hum. Mol. Genet. 24, 3296–3303 (2015).
Zhong, K., Nijsten, T. & Kayser, M. Pigmentation-independent susceptibility loci for actinic keratosis highlighted by compound heterozygosity analysis. J. Invest. Dermatol. 137, 77–84 (2017).
Flohil, S. C. et al. Prevalence of actinic keratosis and its risk factors in the general population: the Rotterdam Study. J. Invest. Dermatol. 133, 1971–1978 (2013).
Barreiro, L. B., Laval, G., Quach, H., Patin, E. & Quintana-Murci, L. Natural selection has driven population differentiation in modern humans. Nat. Genet. 40, 340–345 (2008).
Voight, B. F., Kudaravalli, S., Wen, X. & Pritchard, J. K. A map of recent positive selection in the human genome. PLoS Biol. 4, e72 (2006).
Genomes Project, C. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Mishra, A. & Macgregor, S. VEGAS2: software for more flexible gene-based testing. Twin Res. Hum. Genet. 18, 86–91 (2015).
Mishra, A. & MacGregor, S. A novel approach for pathway analysis of GWAS data highlights role of BMP signaling and muscle cell differentiation in colorectal cancer susceptibility. Twin Res. Hum. Genet. 20, 1–9 (2017).
Visconti, A. et al. Genome-wide association study in 176,678 Europeans reveals genetic loci for tanning response to sun exposure. Nat. Commun. 9, 1684 (2018).
Zhang, M. et al. Genome-wide association studies identify several new loci associated with pigmentation traits and skin cancer risk in European Americans. Hum. Mol. Genet. 22, 2948–2959 (2013).
Chahal, H. S. et al. Genome-wide association study identifies 14 novel risk alleles associated with basal cell carcinoma. Nat. Commun. 7, 12510 (2016).
Choquet, H., Ashrafzadeh, S., Kim, Y., Asgari, M. M. & Jorgenson, E. Genetic and environmental factors underlying keratinocyte carcinoma risk. JCI Insight 5, e134783 (2020).
Rocha, J. The evolutionary history of human skin pigmentation. J. Mol. Evol. 88, 77–87 (2020).
Visser, M., Palstra, R. J. & Kayser, M. Allele-specific transcriptional regulation of IRF4 in melanocytes is mediated by chromatin looping of the intronic rs12203592 enhancer to the IRF4 promoter. Hum. Mol. Genet. 24, 2649–2661 (2015).
Sulem, P. et al. Genetic determinants of hair, eye and skin pigmentation in Europeans. Nat. Genet. 39, 1443–1452 (2007).
Han, J. et al. A genome-wide association study identifies novel alleles associated with hair color and skin pigmentation. PLoS Genet. 4, e1000074 (2008).
Asgari, M. M. et al. Identification of susceptibility loci for cutaneous squamous cell carcinoma. J. Invest. Dermatol. 136, 930–937 (2016).
Chahal, H. S. et al. Genome-wide association study identifies novel susceptibility loci for cutaneous squamous cell carcinoma. Nat. Commun. 7, 12048 (2016).
Law, M. H. et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat. Genet. 47, 987–995 (2015).
Barrett, J. H. et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat. Genet. 43, 1108–1113 (2011).
Jacobs, L. C. et al. A Genome-Wide Association Study identifies the skin color genes IRF4, MC1R, ASIP, and BNC2 influencing facial pigmented spots. J. Investig. Dermatol. 135, 1735–1742 (2015).
Praetorius, C. et al. A polymorphism in IRF4 affects human pigmentation through a tyrosinase-dependent MITF/TFAP2A pathway. Cell 155, 1022–1033 (2013).
Chhabra, Y. et al. Genetic variation in IRF4 expression modulates growth characteristics, tyrosinase expression and interferon-gamma response in melanocytic cells. Pigment Cell Melanoma Res. 31, 51–63 (2018).
Chatzinasiou, F. et al. Comprehensive field synopsis and systematic meta-analyses of genetic association studies in cutaneous melanoma. J. Natl Cancer Inst. 103, 1227–1235 (2011).
Khoruddin, N. A., Noorizhab, M. N., Teh, L. K., Mohd Yusof, F. Z. & Salleh, M. Z. Pathogenic nsSNPs that increase the risks of cancers among the Orang Asli and Malays. Sci. Rep. 11, 16158 (2021).
Spichenok, O. et al. Prediction of eye and skin color in diverse populations using seven SNPs. Forensic Sci. Int. Genet. 5, 472–478 (2011).
Visser, M., Palstra, R. J. & Kayser, M. Human skin color is influenced by an intergenic DNA polymorphism regulating transcription of the nearby BNC2 pigmentation gene. Hum. Mol. Genet. 23, 5750–5762 (2014).
Liyanage, U. E. et al. Combined analysis of keratinocyte cancers identifies novel genome-wide loci. Hum. Mol. Genet. 28, 3148–3160 (2019).
Donnelly, M. P. et al. A global view of the OCA2-HERC2 region and pigmentation. Hum. Genet. 131, 683–696 (2012).
Ratushny, V., Gober, M. D., Hick, R., Ridky, T. W. & Seykora, J. T. From keratinocyte to cancer: the pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Investig. 122, 464–472 (2012).
Padilla, R. S., Sebastian, S., Jiang, Z., Nindl, I. & Larson, R. Gene expression patterns of normal human skin, actinic keratosis, and squamous cell carcinoma: a spectrum of disease progression. Arch. Dermatol. 146, 288–293 (2010).
Oliveira, W. R. P. et al. Skin lesions in organ transplant recipients: a study of 177 consecutive Brazilian patients. Int. J. Dermatol. 58, 440–448 (2019).
Infusino, S. D. et al. Cutaneous complications of immunosuppression in 812 transplant recipients: a 40-year single center experience. G Ital. Dermatol. Venereol. 155, 662–668 (2020).
Ulrich, C. et al. Topical immunomodulation under systemic immunosuppression: results of a multicentre, randomized, placebo-controlled safety and efficacy study of imiquimod 5% cream for the treatment of actinic keratoses in kidney, heart, and liver transplant patients. Br. J. Dermatol. 157, 25–31 (2007).
Jenni, D. & Hofbauer, G. F. Keratinocyte cancer and its precursors in organ transplant patients. Curr. Probl. Dermatol 46, 49–57 (2015).
Brown, P. J. et al. FOXP1 suppresses immune response signatures and MHC class II expression in activated B-cell-like diffuse large B-cell lymphomas. Leukemia 30, 605–616 (2016).
De Silva, P. et al. FOXP1 negatively regulates tumor infiltrating lymphocyte migration in human breast cancer. EBioMedicine 39, 226–238 (2019).
Rosenberg, A. R. et al. Skin cancer precursor immunotherapy for squamous cell carcinoma prevention. JCI Insight 4, e125476 (2019).
Cunningham, T. J. et al. Randomized trial of calcipotriol combined with 5-fluorouracil for skin cancer precursor immunotherapy. J. Clin. Investig. 127, 106–116 (2017).
Liu, F. et al. Genetics of skin color variation in Europeans: genome-wide association studies with functional follow-up. Hum. Genet. 134, 823–835 (2015).
Morgan, M. D. et al. Genome-wide study of hair colour in UK Biobank explains most of the SNP heritability. Nat. Commun. 9, 5271 (2018).
Denny, J. C. et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 31, 1102–1110 (2013).
Ioannidis, N. M. et al. Gene expression imputation identifies candidate genes and susceptibility loci associated with cutaneous squamous cell carcinoma. Nat. Commun. 9, 4264 (2018).
Sarin, K. Y. et al. Genome-wide meta-analysis identifies eight new susceptibility loci for cutaneous squamous cell carcinoma. Nat. Commun. 11, 820 (2020).
Di Zenzo, G., Amber, K. T., Sayar, B. S., Muller, E. J. & Borradori, L. Immune response in pemphigus and beyond: progresses and emerging concepts. Semin. Immunopathol. 38, 57–74 (2016).
Nguyen, V. T., Ndoye, A. & Grando, S. A. Pemphigus vulgaris antibody identifies pemphaxin. A novel keratinocyte annexin-like molecule binding acetylcholine. J. Biol. Chem. 275, 29466–29476 (2000).
Amos, C. I. et al. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Hum. Mol. Genet. 20, 5012–5023 (2011).
Cohen, O. G., Margolis, D. J. & Wehner, M. R. The validity of diagnostic and treatment codes for actinic keratosis in electronic health records. Br. J. Dermatol. 182, 1487–1488 (2020).
Hoffmann, T. J. et al. Next generation genome-wide association tool: design and coverage of a high-throughput European-optimized SNP array. Genomics 98, 79–89 (2011).
Kvale, M. N. et al. Genotyping informatics and quality control for 100,000 subjects in the genetic epidemiology research on adult health and aging (GERA) cohort. Genetics 200, 1051–1060 (2015).
Banda, Y. et al. Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the genetic epidemiology research on adult health and aging (GERA) cohort. Genetics 200, 1285–1295 (2015).
Delaneau, O., Marchini, J. & Zagury, J. F. A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2011).
Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. & Abecasis, G. R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955–959 (2012).
Howie, B., Marchini, J. & Stephens, M. Genotype imputation with thousands of genomes. G3 (Bethesda) 1, 457–470 (2011).
Marees, A. T. et al. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 27, e1608 (2018).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Weiss, S. T. & Shin, M. S. Infrastructure for personalized medicine at partners healthCare. J Pers Med 6, 13 (2016).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Glickman, M. E., Rao, S. R. & Schultz, M. R. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J. Clin. Epidemiol. 67, 850–857 (2014).
Zheng, J. et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33, 272–279 (2017).
Acknowledgements
We thank Dr. Sae Kyu Lee for programming support, and Dr. Wenyu Song, Dr. Lu Chen Weng, and Dr. Hao Limin for statistical support. This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (K24 AR069760 to MA). Data used in this study were provided by the Kaiser Permanente Research Bank (KPRB) from the KPRB collection, which includes the Kaiser Permanente Research Program on Genes, Environment, and Health (RPGEH) and the Genetic Epidemiology Research on Adult Health and Aging (GERA), funded by the National Institutes of Health (RC2 AG036607), the Robert Wood Johnson Foundation, the Wayne and Gladys Valley Foundation, The Ellison Medical Foundation, and the Kaiser Permanente Community Benefits Program. H.C. and M.M.A. are also supported by the National Cancer Institute (NCI) R01CA2416323.
Author information
Authors and Affiliations
Contributions
Y.K. contributed to the conception of the study, acquisition, analysis, interpretation of data, and drafting and revising the work. J.Y. contributed to the analysis and interpretation of data and revising the work. H.H. contributed to the conception of the study, interpretation of data, and revising the work. E.J. contributed to the conception of the study, acquisition, interpretation of data, and revising the work. H.C. contributed to the conception of the study, interpretation of data, drafting, and revising the work. M.M.A. contributed to the conception of the study, acquisition, interpretation of data, drafting and revising the work, and supervision of the study.
Corresponding authors
Ethics declarations
Competing interests
H.C. is an Editorial Board Member for Communications Biology, but was not involved in the editorial review of, nor the decision to publish this article. The authors declare no other competing interests.
Peer review
Peer review information
Communications Biology thanks Luba Pardo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Chiea Chuen Khor and Christina Karlsson Rosenthal. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, Y., Yin, J., Huang, H. et al. Genome-wide association study of actinic keratosis identifies new susceptibility loci implicated in pigmentation and immune regulation pathways. Commun Biol 5, 386 (2022). https://doi.org/10.1038/s42003-022-03301-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-022-03301-3
This article is cited by
-
Advances in cutaneous squamous cell carcinoma
Nature Reviews Cancer (2023)
-
Compatibility between snails and schistosomes: insights from new genetic resources, comparative genomics, and genetic mapping
Communications Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
