
Abstract
Adverse environmental and pathophysiological situations can overwhelm the biosynthetic capacity of the endoplasmic reticulum (ER), igniting a potentially lethal condition known as ER stress. ER stress hampers growth and triggers a conserved cytoprotective signaling cascade, the unfolded protein response (UPR) for ER homeostasis. As ER stress subsides, growth is resumed. Despite the pivotal role of the UPR in growth restoration, the underlying mechanisms for growth resumption are yet unknown. To discover these, we undertook a genomics approach in the model plant species Arabidopsis thaliana and mined the gene reprogramming roles of the UPR modulators, basic leucine zipper28 (bZIP28) and bZIP60, in ER stress resolution. Through a network modeling and experimental validation, we identified key genes downstream of the UPR bZIP-transcription factors (bZIP-TFs), and demonstrated their functional roles. Our analyses have set up a critical pipeline for functional gene discovery in ER stress resolution with broad applicability across multicellular eukaryotes.
Similar content being viewed by others
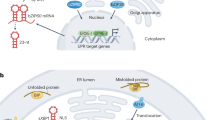


Introduction
Specialized ER membrane-associated sensors perceive ER stress and control growth in physiological conditions and ER stress-causing conditions. The ribonuclease and kinase inositol requiring enzyme 1 (IRE1) is the most conserved ER stress sensor1,2. IRE1 splices the mRNA of a bZIP-transcription factor (TF) (Xbp1 in mammals3,4,5, bZIP60 in plants6, and Hac1 in yeast7). The spliced bZIP-TF mRNA is then translated into an active TF that modulates the expression of UPR genes in the nucleus8,9,10. In yeast, the UPR depends solely on Ire1p; however, in metazoans and plants, the UPR has expanded to additional arms and components within each arm11,12,13,14, likely to suit the complexity of tissue development and growth of multicellular eukaryotes. The additional UPR arm conserved in metazoans and plants relies on ER membrane-tethered bZIP-TFs, namely the Activating Transcription Factor 6 (ATF6) in metazoans15 and bZIP28 in plants1. Upon stress, these bZIP-TFs are transported to the Golgi apparatus for proteolytic activation and release of the TF domain, which is then translocated into the nucleus to control UPR gene expression16,17. The UPR bZIP-TFs bind to promoters of UPR target genes via ER stress-responsive cis-regulatory elements (CREs), which are generally conserved15,18,19,20. The DNA binding of the UPR bZIP-TFs can also be modulated by other TFs and transcriptional regulators21,22,23, underscoring an elevated complexity in the regulation of UPR gene expression.
In the early phase of ER stress responses, the ER stress sensors activate the adaptive phase of the UPR, which involves the production of ER chaperones and foldases to restore the biosynthetic capacity of the ER2,24. During ER stress, growth is reduced25,26,27, but, as ER stress is mitigated, growth is resumed28,29,30. Successful restoration of growth during ER stress recovery requires an intact UPR. Indeed, during ER stress recovery, the loss of either bZIP28 or bZIP60 partially hampers organ growth, specifically the root30,31,32. Noticeably, either bZIP28 or bZIP60 is not essential for seedling growth in physiological conditions nor in persistent ER stress conditions23,30,31,32. Therefore, these UPR bZIP-TFs are individually necessary for growth during ER stress recovery. Although genetic evidence poses that the growth mechanisms controlled by bZIP28 and bZIP60 in ER stress recovery are partially overlapping30, their identity is yet completely unknown. The identification of the effectors that control growth in dependence of the UPR bZIP-TFs would contribute to the understanding of the interplay of stress responses and the growth of multicellular organisms at a mechanistic level. In this work, we addressed this knowledge gap and focused on identifying and characterizing the downstream effectors of the UPR bZIP-TFs in organ growth during ER stress recovery through innovative integration of large-scale time-series RNA sequencing (RNA-seq), chromatin immunoprecipitation sequencing (ChIP-seq), and cistrome analyses into gene regulatory network modeling in the model plant species Arabidopsis thaliana (hereafter Arabidopsis).
Results
bZIP28 and bZIP60 are required for root growth during recovery from ER stress
As a first step to determine growth effectors in ER stress resolution, we sought to set up experimental conditions of recovery from ER stress caused by tunicamycin (Tm), a widely used ER stress-inducing agent, which is highly effective in plant cells33. We used DMSO (Tm-solvent) as mock control. We exposed Arabidopsis Col-0 seedlings to Tm for 6 h and then either transferred them to growth media containing no Tm for ER stress recovery or continuously exposed them to Tm to elicit adaptive ER stress responses (Fig. 1a). Using a high-resolution LC/MS/MS on our samples, we verified a significant decrease of Tm levels during the ER stress recovery time-course (75.4% decrease at 24 h of recovery compared to 0 h) and increased levels of Tm in adaptive ER stress conditions (Fig. 1b). In the progression of ER stress recovery, the levels of induction of spliced bZIP60 (sbZIP60) transcripts, a well-established biomarker for UPR activation10,25, were substantially attenuated (Fig. 1c). Conversely in adaptive ER stress conditions, the sbZIP60 levels were only mildly reduced at 12 and 24 h compared to the 6 h of Tm treatment. These results indicate that we successfully set up the experimental conditions for a time-course of ER stress recovery in which the UPR is significantly attenuated. Based on these experimental premises, we quantitatively followed the growth of Col-0 and single and double loss-of-function mutants of the UPR bZIP-TFs (i.e., bzip28-216, bzip60-210, and bzip28-2 bzip60-134) in an extended time-course of ER stress recovery (Fig. 1d, e and Supplementary Fig. 1a, b). While bzip28-2 bzip60-1 did not show growth recovery, consistent with previous findings21,30, bzip28-2 and bzip60-2 reestablished growth: the total biomass of bzip60-2 was 72% compared to Col-0, and was significantly higher than that of bzip28-2 and bzip28-2 bzip60-1. We observed a significantly higher relative growth rate (Tm/DMSO) of the primary root of bzip60-2 compared to bzip28-2, yet lower compared to Col-0. In physiological conditions, these mutants did not show significant differences in root growth, while IRE1 loss-of-function mutations showed significant root growth defects (Supplementary Fig. 1c, d), as reported earlier26,34. These results support the notion that bZIP28 and bZIP60 are primarily required for the actuation of partially overlapping genetic pathways for root growth in ER stress recovery conditions30,31,32.

a Scheme of the experimental design for ER stress recovery assay. Seedlings were germinated in normal growth conditions for 5 days and then treated for 6 h with either Tm or DMSO (mock control). Following the drug treatment, seedlings were transferred to drug-free growth media to allow ER stress to subside and growth to restore. b Quantitative LC/MS/MS analysis of Tm contents in Col-0 whole seedlings subjected to 6 h of Tm (or DMSO) treatment followed by ER stress recovery or extended ER stress (adaptive ER stress) as described in (a). n (number of biological replicates) = 3 (~50 seedlings per replicate). c Relative expression levels of sbZIP60 (Log2[Tm/DMSO]) at each time-point under each condition. n = 3 (12 seedlings per replicate). d Relative growth rate of the primary root of Col-0, bzip28-2, bzip60-2, and bzip28-2 bzip60-1 in ER stress recovery as described in (a). The relative growth rate of the primary root was monitored during ER stress recovery. n = 5 (6 seedlings per replicate). e Representative images of the primary root of Col-0, bzip28-2, bzip60-2, and bzip28-2 bzip60-1 in ER stress recovery taken at 8 days in recovery from ER stress. Scale bar (black line) = 1.4 cm. *P < 0.05, **P < 0.01, ***P < 0.001 (two-tailed Student’s t-test). The source data of b–d is provided in Supplementary Data 6.
bZIP28 and bZIP60 initiate broad gene reprogramming in ER stress recovery
We next aimed to identify the downstream effectors of bZIP28 and bZIP60 in growth restoration during ER stress resolution using a genomics approach. We reasoned that the initial phase of ER stress recovery would be critical to actuate transcriptional regulations by bZIP28 and bZIP60 to restore organ growth. Therefore, we performed RNA-seq analyses in Col-0, bzip28-2, and bzip60-2 collected at 0, 12, and 24 h of ER stress recovery, using the corresponding DMSO-treated samples as additional controls in each genotype at each time-point (Supplementary Data 1). From a highly reproducible dataset (Supplementary Fig. 2a, b), we identified differentially expressed genes (DEGs) in each genotype at each time-point (Fig. 2a and Supplementary Data 2). The number of DEGs was relatively low at 0 h (576, Col-0; 629, bzip28-2; 552, bzip60-2) but substantially increased at 12 h, and was significantly higher in both bzip28-2 (4076; P = 1.3 × 10−10) and bzip60-2 (4254; P = 3.2 × 10−10) compared to Col-0 (2484). Although the number of DEGs (2930) in Col-0 increased at 24 h compared to 12 h, it decreased in bzip28-2 (3162) and in bzip60-2 (2316) at 24 h to significantly different levels compared to Col-0 (bzip28-2, P = 6.7 × 10−15; bzip60-2, P = 6.6 × 10−15). About 20% of the DEGs were common to the bzip28-2 and bzip60-2 backgrounds and 6–7% were unique to each background (Supplementary Fig. 2c).

a Numbers of DEGs in each genotype at each time-point (|Log2FC| > 1, Adjusted P > 0.05). Statistical differences of the number change between each mutant and Col-0 were calculated by Fisher’s exact test and indicated in the main text. b Numbers of DDEGs at each time-point. c, d Temporal dynamic enrichments of CREs on 1-kb promoters of DDEGs. The upper heatmap (c) shows enrichments of the canonical ER stress-responsive CREs on the promoters of DDEGs. ERSE-I, CCAAT-N10-CACG. ERSE-II, ATTGG-N2-CACG. UPRE-I, TGACGT-GG/A, UPRE-II, GATGACGCGTAC. UPRE-III, TCATCG. The lower heatmap (d) shows enrichments of de novo motifs on the promoters of DDEGs. The motifs are displayed on the left of each row. The best match to a transcription factor family for each motif is displayed to the right of each row. The median number of DDEGs across all pairwise comparisons is 201, and we randomly selected 201 DEGs and included the analysis as a control set. This analysis revealed multiple TF motifs significantly enriched. The source data is provided in Supplementary Data 6.
We observed that some of the UPR biomarker genes were regulated in a genotype-dependent manner (Supplementary Fig. 2d–f). For example, while substantially induced in all genotypes, the expression induction of ER-resident J protein 3B (ERdj3B) was significantly attenuated in bzip28-2. Therefore, we next performed genome-scale pairwise comparisons (two-tailed Student’s t-test, P < 0.01) of expression Log2FC (Tm/DMSO) of each DEG (a total of 6670 genes) at each time-point across the genotypes (Col-0 vs. bzip28-2, Col-0 vs. bzip60-2, and bzip28-2 vs. bzip60-2) (Supplementary Data 2). Overall, we identified 875 and 904 DEGs regulated in a bZIP28- and the bZIP60-dependent manner in at least one time-point, respectively (Fig. 2b). In addition, 362 DEGs were significantly different between bzip28-2 and bzip60-2. Hereafter, we called these genotype-dependently regulated DEGs as differentially-differentially expressed genes (DDEGs). Intriguingly, the number of DDEGs was relatively small at 0 h (50, bZIP28-dependent; 52, bZIP60-dependent), but, for bZIP28 and bZIP60 respectively, it increased to 645 and 653 at 12 h and declined to 216 and 218 at 24 h. Thus, the responses of genes to the lack of functional bZIP28 or bZIP60 followed a temporal and partially non-overlapping profile during ER stress recovery.
The observed gene responses were most likely controlled by activities of two canonical CREs, the ER stress-responsive element-I (ERSE-I)15 and unfolded protein response element-I (UPRE-I)18, as supported by the evidence that these CREs were predominantly enriched in the promoters of DDEGs compared to the other UPR CREs (i.e., ERSE-II35, UPRE-II20, and UPRE-III12) (Fig. 2c). Through de novo motif analyses, we also identified a variety of TF motifs temporally enriched in the gene promoters (Fig. 2d). For example, the binding motif (ACAAAAAAAA) of C2H2 Zinc Finger TF Indeterminate (ID)-Domain11 (IDD11) was highly enriched in promoters of both bZIP28- and bZIP60-dependently regulated genes (i.e., Col-0 vs. bzip28-2; Col-0 vs. bzip60-2) at 12 h, indicating that IDD11 could directly bind to promoters of these DDEGs. Also, the binding motif (5′-CCGGCG-3′) of an ERF/AP2 TF (AT3G58630) was exclusively enriched in promoters of bZIP28-dependently regulated genes while the binding motif (5′-GAAGAAGAAGA-3′) of NTM1-LIKE 8 (NTL8), a NAM, ATAF1/2, and CUC (NAC) TF, was enriched in the promoter of bZIP60-dependently regulated genes at 12 h. The enrichment of these CREs in the gene promoters at 12 h strongly suggests that the regulatory dynamics of bZIP28 and bZIP60 may be associated with activities of other TFs that could operate as coregulators or downstream of bZIP28 and/or bZIP60 by bindings to non-canonical UPR CREs.
To funnel the results obtained thus far towards the identification of genes involved in organ growth in dependence of bZIP28 and/or bZIP60 during ER stress recovery, we performed a weighted gene coexpression network analysis (WGCNA)36, which categorized the 6670 DEGs based on expression similarity across time-points and genotypes (Fig. 3a). This approach enabled us to identify 14 coexpression modules that were color-coded and ranged in size from 25 (light green) to 890 genes (turquoise). Then, to identify the biological functions of each coexpression module we performed Gene Ontology (GO) enrichment analysis and compared the GO categories enriched in each coexpression module (Fig. 3b and Supplementary Data 3). Each coexpression module exhibited strong enrichment of distinct GO categories associated with important biological processes such as metabolic pathways, hormone responses, and stress responses, pointing to an extensive level of the biological relevance of gene regulation in the UPR. Among them, the pink module (354 genes) showed a unique expression profile of the eigengene in each genotype (i.e., increased expression in Col-0 over time but variable expression in bzip28-2 and bzip60-2 at different levels), and importantly, it exhibited exclusive enrichment of root development-related pathways (Fig. 3b). The genotype-specific expression of the eigengene and exclusive enrichment of the GO terms associated with root development (several of the annotations have been experimentally validated12,37,38) in the pink model are in the line with the non-completely overlapping growth phenotype of the primary root in bzip28-2 and bzip60-2 (Fig. 1d, e and Supplementary Fig. 1b). Therefore, we decided to focus on the pink module for functional characterization.

a All DEGs identified were subjected to WGCNA to detect modules of coexpressed genes based on the expression similarity. Each graph shows the expression of the module eigengene, which can be considered as the representative expression in the respective coexpression module. The y-axis indicates Log2 expression values relative to the mean expression across all samples. The x-axis indicates time-points in each genotype. The modules are named according to their color created through WGCNA. The number of genes in each module is in parentheses. b GO analysis of genes in each coexpression module (adjusted P < 0.05, Hypergeometric test). The respective module is indicated by the color bar above the heatmap. GO terms strongly enriched in genes of the pink module are marked by a pink box. A full list of GO terms is provided in Supplementary Data 3. The source data is provided in Supplementary Data 6.
Gene regulatory network construction based on coexpression networks coupled with DNA-binding profiles
We next aimed to establish the in vivo temporal DNA-binding interactomes of bZIP28 and bZIP60 as a strategy to identify direct targets of these TFs in ER stress recovery. To do so, we performed ChIP-seq analyses at 0, 12, and 24 h of ER stress recovery in stable transgenic lines (Col-0 background) expressing a green fluorescent protein (GFP)-tagged fusion to either bZIP28 (35Spro:GFP-bZIP28/Col-0)39 in which the transcripts of bZIP28 were not overexpressed compared to Col-0 (rather constitutively expressed in both mock and Tm treatments) or a bZIP60 in bzip60-2 (bZIP60pro:GFP-bZIP60/bzip60-2)40, which successfully rescued the root the phenotype and gene expression profiles of bzip60-2 (Supplementary Fig. 3a–d). ChIP-quantitative PCR (ChIP-qPCR) results showed that GFP-bZIP28 and GFP-bZIP60 bound to ERSE-I15,18, which is present in the promoter regions of BiP3 and ERdj3B (fragments A and B; Supplementary Fig. 3e, f) specifically in Tm treatment, but not in DMSO treatment. This observation is in line with the notion that bZIP28 and bZIP60 operate in the first layer of the UPR upon ER stress-specific protease cleavage16 and unconventional splicing by IRE110, respectively, for activation as TFs. However, these TFs did not bind to an Erdj3B promoter region without the ERSE-I (fragment C) or in the ChIP mock controls (i.e., no antibody). These DNA-binding activities were unlikely to be driven by the GFP tag. This is supported by the evidence from ChIP assays in a transgenic line expressing a yellow fluorescent protein (YFP, a variant of GFP with three-point mutations in the chromophore)41. Specifically, YFP was enriched on the three loci only at background levels despite a constitutive YFP expression, which was 42 times higher than the expression of endogenous sbZIP60 in Tm-treated conditions (Supplementary Fig. 3f, g). Although bZIP28 was expressed at similar levels in 35Spro:GFP-bZIP28/Col-0 and Col-0 (Supplementary Fig. 3a), we could not exclude a possibility that the constitutive expression of bZIP28 may lead to some spurious binding of bZIP28 to the genome. Thus, we subjected our results to a rigorous filtering protocol using an analysis pipeline developed based on existing guidelines42,43 with a key modification: any peaks found in both Tm-treated samples and the corresponding DMSO-treated samples, which were processed in parallel, were removed (see Methods). This step was added in the analysis pipeline to ensure that the resulting bZIP28 binding peaks were specific to Tm treatment. Using this approach, we captured a high-confident set of 676 and 467 UPR-specific peaks of bZIP28 and bZIP60, respectively, the majority of which were located in the proximal gene promoters (i.e., within 1-kb of the transcription start site) (Supplementary Fig. 4a, b), consistent with the binding behavior of other TFs in Arabidopsis44. We observed temporal changes of bZIP28- and bZIP60-binding signals (Input-normalized ChIP-seq signals), whereby an initial binding peak at 0 h was followed by a substantial decline at 12 h, and another peak at 24 h of ER stress recovery (Supplementary Fig. 4c, d). De novo motif analysis identified that ERSE-I was one of the top-scoring motifs (E-value = 3.62 × 10−169 in bZIP28 UPR-specific binding peaks; E value = 7.1 × 10−103 in bZIP60 UPR-specific binding peaks), and that ERSE-I was centered at the summit of these binding peaks (Supplementary Fig. 4e, f), validating the high quality of our data. Intriguingly, the analysis also found other TF-binding motifs exclusively enriched in either bZIP28 or bZIP60 UPR-specific binding peaks (Supplementary Fig. 4g–i). The results support the notion that ERSE-I is the canonical ER stress-responsive CRE acting for DNA-binding activities of both bZIP28 and bZIP606,19, but also reveal potential coregulators exclusive to either bZIP28 or bZIP60 in ER stress recovery. We then mapped bZIP28 and bZIP60 UPR-specific binding peaks to the potential target genes and found a total of 440 bZIP28- and 356 bZIP60-bound genes (Supplementary Fig. 4g and Supplementary Data 4). Among these genes, 162 genes were bound by both bZIP28 and bZIP60 (significant overlap validated by hypergeometric test, P = 7.7 × 10−203), including BiP3 and Erdj3B, and showed a strong enrichment of GO terms associated with ER stress. Interestingly, the genes exclusively bound by bZIP28 were enriched with GO terms associated with biotic stress-related processes. In contrast, the genes exclusively bound by bZIP60 were enriched with GO terms associated with abiotic stress-related processes (Supplementary Data 5). Overall, these results are consistent with a partially overlapping function of bZIP28 and bZIP60 in ER stress recovery. Importantly also, they provide genome-scale evidence that bZIP28 and bZIP60 not only bind to known genes involved in ER stress responses but also that they interact with genes involved in non-completely overlapping stress pathways during ER stress recovery.
To couple the calculated coexpression network connectivity (i.e., the strength of interaction) with a regulatory relationship to bZIP28 and bZIP60, the DNA-binding profiles of bZIP28 and bZIP60 were integrated into the pink module, revealing that the pink module consists of two subnetworks (hereafter named as pink-sub1 and pink-sub2) (Fig. 4a). In pink-sub1, bZIP28 and bZIP60 bound only to the promoters of three genes and one gene, respectively, supporting the possibility that the sub-module includes 239 genes indirectly regulated by bZIP28 and bZIP60. In contrast, 25% of genes in pink-sub2 were bound exclusively by bZIP60, supporting the likelihood of a higher level of direct regulation by bZIP60 of the genes in this sub-module compared to pink-sub1. Therefore, the ChIP-seq results support the RNA-seq data that, although the entire pink module is controlled by both UPR bZIP-TFs, each of these bZIP-TF contributes to controlling the expression of the genes in each subnetwork to a different extent (Fig. 3a). Consistent with these observations, most genes in pink-sub1 were downregulated while pink-sub2 genes were predominantly upregulated during ER stress recovery (Fig. 4b), supporting distinctive gene regulation signature between pink-sub1 and pink-sub2. Then to experimentally characterize pink-sub1 and pink-sub2, as a first step we used a stringent protocol to predict the regulatory-hub genes (i.e., the genes that are most connected with other genes in a network)45 in each subnetwork. We considered two main criteria: ranking in the top ≥10% of network degree (i.e., the number of connections) and having a known phenotype of loss-of-function mutations or expected role in roots, cell development, and/or ER stress processes. According to these criteria, out of the genes in the top ≥10% of network degree (out of 242 genes used for the network construction), we selected Can of Worms1 (COW1) (ranked in the top 4.0%), which encodes a phosphatidylinositol transfer protein essential for root hair tip growth37,46, and root cell wall formation-related Leucine-rich repeat/Extensin1 (LRX1)37,47 (ranked in the top 1.6%), as major hub genes in pink-sub1. We also selected the Integral membrane Yip1 family protein 1a (YIP1a), whose yeast and mammalian homologs function in ER-Golgi transport48,49, with the highest network degree in pink-sub2 (out of 43 genes used for the network construction). Noticeably in pink-sub2, our ChIP-seq analysis identified a known root growth-related direct target of bZIP60, NAC-domain containing protein103 (NAC103)12 (ranked in the top 9.3%), as being bound by bZIP60 (Supplementary Data 4), lending support to the validity of our approach.

a A gene regulatory network in the pink module was constructed based on the coexpression network connectivity coupled with the ChIP-seq data of bZIP28 and bZIP60. The node size indicates the degree of coexpression connections. Edge thickness indicates network weight, which indicates connection strength. Pink-sub1 and 2 were indicated by blue and red circles, respectively. b Expression Log2FC of genes that belong to pink-sub1 or pink-sub2 in ER stress recovery. Genes were hierarchically clustered on the expression pattern. Candidates of regulatory-hub genes are indicated in the heatmaps. The source data of b is provided in Supplementary Data 6.
Functional characterization of the gene regulatory network in ER stress recovery
Next, except for NAC103 for which a null T-DNA allele is not available, we isolated the respective loss-of-function T-DNA mutants of COW1 (cow1-3), LRX1 (lrx1-4), and Yip1a (yip1a-1 and yip1a-2), which we confirmed being null alleles (Supplementary Fig. 5a–d). We then performed ER stress recovery assays to measure the relative growth (Tm-treated/DMSO-treated) of the primary root (Fig. 5a). In physiological conditions of growth, the primary root growth of these mutants and Col-0 was similar (Supplementary Fig. 6). In conditions of ER stress recovery, while lrx1-4 showed no growth alteration, cow1-3, yip1-1, and yip1a-2 exhibited a significant reduction in the relative growth rate of the primary root compared to Col-0 (Fig. 5a). Although the reduction was not as severe as bzip28-2 or bzip60-2, likely due to functional redundancy with other genes within the pink module, these results experimentally validate that, among the pink module genes, COW1, which is bound by neither bZIP28 or bZIP60 and YIP1a, which is bound exclusively by bZIP60, have an important role in root growth in recovery from ER stress. We next aimed at consolidating these observations by testing an epistatic relationship of COW1 and YIP1a with bZIP28 and bZIP60 in ER stress recovery. Therefore, we generated the following high-order mutants: cow1-3 bzip28-2, cow1-3 bzip60-2, yip1a-1 bzip28-2, yip1a-1 bzip60-2, and cow1-3 bzip28-2 bzip60-1. The yip1a-1 bzip28-2 bzip60-1 triple mutant was not recovered due to lethality. We tested the ability of the high-order mutants to reestablish root growth in recovery from ER stress. Consistent with an epistatic relationship with bZIP28 and bZIP60, we found that cow1-3 bzip28-2 and yip1a-1 bzip28-2 displayed a significantly higher relative root growth recovery compared to bzip28-2. Furthermore, the growth defect of bzip60-2 was suppressed by the cow1 mutation (cow1-3 bzip60-2) and yip1a mutation (yip1a-1 bzip60-2). A direct comparison of the genetic effects between cow1-3 and yip1a-1 on pink-sub1 and pink-sub2, respectively, is not possible because the relative root growth defect of bzip60-2 (Tm-treated/DMSO-treated, 0.204) is not as severe as bzip28-2 (Tm-treated/DMSO-treated, 0.076) and the network size of pink-sub2 (43 genes) is substantially smaller than pink-sub1 (242 genes). Nonetheless, our epistasis results are consistent with our network predictions that bZIP28 is more tightly associated with pink-sub1, in which COW1 appears as a regulatory-hub, compared to pink-sub2, and that pink-sub2 depends largely on bZIP60 (Fig. 4a).

a Relative growth of primary root of T-DNA mutants of the selected regulatory-hub gene candidate (cow1-3, yip1a-1, yip1a-2, and lrx1-4) and their higher-order mutants crossed with bzip28-2, bzip60-2, or bzip28-2 bzip60-1 in ER stress recovery as described in Fig. 1a. n = 3–5 (6 seedlings per replicate). b Suppression of bzip28-2 bzip60-1 by cow1-3 in chronic ER stress. Seeds germinated directly on the growth media in split plates of which one half contained Tm (15, 25, or 50 ng/mL) while the other half contained DMSO only (the corresponding mock control). The relative growth rate was measured 11 days after growth. n = 4–5 (3–5 seedlings per replicate). Scale bar (black line) = 1.2 cm. In boxplots, each box is bounded by the lower and upper quartiles from which the whisker extends 1.5× the interquartile range, and central bars represent the medians. Representative pictures of 50 ng/mL Tm plates are shown at the right. *P < 0.05, **P < 0.01, ***P < 0.001 (two-tailed Student’s t-test). The source data is provided in Supplementary Data 6.
Because cow1-3 recovered the root growth phenotype linked to the individual loss of bZIP28 and bZIP60 in ER stress recovery conditions, we next aimed to test whether the loss of COW1 could recover the sub-lethal phenotype of the bzip28-2 bzip60-1 double mutant. We found that the cow1-3 bzip28-2 bzip60-1 triple mutant partially recovered the sub-lethality of bzip28-2 bzip60-1 mutant in ER stress conditions, further implying that COW1 functions in regulating root growth through the process regulated by bZIP28 and bZIP60 (Fig. 5a). We then extended these findings to test the role of COW1 in chronic ER stress conditions (direct germination and growth on Tm-containing growth media) (Fig. 5b). In chronic ER stress, bZIP28 and bZIP60 redundantly regulate survival strategies (i.e., normal growth in bzip28-2 and bzip60-2, but lethality in bzip28-2 bzip60-1)13,30. Consistently, the relative growth rate of bzip28-2 bzip60-1 primary root was significantly reduced compared to Col-0 under all conditions tested, including 50 ng/mL Tm-containing media on which the root growth of bzip28-2 bzip60-1 was stunted soon after germination. Conversely, cow1-3 bzip28-2 bzip60-1 showed significantly higher relative root growth than bzip28-2 bzip60-1 in these conditions, demonstrating a remarkable genetic suppression of the bzip28-2 bzip60-1 ER stress phenotype by cow1-3 also in chronic ER stress. Taken together, our network analysis functionally characterized the temporal dynamics of gene regulation by bZIP28 and bZIP60 for reestablishing root growth in ER stress recovery and identified key effector genes acting, directly and indirectly, downstream of these UPR bZIP-TFs.
Discussion
ER stress resolution is critical for the survival of the cell and the growth of the entire organism. Owing mostly to the fact that functional studies of the UPR have been mainly conducted in unicellular organisms and cultured cells2,50, the fundamental question of how tissue growth is controlled by the UPR is still largely unanswered. In this work, we undertook a multi-omics approach based on combining RNA-seq, ChIP-seq, and cistrome analyses into functional gene network analyses in Arabidopsis. Our approach has allowed dissecting the gene regulatory networks operated by bZIP28 and bZIP60 and identified their downstream genes that are functionally involved in organ growth during ER stress recovery, paving the ground to UPR studies also in other multicellular organisms.
Earlier studies indicated that individually bZIP28 and bZIP60 are functionally redundant in gene expression under adaptive UPR28 and in survival to chronic ER stress26,34. However, a quantitative analysis on a handful of UPR genes in adaptive UPR suggested the existence of an overlapping transcriptional regulation by bZIP28 and bZIP60, and a different degree of dependence of gene expression on either UPR bZIP-TF30. The same study also indicated that in conditions of ER stress recovery the bzip28-2 and bzip60-2 loss-of-function mutants had reduced growth restoration of the primary root compared to Col-0, implying that these UPR bZIP-TFs control the expression of overlapping and unique genes involved in stress recovery and associated organ growth restoration. Our work extends these observations by providing experimental evidence for partially overlapping gene regulatory activities of bZIP28 and bZIP60 on a genome-wide scale. In addition, we established that bZIP28 and bZIP60 temporally regulate gene-regulatory modules for distinct biological processes in the progress of ER stress resolution. Such a temporal gene regulation is likely achieved by the combinatorial effects of shared and unique regulatory activities of bZIP28 and bZIP60 with other TFs or transcriptional regulators. This hypothesis is supported by multiple lines of evidence. First, non-canonical UPR CREs were significantly enriched in the promoters of DDEGs (Fig. 2d), indicating that most likely bZIP28 and bZIP60 directly regulate other TFs that regulate genes in the pink module. Indeed, 16 genes among 354 pink module genes encode TFs, suggesting potential transcriptional cascade effects. Another possibility is that bZIP28 and bZIP60 generate an environment or conditions in which the expression of these genes is activated (e.g., disruption of metabolic signaling). Second, Our characterization of the UPR-specific binding peaks of bZIP28 and bZIP60 showed an exclusive enrichment of de novo motifs statistically similar with DNA-binding motifs of TFs involved in other stress pathways (e.g., MYB96: abscisic acid signaling51, WRKY75: phosphate deprivation and senescence52,53,54, PHL4: phosphate starvation55). It is intriguing that not only the genes encoding MYB96 and WRKY75, which were predicted to be coregulators of bZIP28 (Supplementary Fig. 4g, h), were differentially regulated during ER stress, their expression changes were even significantly stronger in bzip60-2 (Supplementary Fig. 4i). Moreover, the UPR-specific binding peaks of both bZIP28 and bZIP60 had a strong enrichment of ERSE-I, for the binding of both UPR bZIP-TFs, as well as an identical temporal enrichment pattern of bZIP28- and bZIP60-binding signals (Supplementary Fig. 4c–f). We also found distinctive GO terms enriched for the exclusive binding targets of the two UPR bZIP-TFs (i.e., biotic stress for bZIP28 and abiotic stress for bZIP60) (Supplementary Fig. 4j). Therefore, our findings suggest that bZIP28 and bZIP60 interface with gene regulation in the UPR and other stress pathways potentially via co-regulation with other TFs. We speculate that this may facilitate resource allocations from other pathways to respond to ER stress and restore growth in specific growth conditions. Activation of the plant UPR in response to biotic and biotic stress has been verified56,57,58,59, but the underlying signaling pathways are largely unknown. The identification of TFs operating in other stress pathways in recovery from ER stress presented in this work provides the opportunity to define the intersection of ER stress with other forms of stress at a mechanistic level.
Although it is known that bZIP28 and to a minor extent bZIP6030 control necessary pathways for primary root growth, prior to our work, their effectors were not known. Because growth restoration impinges on coordinating a large number of processes, including cell division and elongation, tissue formation, and differentiation60, we expected that such effectors would be TFs, protein transporters, cell wall modifying enzymes, and many other proteins involved in growth. To rationalize which of the effectors could have a functional role in organ growth in ER stress resolution, we resolved in generating gene regulatory networks. Our network analyses revealed that the temporally regulated combinatorial effects of overlapping and distinctive roles between the two UPR pathways are based on the broad array of coexpression modules specialized for distinct biological processes. The analyses predicted that one of such modules, the pink module, is populated by genes involved in root organogenesis and that COW1 and YIP1a function as two regulatory-hub genes. We functionally validated this prediction by demonstrating that the loss of COW1 and Yip1a partially restore the reduced root growth phenotype of the loss-of-function mutations of bZIP28 and bZIP60 in ER stress recovery. Furthermore, the loss of COW1 in the bzip28-2 bzip60-1 background unambiguously reverted the lethal phenotype of this UPR bZIP-TF double null mutant. While this epistatic relationship underscores that COW1 serves as a life-or-death effector in the UPR, our data do not exclude a possibility that other gene hubs in the organogenesis module may function partially redundantly or independently from the hub genes characterized in this work. Interestingly, null mutants of yeast phosphatidylinositol transfer protein (Sec14p), a COW1 homolog, display a lethal phenotype61 which is rescued by expression of COW1 lipid-binding domain62. Defects in SEC14 proteins also cause human diseases, including retinal degeneration and cancer63, which are conditions linked to the UPR64. In addition to the pink module, other coexpression modules found here are a valuable premise to broaden our view of the impact of the UPR on other important biological processes. For example, the brown and yellow modules are strongly associated with biotic stress (Fig. 3b), supporting the previous report that the UPR is modulated by key regulators in biotic stress21,56.
In conclusion, our work addressed the fundamental open question of how growth programs are coordinated during ER stress recovery. Through a multi-omics pipeline that we developed in the model plant Arabidopsis, we identified temporal gene regulatory dynamics of two master transcriptional regulators in ER stress recovery, pinpointed the regulatory-hub genes in a gene regulatory network underlying organ growth, and functionally validated effectors predicted in our network analyses. Our work provides critical insights on the gene regulatory landscape during ER stress recovery with broad applicability across multicellular eukaryotes.
Methods
Plant materials
A. thaliana ecotype Columbia-0 (Col-0) was used as the WT control. The following mutants and transgenic lines, which are in Col-0 background except for bZIP60pro:GFP-bZIP60 (bzip60-2 background), were used in this study: bzip28-2 (SALK_132285), bzip60-2 (SAIL_283_B03), bzip28-2 bzip60-1 (SALK_132285 SALK_050203), ire1a ire1b (WiscDsLox420D09 SAIL_238_F07), ire1a ire1b bzip60-2, 35Spro:GFP-bZIP28 and bZIP60pro:GFP-bZIP60, cow1-3 (SALK_002124), lrx1-4 (SALK_057038), yip1a-1 (SALK_008449), yip1a-2 (GK-087C09-012440), and nac103 (SALK_053122). All T-DNA mutants and high-order mutants used in this study were confirmed to be homozygous before the analyses.
Plant growth condition
Surface-sterilized seeds were plated on half-strength Linsmaier Skoog (LS) medium (Caisson Labs, Ontario, Canada) supplemented with 1% sucrose (Sigma-Aldrich, St. Louis, MO, USA), and 1.2% Agar (Acumedia, Lansing, MI, USA). Appropriate antibiotics was also added for screening transgenic lines. After stratification in the dark at 4 °C for 2 days, seeds were transferred to a controlled growth chamber with 80 μmol m−2 s−1 under 16 h light:8 h dark with 22 °C.
ER stress assays
For ER stress recovery assay, 5-day-old seedlings were transferred to half-strength LS liquid buffer containing either 0.5 μg/mL Tm (Sigma-Aldrich, St. Louis, MO, USA) or DMSO-only as mock, and treated for 6 h. After the drug treatment, seedlings were transferred to 100 × 100 square Petri dishes with grid (Fisher Scientific, Hampton, NH, USA) containing half-strength LS medium (Caisson Labs, Ontario, Canada) supplemented with 1% sucrose (Sigma-Aldrich, St. Louis, MO, USA), 1.2% Agar (Acumedia, Lansing, MI, USA). Each square plate was split into two portions, of which each side accommodated either Tm-treated seedlings or DMSO-treated seedlings, which served as a biological replicate (n = 6 per replicate). Then, the seedlings grew vertically under the normal growth condition. For adaptive ER stress assay, 5-day-old seedlings were transferred to half-strength LS liquid buffer containing either 0.5 μg/mL Tm (Sigma-Aldrich, St. Louis, MO, USA) or DMSO as mock, and treated for 12 and 24 h. For chronic ER stress assay, seeds were plated on half-strength LS medium (Caisson Labs, Ontario, Canada) supplemented with 1% sucrose (Sigma-Aldrich, St. Louis, MO, USA), 1.2% Agar (Acumedia, Lansing, MI, USA), and 15, 25, 50 ng/mL Tm (Sigma-Aldrich, St. Louis, MO, USA) or DMSO only as mock.
Phenotyping analysis
For measuring the primary root length of seedlings, plates were photo-scanned at the same time of the day. The length of primary roots was analyzed using ImageJ software (https://imagej.nih.gov). For measuring dry weight, six whole seedlings of each genotype for each treatment were collected 8 days after stress (DAS) and pooled as a biological replicate (n = 5). The dry weight was measured after desiccation for 24 h at 80 °C. All experiments were independently replicated with consistent results.
LC/MS/MS for Tm measurements
Following the 6 h of initial treatment with Tm or DMSO, 5-day-old seedlings were subjected to either ER stress recovery (12 and 24 h) or adaptive ER stress assay (12 and 24 h), as detailed above. Tm is a mixture of four homologs A, B, C, and D (Sigma-Aldrich, St. Louis, MO, USA). The Tm 079M4004V batch (cat# T7765) used in this experiment had 8.57% Tm A, 29.91% Tm B, 47.91% Tm C, and 12.18% Tm D. Approximately 30 seedlings were collected for each treatment at each time-point as one of three biological replicates, weighed and then ground in liquid nitrogen. Tm is readily soluble in 80% methanol (https://www.sigmaaldrich.com), which efficiently penetrates plant cell walls/membranes to extract many metabolites65,66,67. Therefore, each ground sample was incubated overnight in 1 mL methanol-based Extraction Buffer (0.1% formic acid, 80% methanol, and 100 nM telmisartan, as an internal standard across samples). Samples were shortly vortexed and centrifuged at 12,000 × g at 4 °C for 10 min. The supernatant of each sample was transferred to a new microcentrifuge tube for the measurement. A standard curve of Tm was prepared by serial dilution in Extraction Buffer to give total Tm concentrations of 0.00122, 0.00488, 0.01953, 0.07813, 0.3125, 1.25, and 5 µM. The endogenous level of Tm contents (A, B, C, and D) in each sample was quantified by LC/MS/MS using a Waters Quattro Premier XE UPLC/MS/MS at the Research Technology Support Facility Mass Spectrometry and Metabolomics Core at Michigan State University. A 10 µL volume of extract was injected onto a Supelco Ascentis Express C18 HPLC column (2.1 × 50 mm, 2.7 µm particle size) maintained at 50 °C. Compounds were separated according to the following gradient run at 0.4 mL/min: initial conditions were 90% mobile phase A (water + 0.1% formic acid) and 10% mobile phase B (methanol) and held for 1 min, ramp to 50% A/50% B at 2 min, then ramp to 100% B at 3.5 min, followed by a ramp back to 90% A/10% B at 4 min and hold at the initial conditions until 5 min. Compounds were ionized by electrospray ionization operating in negative ion mode. The capillary voltage was set to 1.0 kV, the source temperature was 120 °C, the desolvation temperature was 350 °C, the cone gas low was 50 L/h, and the desolvation gas flow was 800 L/h. MS/MS data were acquired using a multiple-reaction-monitoring (MRM) method using the following parameters: Tm A (815.2 > 294.1), Tm B (829.2 > 308.1), Tm C (843.36 > 322.1), and Tm D (857.2 > 336.1), telmisartan (513.2 > 287). All compounds used a cone voltage of 38 V and collision energy of 38 V and dwell time of 0.1 s. Peak areas were integrated using the Quanlynx tool in Masslynx, and concentrations were calculated using the calibration curve with telmisartan used as the internal standard then normalized to the fresh weight of the extracted plant material.
RNA extraction and qRT-PCR analysis
Whole seedlings were harvested at 0, 12, and 24 h of ER stress recovery in three biological replicates (n = 12 per each replicate), and immediately frozen in liquid nitrogen. The frozen samples were ground to a fine powder in liquid nitrogen using a Retch MM400 Mixer Mill with zirconium oxide balls. Total RNA was extracted using the NucleoSpin RNA Plant kit (MACHEREY-NAGEL, Düren, Germany) according to the manufacturer’s instructions. cDNA was synthesized from 1 µg of DNaseI-treated total RNA using iScript cDNA Synthesis Kit (BIO-RAD, Hercules, CA, USA) according to the manufacturer’s instruction. For qRT-PCR, Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) was used in the presence of gene-specific primers and template cDNAs in an ABI7500 (Applied Biosystems, Foster City, CA, USA).
RNA-seq analysis
RNA-seq libraries were constructed using the Illumina TruSeq Stranded mRNA Library (Illumina, San Diego, CA, USA) and sequenced in single-end mode on the Illumina HiSeq4000 platform (50-nt) at the Research Technology Support Facility Genomics Core at Michigan State University. The quality of raw reads was evaluated using FastQC (version 0.11.5). Reads were cleaned for quality and adapters with Cutadapt (version 1.8.1)68 using a minimum base quality of 20 retaining reads with a minimum length of 30 nucleotides after trimming. Quality-filtered reads were aligned to the Col-0 reference genome (TAIR10) using Bowtie (version 2.2.4)69 and TopHat (version 2.0.14)70 with a 10-bp minimum intron length and 15,000-bp maximum intron length. Fragments per kilobase exon model per million mapped reads (FPKM) were calculated using TAIR10 gene model annotation with Cufflinks (version 1.3.0)71. Per-gene read counts were measured using HTSeq (version 0.6.1p1)72 in the union mode with a minimum mapping quality of 20 with stranded = reverse counting. Differential gene expression analysis was performed in each sample relative to the mock control using DESeq2 (version 1.16.1)73 within R (version 3.4.0). Genes of which the total count is <100 were not included in the analysis. DEGs were obtained based on adjusted P value <0.01 and absolute Log2FC > 1. Among DEGs identified in one or more genotypes at one or more time-points (n = 6670), DDEGs were obtained based the pairwise comparison of each DEG between Col-0 and bzip28-2, Col-0 and bzip60-2, or bzip28-2 and bzip60-2 with two-tailed Student t-test P < 0.01. For visualization purposes, tdf files of each replicate file were generated using igv tools (version 2.3.26) with the command “count” and loaded to Integrative Genome Browser (version 2.5.0)74. GO enrichment analysis was performed using agriGO (version 2.0) (http://systemsbiology.cau.edu.cn/agriGOv2/)75 with a false-discovery rate adjusted P < 0.05 (hypergeometric test) as a cutoff. Biological process GO categories were analyzed and the heatmap of GO analysis was produced using R package ggplot2. To validate the RNA-seq profiling, we performed quantitative RT-PCR (qRT-PCR) for 11 genes including seven known UPR biomarker genes and four downstream genes identified or will be identified below. We generated a total of 93 data points (Log2[Tm/DMSO]) across the time-points and genotypes (Supplementary Fig. 2b), which significantly correlated with those measured by the RNA-seq analysis (PCC = 0.947, P < 2.2 × 10−16).
Coexpression analysis
Coexpression network was built using the WGCNA R package36. All DEGs obtained from the DESeq2 analysis with the strict criteria (total read count across six samples [three for Tm-treated and three for DMSO-treated] >100, adjusted P < 0.01 and absolute Log2-transformed fold change >1) were considered. Log2-transformed DESeq2-normalized fold-changes (Tm/DMSO) served as input. Through empirical optimizations, a soft-power threshold of 18 was selected to create a signed network of a Spearman correlated matrix. Topographical overlap matrices (TOMs) were constructed using TOMsimilarityFromExpr() function with default parameter. The TOM scores were used as edge weights in the analysis. Coexpression modules were constructed through hierarchical clustering the TOM distance using flashClust() function with (method = “average”). Modules were derived using the cutreeDynamic() function with 25 of minimum module size, and similar modules were merged into a single module using mergeCloseModules() function with (cutHeight = 0.05). Network visualization was achieved using the Cytoscape software (version 3.6.1) with a cutoff of the weight parameter obtained from the WGCNA, set at 0.2.
Chromatin immunoprecipitation (ChIP) assays, ChIP-qPCR, ChIP-seq library preparation
ChIP was performed using a modified protocol based on general methods previously described76. Whole seedlings harvested at 0, 12, and 24 h of ER stress recovery in three biological replicates were completely submerged in freshly prepared pre-chilled crosslinking buffer (0.4 M Sucrose, 10 mM Tris-HCl pH 8.0, 1 mM EDTA, 1 mM PMSF, and 1% Formaldehyde) in 50 mL conical tubes. The samples were subjected to a vacuum infiltration for 15 min and another for 5 min with glycine (125 mM final concentration). After the crosslinking buffer was removed, the crosslinked tissues were briefly rinsed with prechilled sterilized water, dried by gently blotting between paper towels, immediately frozen in the liquid nitrogen, and stored at −80 °C for the next steps. The frozen tissues were ground to a fine powder in liquid nitrogen using a prechilled motor and pestle. The tissue powder was homogenized in 30 mL of pre-chilled nuclei isolation buffer A (0.4 M Sucrose, 10 mM Tris-Cl pH 8.0, 1 mM PMSF, 5 mM β-Mercaptoethanol and 1× Protease inhibitor cocktail [Sigma-Aldrich, St. Louis, MO, USA; cat# P2714]) at 4 °C for 30 min. Homogenized tissue powder was filtered through Miracloth and then centrifuged at 4 °C with 2880 g for 20 min. After centrifugation, the supernatant was discarded and pellets were resuspended in 1 mL of pre-chilled nuclei isolation buffer B (0.25 M Sucrose, 10 mM MgCl2, 1% Triton X-100, 10 mM Tris-Cl pH 8.0, 1 mM PMSF, 5 mM β-Mercaptoethanol and 1× Protease inhibitor cocktail [Sigma-Aldrich, St. Louis, MO, USA; cat# P2714]). The suspension was centrifuged at 4 °C with 12,000 g for 10 min. After centrifugation, the supernatant was discarded, and pellets were resuspended in 300 µL of pre-chilled nuclei isolation buffer C (1.7 M Sucrose, 2 mM MgCl2, 0.15% Triton X-100, 10 mM Tris-Cl pH 8.0, 1 mM PMSF, 5 mM β-Mercaptoethanol and 1× Protease inhibitor cocktail [Sigma-Aldrich, St. Louis, MO, USA; cat# P2714]). The suspension was added to the top of 1.5 mL cold nuclei isolation buffer C and centrifuged at 4 °C with 16,000 g for 1 h. After centrifugation, the pellet was resuspended in 310 µL of pre-chilled nuclei lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1% SDS, 0.1% Na deoxycholate, 1% Triton X-100, 1 µg/mL Pepstatin A [Sigma-Aldrich, St. Louis, MO, USA; cat# P5318] and 1× Protease inhibitor cocktail [Sigma-Aldrich, St. Louis, MO, USA; cat# P2714]). After isolation, chromatin was fragmented using Covaris M220 sonicator (Covaris, Woburn, MA, USA) with settings of three cycles of PIP-50, duty factor 20% time-70s at 4 °C. Immunoprecipitation (IP) was performed with a polyclonal anti-GFP antibody (Abcam, Cambridge, UK; cat# ab290; 1/200 dilution). For each ChIP sample, a mock (no antibody) and input (no IP) were included for control experiments. About 2 µL purified DNA of ChIP, mock and input samples, which was diluted by fourfold, was used for qPCR analysis in an ABI7500 machine (Applied Biosystems, Foster City, CA, USA) using Fast SYBR Green Master Mix (Life Technologies, Carlsbad, CA, USA). The enrichment of amplicons from ChIP DNA samples was normalized relative to one of the corresponding input DNA samples. Three technical replicates were assayed for each of the three biological replicates. The list of primers used in ChIP-qPCR is provided in Supplementary Table 1. The final purified ChIP and input DNAs were quantified using the Qubit fluorometer (Thermo Fisher Scientific, Carlsbad, CA, USA), and subjected to ChIP-seq libraries construction in two biological replicates using the NEBNext Ultra II DNA Library Prep Kit (New England BioLabs, Beverly, MA, USA) according to manufacturer’s protocol. The suitable size distribution of libraries was confirmed using the 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA). Multiplexed libraries were sequenced in single-end mode on the Illumina HiSeq4000 platform (50-nt) at the Research Technology Support Facility Genomics Core at Michigan State University.
ChIP-seq data analysis
The guideline and analysis pipeline provided by the ENCODE project42 was adapted to our ChIP-seq analysis with several modifications optimized for the dataset. The quality of raw ChIP-seq reads was evaluated using FastQC (version 0.11.5). Reads were cleaned for quality and adapters with Cutadapt (version 1.8.1)68 using a minimum base quality of 20 retaining reads with a minimum length of 30 nucleotides after trimming. Quality-filtered reads were aligned to the Col-0 reference genome (TAIR10) using Bowtie (version 1.1.2)77 with parameters “-n 2 -m 3 -k 1 –threads 7 –best –chunkmbs 256 -q”. Duplicated reads were removed using Samtools (version 1.8)78. Peak calling was performed using MACS2 (version 2.1.2)79 in individual samples with input samples pooled for each of DMSO- and Tm-treated samples and with a relaxed threshold of P value (–p value = 1e-2), as recommended by the IDR pipeline (https://sites.google.com/site/anshulkundaje/projects/idr). Peaks across replicates with an IDR <0.05 were retained for further analysis. To obtain binding peaks with high-confidence, we applied two parameters to IDR-filtered peaks; (1) a peak called in Tm samples that was overlapped with a peak in the corresponding DMSO-treated samples by >30% was eliminated, (2) Among the peaks that were overlapped with peaks in DMSO-treated sample, if its P float (eighth column in the IDR output file) in the Tm-treated sample was higher than the corresponding peak in the DMSO-treated sample by greater than threefold, the peak was retained and named as a UPR-specific binding peak. UPR-specific binding peaks obtained at each time-point were merged into a single list for further analysis and were annotated using the ChIPseeker and the GenomicFeatures R Package (version 3.4.0)80. For visualization purposes, bigwig files (using pooled data across biological replicates) were generated by deepTools suite (https://deeptools.readthedocs.io/en/develop/)81 with the command “bamCoverage”; read coverage was normalized as RPKM (Reads Per Kilobase per Million reads). ChIP-seq tracks were visualized in Integrative Genome Browser (version 2.5.0)74. ChIP-seq metaplot was generated from the merged single file by deepTools suite with the commands “bamCompare” and “plotProfile”. UPR-specific binding peaks were mapped to the vicinity of a coding sequence (<1-kb), generating a total of 440 bZIP28- and 356 bZIP60-bound genes, respectively. GO enrichment analysis was performed as described in RNA-seq analysis.
Motif discovery and enrichment analysis
All motif analyses presented in this study were performed using various tools of the MEME suite (version 5.0.5) (http://meme-suite.org) with default parameters and modifications indicated elsewhere. Random model letter frequencies were calculated based on GC content in the Arabidopsis genome (https://genomevolution.org/wiki/index.php/Plant_genome_GC_content) and used for the following analyses. 1-kb upstream sequence of the transcription start sites (TSS), hereafter called 1-kb promoter, in the TAIR10 Arabidopsis genome annotation was used. Enrichment of canonical CREs in response to ER stress on the promoters of DDEGs was investigated using AME82. For this analysis, one mismatch was allowed for each CRE except for UPRE-I. De novo motif discovery in 1-kb promoter of DDEGs was performed using MEME83. To eliminate any de novo motifs not specific to DDEGs, 201, which is the median number of DDEGs across all pairwise comparisons, DEGs were randomly selected and served as a control for the analyses. The similarity of enriched motifs with the Plant Cistrome Database84 was assessed using TOMTOM85. De novo motif discovery in UPR-specific binding peaks of bZIP28 and bZIP60 from the ChIP-seq analyses was carried out using MEME-ChIP86. For this analysis, UPR-specific binding peaks obtained at each time-point were combined, creating union sets of peaks for bZIP28 and bZIP60. Then, 250-bp sequence upstream and downstream of the summit in each of the combined peaks was obtained from the TAIR10 Arabidopsis genome annotation; for peaks that had multiple sub-peaks, one that had the highest of -Log10P was selected. The enrichment distribution of ERSE-I in UPR-specific binding peaks was analyzed using CentriMo87.
Statistics and reproducibility
For experimental analyses, a two-tailed Student’s t-test was used to obtain P values in this study using the R function and values of P < 0.05 were considered statistically significant. The number of samples is indicated in the respective Figure legends: n (number of biological replicates) = 3 (~50 seedlings per replicate), Fig. 1b; n = 3 (12 seedlings per replicate), Fig. 1c and Supplementary Fig. 3a, d, g; n = 5 (6 seedlings per replicate), Fig. 1d and Supplementary Figs. 1b, 3b; n = 3–5 (6 seedlings per replicate), Fig. 5a; n (number of biological replicates) = 4–5 (3–5 seedlings per replicate), Fig. 5b; n = 10 (2 seedlings per replicate), Supplementary Fig. 1c; n = 3 (~100 seedlings per replicate), Supplementary Fig. 2f; n = 6–8, Supplementary Fig. 6. No data points were removed from the statistical analyses as outliers. Results were confirmed in at least two independent experiments.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data supporting the findings of this study are available within this paper and its Supplementary Information files. RNA-seq and ChIP-seq data supporting the finding of this study have been deposited in the NCBI Gene Expression Omnibus and are accessible through the GEO series accession code GSE146723. The source data, which consists of an excel spreadsheet, are provided in Supplementary Data 6.
Code availability
The bioinformatic scripts used in this study are available on GitHub (https://github.com/DaeKwan-Ko/UPR-GRN.git).
References
Pastor-Cantizano, N., Ko, D. K., Angelos, E., Pu, Y. & Brandizzi, F. Functional diversification of ER stress responses in Arabidopsis. Trends Biochem. Sci. 45, 123–136 (2020).
Walter, P. & Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 (2011).
Liou, H.-C. et al. A new member of the leucine zipper class of proteins that binds to the HLA DR alpha promoter. Science 247, 1581–1584 (1990).
Calfon, M. et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 (2002).
Yoshida, H. et al. Endoplasmic reticulum stress-induced formation of transcription factor complex ERSF including NF-Y (CBF) and activating transcription factors 6alpha and 6beta that activates the mammalian unfolded protein response. Mol. Cell Biol. 21, 1239–1248 (2001).
Iwata, Y. & Koizumi, N. An Arabidopsis transcription factor, AtbZIP60, regulates the endoplasmic reticulum stress response in a manner unique to plants. Proc. Natl Acad. Sci. USA 102, 5280–5285 (2005).
Cox, J. S., Shamu, C. E. & Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206 (1993).
Lee, A.-H., Iwakoshi, N. N. & Glimcher, L. H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459 (2003).
Acosta-Alvear, D. et al. XBP1 controls diverse cell type-and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66 (2007).
Deng, Y. et al. Heat induces the splicing by IRE1 of a mRNA encoding a transcription factor involved in the unfolded protein response in Arabidopsis. Proc. Natl Acad. Sci. USA 108, 7247–7252 (2011).
Mori, K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J. Biochem. 146, 743–750 (2009).
Sun, L. et al. The plant-specific transcription factor gene NAC103 is induced by bZIP60 through a new cis-regulatory element to modulate the unfolded protein response in Arabidopsis. Plant J. 76, 274–286 (2013).
Yang, Z. T. et al. A plasma membrane‐tethered transcription factor, NAC 062/ANAC 062/NTL 6, mediates the unfolded protein response in Arabidopsis. Plant J. 79, 1033–1043 (2014).
Fink, E. E. et al. XBP1-KLF9 axis acts as a molecular rheostat to control the transition from adaptive to cytotoxic unfolded protein response. Cell Rep. 25, 212–223.e214 (2018).
Yoshida, H., Haze, K., Yanagi, H., Yura, T. & Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749 (1998).
Liu, J. X., Srivastava, R., Che, P. & Howell, S. H. An endoplasmic reticulum stress response in Arabidopsis is mediated by proteolytic processing and nuclear relocation of a membrane-associated transcription factor, bZIP28. Plant Cell 19, 4111–4119 (2007).
Haze, K., Yoshida, H., Yanagi, H., Yura, T. & Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799 (1999).
Oh, D. H., Kwon, C. S., Sano, H., Chung, W. I. & Koizumi, N. Conservation between animals and plants of the cis-acting element involved in the unfolded protein response. Biochem. Biophys. Res. Commun. 301, 225–230 (2003).
Liu, J. X. & Howell, S. H. bZIP28 and NF-Y transcription factors are activated by ER stress and assemble into a transcriptional complex to regulate stress response genes in Arabidopsis. Plant Cell 22, 782–796 (2010).
Hayashi, S., Takahashi, H., Wakasa, Y., Kawakatsu, T. & Takaiwa, F. Identification of a cis-element that mediates multiple pathways of the endoplasmic reticulum stress response in rice. Plant J. 74, 248–257 (2013).
Lai, Y. S. et al. Salicylic acid-independent role of NPR1 is required for protection from proteotoxic stress in the plant endoplasmic reticulum. Proc. Natl Acad. Sci. USA 115, E5203–E5212 (2018).
Nawkar, G. M. et al. HY5, a positive regulator of light signaling, negatively controls the unfolded protein response in. Proc. Natl Acad. Sci. USA 114, 2084–2089 (2017).
Meng, Z., Ruberti, C., Gong, Z. & Brandizzi, F. CPR5 modulates salicylic acid and the unfolded protein response to manage tradeoffs between plant growth and stress responses. Plant J. 89, 486–501 (2017).
Ron, D. & Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 (2007).
Nagashima, Y. et al. Arabidopsis IRE1 catalyses unconventional splicing of bZIP60 mRNA to produce the active transcription factor. Sci. Rep. 1, 29 (2011).
Chen, Y. & Brandizzi, F. AtIRE1A/AtIRE1B and AGB1 independently control two essential unfolded protein response pathways in Arabidopsis. Plant J. 69, 266–277 (2012).
Sun, L. et al. The lumen-facing domain is important for the biological function and organelle-to-organelle movement of bZIP28 during ER stress in Arabidopsis. Mol. Plant 6, 1605–1615 (2013).
Mishiba, K. et al. Defects in IRE1 enhance cell death and fail to degrade mRNAs encoding secretory pathway proteins in the Arabidopsis unfolded protein response. Proc. Natl Acad. Sci. USA 110, 5713–5718 (2013).
McCormack, M. E., Liu, X., Jordan, M. R. & Pajerowska-Mukhtar, K. M. An improved high-throughput screening assay for tunicamycin sensitivity in Arabidopsis seedlings. Front. Plant Sci. 6, 663 (2015).
Ruberti, C., Lai, Y. & Brandizzi, F. Recovery from temporary endoplasmic reticulum stress in plants relies on the tissue-specific and largely independent roles of bZIP28 and bZIP60, as well as an antagonizing function of BAX-Inhibitor 1 upon the pro-adaptive signaling mediated by bZIP28. Plant J. 93, 155–165 (2018).
Kim, J. S., Yamaguchi-Shinozaki, K. & Shinozaki, K. ER-anchored transcription factors bZIP17 and bZIP28 regulate root elongation. Plant Physiol. 176, 2221–2230 (2018).
Bao, Y., Bassham, D. C. & Howell, S. H. A functional unfolded protein response is required for normal vegetative development. Plant Physiol. 179, 1834–1843 (2019).
Iwata, Y., Sakiyama, M., Lee, M.-H. & Koizumi, N. Transcriptomic response of Arabidopsis thaliana to tunicamycin-induced endoplasmic reticulum stress. Plant Biotechnol. 27, 161–171 (2010).
Deng, Y., Srivastava, R. & Howell, S. H. Protein kinase and ribonuclease domains of IRE1 confer stress tolerance, vegetative growth, and reproductive development in Arabidopsis. Proc. Natl Acad. Sci. USA 110, 19633–19638 (2013).
Kokame, K., Kato, H. & Miyata, T. Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J. Biol. Chem. 276, 9199–9205 (2001).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559 (2008).
Bruex, A. et al. A gene regulatory network for root epidermis cell differentiation in Arabidopsis. PLoS Genet. 8, e1002446 (2012).
Boron, A. K. et al. Proline-rich protein-like PRPL1 controls elongation of root hairs in Arabidopsis thaliana. J. Exp. Bot. 65, 5485–5495 (2014).
Srivastava, R., Chen, Y., Deng, Y., Brandizzi, F. & Howell, S. H. Elements proximal to and within the transmembrane domain mediate the organelle-to-organelle movement of bZIP28 under ER stress conditions. Plant J. 70, 1033–1042 (2012).
Parra-Rojas, J., Moreno, A. A., Mitina, I. & Orellana, A. The dynamic of the splicing of bZIP60 and the proteins encoded by the spliced and unspliced mRNAs reveals some unique features during the activation of UPR in Arabidopsis thaliana. PLoS ONE 10, e0122936 (2015).
Withers, J. et al. Transcription factor-dependent nuclear localization of a transcriptional repressor in jasmonate hormone signaling. Proc. Natl Acad. Sci. USA 109, 20148–20153 (2012).
Landt, S. G. et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831 (2012).
Bailey, T. et al. Practical guidelines for the comprehensive analysis of ChIP-seq data. PLoS Comput. Biol. 9, e1003326 (2013).
Yu, C. P., Lin, J. J. & Li, W. H. Positional distribution of transcription factor binding sites in Arabidopsis thaliana. Sci. Rep. 6, 25164 (2016).
Guelzim, N., Bottani, S., Bourgine, P. & Képès, F. Topological and causal structure of the yeast transcriptional regulatory network. Nat. Genet. 31, 60–63 (2002).
Böhme, K. et al. The Arabidopsis COW1 gene encodes a phosphatidylinositol transfer protein essential for root hair tip growth. Plant J. 40, 686–698 (2004).
Baumberger, N., Ringli, C. & Keller, B. The chimeric leucine-rich repeat/extensin cell wall protein LRX1 is required for root hair morphogenesis in Arabidopsis thaliana. Genes Dev. 15, 1128–1139 (2001).
Kano, F. et al. Yip1A regulates the COPI-independent retrograde transport from the Golgi complex to the ER. J. Cell Sci. 122, 2218–2227 (2009).
Jin, C. et al. Human Yip1A specifies the localization of Yif1 to the golgi apparatus. Biochem. Biophys. Res. Commun. 334, 16–22 (2005).
Mitra, S. & Ryoo, H. D. The unfolded protein response in metazoan development. J. Cell Sci. https://doi.org/10.1242/jcs.217216 (2019).
Seo, P. J. et al. The MYB96 transcription factor mediates abscisic acid signaling during drought stress response in Arabidopsis. Plant Physiol. 151, 275–289 (2009).
Hossain, M. A. et al. Identification of novel components of the unfolded protein response in Arabidopsis. Front. Plant Sci. 7, 650 (2016).
Devaiah, B. N., Karthikeyan, A. S. & Raghothama, K. G. WRKY75 transcription factor is a modulator of phosphate acquisition and root development in Arabidopsis. Plant Physiol. 143, 1789–1801 (2007).
Guo, P. et al. A tripartite amplification loop involving the transcription factor WRKY75, salicylic acid, and reactive oxygen species accelerates leaf senescence. Plant Cell 29, 2854–2870 (2017).
Rubio, V. et al. A conserved MYB transcription factor involved in phosphate starvation signaling both in vascular plants and in unicellular algae. Genes Dev. 15, 2122–2133 (2001).
Moreno, A. A. et al. IRE1/bZIP60-mediated unfolded protein response plays distinct roles in plant immunity and abiotic stress responses. PLoS ONE 7, e31944 (2012).
Deng, Y. et al. IRE 1, a component of the unfolded protein response signaling pathway, protects pollen development in Arabidopsis from heat stress. Plant J. 88, 193–204 (2016).
Nagashima, Y., Iwata, Y., Ashida, M., Mishiba, K. & Koizumi, N. Exogenous salicylic acid activates two signaling arms of the unfolded protein response in Arabidopsis. Plant Cell Physiol. 55, 1772–1778 (2014).
Zhang, S. S. et al. Tissue-specific transcriptomics reveals an important role of the unfolded protein response in maintaining fertility upon heat stress in Arabidopsis. Plant Cell 29, 1007–1023 (2017).
Motte, H., Vanneste, S. & Beeckman, T. Molecular and environmental regulation of root development. Annu Rev. Plant Biol. 70, 465–488 (2019).
Phillips, S. E. et al. Yeast Sec14p deficient in phosphatidylinositol transfer activity is functional in vivo. Mol. Cell 4, 187–197 (1999).
Vincent, P. et al. A Sec14p-nodulin domain phosphatidylinositol transfer protein polarizes membrane growth of Arabidopsis thaliana root hairs. J. Cell Biol. 168, 801–812 (2005).
Bankaitis, V. A., Mousley, C. J. & Schaaf, G. The Sec14 superfamily and mechanisms for crosstalk between lipid metabolism and lipid signaling. Trends Biochem. Sci. 35, 150–160 (2010).
Li, J., Wang, J. J., Yu, Q., Wang, M. & Zhang, S. X. Endoplasmic reticulum stress is implicated in retinal inflammation and diabetic retinopathy. FEBS Lett. 583, 1521–1527 (2009).
Mansfeld, B. N., Colle, M., Kang, Y., Jones, A. D. & Grumet, R. Transcriptomic and metabolomic analyses of cucumber fruit peels reveal a developmental increase in terpenoid glycosides associated with age-related resistance to. Hortic. Res. 4, 17022 (2017).
Forster, G. M. et al. A comparative study of serum biochemistry, metabolome and microbiome parameters of clinically healthy, normal weight, overweight, and obese companion dogs. Top. Companion Anim. Med. 33, 126–135 (2018).
Ghosh, B., Westbrook, T. C. & Jones, A. D. Comparative structural profiling of trichome specialized metabolites in tomato. Metabolomics 10, 496–507 (2014).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Tian, T. et al. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx382 (2017).
Saleh, A., Alvarez-Venegas, R. & Avramova, Z. An efficient chromatin immunoprecipitation (ChIP) protocol for studying histone modifications in Arabidopsis plants. Nat. Protoc. 3, 1018–1025 (2008).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Yu, G., Wang, L. G. & He, Q. Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Buske, F. A., Bodén, M., Bauer, D. C. & Bailey, T. L. Assigning roles to DNA regulatory motifs using comparative genomics. Bioinformatics 26, 860–866 (2010).
Bailey, T. L. & Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36 (1994).
O’Malley, R. C. et al. Cistrome and epicistrome features shape the regulatory DNA landscape. Cell 165, 1280–1292 (2016).
Gupta, S., Stamatoyannopoulos, J. A., Bailey, T. L. & Noble, W. S. Quantifying similarity between motifs. Genome Biol. 8, R24 (2007).
Machanick, P. & Bailey, T. L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011).
Bailey, T. L. & Machanick, P. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res. 40, e128 (2012).
Acknowledgements
This study was supported primarily by the National Institutes of Health (R35-GM136637) with contributing support from by the Great Lakes Bioenergy Research Center, US Department of Energy, Office of Science, Office of Biological and Environmental Research (DE-SC0018409), Chemical Sciences, Geoscience, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, US Department of Energy (DE-FG02-91ER20021), and MSU AgBioResearch (MICL02598). We thank the Research Technology Support Facility Genomics Core and Mass Spectrometry and Metabolomics Core facilities at Michigan State University for the next-generation sequencing and quantitative LC/MS/MS, respectively. We also thank Cristina Ruberti for preparing samples for RNA-seq analysis.
Author information
Authors and Affiliations
Contributions
D.K.K. and F.B. conceived the original experiment design and research plan; F.B. supervised the project and experiments; D.K.K. performed experiments and data analyses; D.K.K. and F.B. wrote the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review information
Communications Biology thanks Karolina Mukhtar and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editors: José Estevez and George Inglis. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ko, D.K., Brandizzi, F. Advanced genomics identifies growth effectors for proteotoxic ER stress recovery in Arabidopsis thaliana. Commun Biol 5, 16 (2022). https://doi.org/10.1038/s42003-021-02964-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02964-8
This article is cited by
-
Dynamics of ER stress-induced gene regulation in plants
Nature Reviews Genetics (2024)
-
Transcriptome analysis revealed that Arabidopsis model plant invokes the activation of heat shock proteins and ER stress response against cesium stress
Plant Biotechnology Reports (2024)
-
An IRE1-proteasome system signalling cohort controls cell fate determination in unresolved proteotoxic stress of the plant endoplasmic reticulum
Nature Plants (2023)
-
Transcriptional competition shapes proteotoxic ER stress resolution
Nature Plants (2022)
-
P58IPK facilitates plant recovery from ER stress by enhancing protein synthesis
Plant Biotechnology Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
