
Abstract
Fluorescence lifetime imaging microscopy (FLIM) is a powerful tool to quantify molecular compositions and study molecular states in complex cellular environment as the lifetime readings are not biased by fluorophore concentration or excitation power. However, the current methods to generate FLIM images are either computationally intensive or unreliable when the number of photons acquired at each pixel is low. Here we introduce a new deep learning-based method termed flimGANE (fluorescence lifetime imaging based on Generative Adversarial Network Estimation) that can rapidly generate accurate and high-quality FLIM images even in the photon-starved conditions. We demonstrated our model is up to 2,800 times faster than the gold standard time-domain maximum likelihood estimation (TD_MLE) and that flimGANE provides a more accurate analysis of low-photon-count histograms in barcode identification, cellular structure visualization, Förster resonance energy transfer characterization, and metabolic state analysis in live cells. With its advantages in speed and reliability, flimGANE is particularly useful in fundamental biological research and clinical applications, where high-speed analysis is critical.
Similar content being viewed by others


Introduction
Using fluorescence decay rate as the contrast mechanism, fluorescence lifetime imaging microscopy (FLIM) is a powerful quantitative tool for studying cell and tissue biology1,2,3, allowing us to monitor the pH4, viscosity5, temperature6, oxygen content7, metabolic state8 and functional property of a biomarker9 inside live cells or tissues. Depending on the intrinsic property of fluorophore, FLIM images are not skewed by fluorophore concentration and excitation power, eliminating the biases introduced by the traditional intensity-based images2. Combined with the Förster resonance energy transfer (FRET) sensors10, FLIM can probe Ca2+ concentration11, glucose concentration12, and protein-protein interactions13, without the need to measure acceptor’s fluorescence14. Whereas FLIM offers many unique advantages in quantifying molecular interactions15 and chemical environments16 in biological or chemical samples, fluorescence lifetime analysis is a slow process with results often impaired by fitting errors. Adopted from disparate disciplines, various fluorescence lifetime estimation methods, such as curve fitting (least-squares fitting17, maximum likelihood estimation18, global analysis19, and Bayesian analysis20), phasor analysis21,22, and deconvolution analysis (stretched exponential analysis23 and Laguerre deconvolution24) have been developed to infer the lifetime of interest. However, these methods are often limited by long computation times, poor accuracy particularly in the low-light conditions, and invalid initial assumptions of decay parameters. Although deep learning methods, such as artificial neural network (ANN)25 or convolutional neural network (CNN)26,27, have been employed to achieve rapid fluorescence lifetime analysis in the medium-photon-count conditions (200–500 photon counts per pixel), other deep learning algorithms may further improve the reliability in analyzing the low-photon-count (100–200 photon counts per pixel) or even ultralow-photon-count data (50–100 photon counts per pixel) for live-cell imaging.
Here we demonstrate a new fluorescence lifetime imaging method based on Generative Adversarial Network Estimation (flimGANE) that can provide fast, fit-free, accurate, and high-quality FLIM images even under the photon-starved conditions (50–200 photon counts per pixel). GAN is one of the frameworks for evaluating generative models via an adversarial process28, which has been adopted to improve astronomical images29,30, transform images across different modalities29,31, and design drugs that target specific signaling molecules32. While GAN-based algorithms have recently drawn much attention for inferring photo-realistic natural images33, they have not been used to generate high-quality FLIM images based on the fluorescence decays collected by a laser scanning confocal microscope. Our flimGANE method is adapted from the Wasserstein GAN algorithm34 (WGAN), where the generator (G) is trained to produce an “artificial” high-photon-count fluorescence decay histogram based on a low-photon-count input, while the discriminator (D) distinguishes the artificial decay histogram from the ground truth (which can be a simulated dataset or a decay histogram collected under strong excitation). As a minimax two-player game, the training procedure for G is to maximize the probability of D making a mistake28, eventually leading to the production of very realistic, artificial high-photon-count decay histograms that can be used to generate a high-quality FLIM image. Using a well-trained generator (G) and an estimator (E), we can reliably map a low-quality decay histogram to a high-quality counterpart, and eventually to the three lifetime parameters (α1, τ1, and τ2) within 0.32 ms/pixel using a CPU. Without the need of curve fitting based on initial guesses, our flimGANE method is up to 258-fold faster than the time-domain least-squares estimation (TD_LSE35,36) and 2,800-fold faster than the time-domain maximum likelihood estimation (TD_MLE37,38) in generating a 512 × 512 FLIM image. While almost all commercial FLIM analysis tools are based on TD_LSE, using the least-squares estimator to analyze Poisson-distributed data is known to lead to biases39, making TD_MLE the gold standard for FLIM analysis by many researchers18. Our flimGANE can provide similar FLIM image quality as TD_MLE, but much faster. Recently, field-programmable gate array (FPGA)-based MLE has been demonstrated for lifetime analysis, reaching an ultrahigh analysis speed40. However, FPGA-MLE needs much more effort in hardware development and programing to be implemented in an existing optical system.
Overcoming a number of hardware limitations in the classical analog frequency-domain approach, the digital frequency-domain (DFD) lifetime measurement method has substantially increased the FLIM acquisition speed21,22,41. The acquired DFD data at each pixel, termed a cross-correlation phase histogram, can lead to a phasor plot with multiple harmonic frequencies41. From such a phasor plot, modulation ratio and phase angle at each harmonic frequency can be obtained, which are then fitted with a least-squares estimator (LSE) to generate a lifetime at each pixel (termed the DFD_LSE method). Our flimGANE not only runs nearly 12-fold faster than DFD_LSE but also produces more accurate quantitative results in all photon-count conditions. Sharper structural images of Convallaria and live HeLa cells are also achieved by flimGANE. Whereas the lowest number of photons needed for reliable estimation of a fluorescence lifetime by TD_MLE is about 100 photons42, flimGANE performs consistently well with a photon count as low as 50 per pixel in our simulations. Moreover, flimGANE improves the energy transfer efficiency estimate of a glucose FRET sensor, leading to a more accurate glucose concentration measurement in live HeLa cells. Providing both efficiency and reliability in analyzing low-photon-count decays, our flimGANE method represents an important step forward towards real-time FLIM.
Results
Training the generative adversarial network in flimGANE
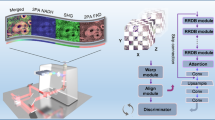
Based on the Wasserstein GAN framework (see Supplementary Methods), flimGANE is designed to analyze one- or two-component fluorescence decays under the photon-starved conditions (Fig. 1). There are two ways to generate a dataset of ground-truth lifetime histograms for training G and D—either by creating a decay dataset using Monte Carlo (MC) simulations or by acquiring an experimental dataset from standard organic fluorophores under high excitation power. The inputs of G are degraded data from the ground truths, which can be obtained by running simulations at a low-emission rate or by recollecting experimental data under low excitation power.

a–c Schematic of the deep learning framework for flimGANE architecture. a Generator (G) is used to transform the acquired decay curve into an artificial high-photon-count decay. It comprises two CNN blocks, each of which is made up of one convolutional layer followed by an average pooling layer of stride four. The CNN section is followed by a flatten layer. Then a multi-task layer converts data into virtual lifetime parameters, followed by two fully connected layers. Skip connection is used to pass data between layers of the same level. b Discriminator (D) consists of four fully connected layers. c Estimator (E) comprises a partially and a fully connected layers followed by a multi-task layer to map the high-photon-count decay curve into lifetime parameters. d Comparison of FLIM images generated by different methods (n = 1340 pixels). e Comparison of squared errors by different methods. Under the ultra-low-photon-count condition (50 counts per pixel), flimGANE provides the best estimation. Under the low-photon-count condition (100 counts), TD_LSE and DFD_LSE fail to generate accurate FLIM images. Under the high-photon-count condition (1500 counts), all FLIM images match well with the ground truth. TD_LSE result exhibits no significant difference from DFD_LSE result under the low-photon-count condition based on the two-tailed t test. f flimGANE successfully characterizes the apparent lifetimes of the two-dye mixtures (stock solution: 3 μM Cy5-NHS ester and 7 μM Atto633 in DI water). The mean values obtained from Gaussian fitting are indicated as white solid circles.
We started our network training using an MC simulation dataset (Supplementary Fig. 1). A Python program was employed to simulate the photon collection process in the counting device with 256-time bins, following the probability mass function (pmf) numerically calculated by the convolution of an experimentally obtained instrument response function (IRF) and a theoretical two-component decay model (α1, τ1, 1–α1, and τ2) at a selected emission rate (rate)43. Depending on the fluorophores that users want to image, proper α1, τ1, τ2 and rate parameters that span the range of interest could be selected (Supplementary Tables 1 and 2), generating about 600 normalized ground truths and 300,000 degraded decays for training G and D. The adversarial network training was completed in 6.1 h (Fig. 1a, b; Supplementary Fig. 2). The normalized degraded decay was transformed into the normalized “ground-truth mimicking” histogram, termed Goutput, with one-to-one correspondence (Supplementary Fig. 3), within 0.17 ms. Such a Goutput was indistinguishable by D from the ground truth dataset. E, which was separately trained on the ground truths and completed in 0.1 h, was then employed to extract the key lifetime parameters (α1, τ1, and τ2) from the Goutput within 0.15 ms (Fig. 1c; Supplementary Fig. 2). In this process, flimGANE did not take information from the surrounding pixels into the current pixel.
To demonstrate the reliability of our flimGANE method, we created ten sets of 14 × 47 “UTBME” FLIM images in silico (independently generated, not used in the training process) at three-photon emission rates (50, 100, and 1500 photons per pixel). At 1500 photons per pixel, all four methods (TD_LSE, TD_MLE, DFD_LSE and flimGANE) generated high-fidelity FLIM images (based on the apparent lifetime, τα = α1τ1 + (1 − α1)τ2), with mean-squared errors (MSE) less than 0.10 ns2 (n = 1,340 pixels). At 150 photons per pixel, flimGANE had similar performance as TD_MLE (MSE were both less than 0.20 ns2); however, flimGANE clearly outperformed TD_LSE, TD_MLE, and DFD_LSE at 50 photons per pixel (0.21 vs. 0.91, 0.36 and 1.65 ns2, respectively; Fig. 1d, e, Supplementary Figs. 4–6, and Supplementary Tables 3–6). CPU-based speed analysis showed that flimGANE was up to 258 and 2,800 times faster than TD_LSE and TD_MLE, respectively (flimGANE—0.32 ms per pixel, TD_LSE—82.40 ms, TD_MLE—906.37 ms; Supplementary Table 7). While DFD_LSE offered a decent speed in generating FLIM images (3.94 ms per pixel), its accuracy was worse than that of flimGANE (Figs. 2–5). In contrast, being a computationally intensive method, TD_MLE offered the accuracy, but not the speed. Only flimGANE could provide both speed and accuracy in generating FLIM images. In addition, the MLE method became unreliable in the ultra-low-photon-count condition (50–100 photons per pixel), while flimGANE still provided a reasonable result.

a Intensity contrast images show 3-µm fluorescent beads with various brightness. b FLIM images obtained from TD_LSE, TD_MLE, DFD_LSE, and flimGANE discriminate fluorescence lifetime barcode beads. c Mean lifetimes versus brightness plot of 97 beads shows the intensity differences of the three barcodes and the cutoff lifetimes (2.15 and 2.95 ns) for barcode identification. d Barcode classification results by the four methods indicate that only flimGANE can correctly identify the three barcodes and restore the correct barcode ratio (1:1:1). e Classification results of all 97 beads show that many barcode_2 beads are misidentified as barcode_1 beads by TD_LSE, TD_MLE, and DFD_LSE, while the identification of barcode_3 beads is more reliable among these methods (except for TD_LSE, which performs poorly under the low-light condition).

a Images of Convallaria acquired at two different intensity levels (left: 50–150 counts per pixel and right: 200–400 counts). b FLIM image generated by flimGANE is sharper and provides more structural details than image from DFD_LSE. c Lifetime histograms obtained from TD_LSE, TD_MLE, DFD_LSE, and flimGANE show that flimGANE histogram most resembles that of the standard (TD_MLE at the medium-photon-count condition). d A zoom-in of red box in b. flimGANE clearly reveals more structural details than other methods. e Intensity contrast images of plasma membrane of live HeLa cells in red channel (685/40 nm) under two photon-count conditions. Dash lines represent the contours of live cells. f Intensity contrast images of nuclei of live HeLa cells in blue channel (494/34 nm) under two photon-count conditions. g 2D scatter plots of lifetime acquired at two photon-count conditions. flimGANE provided more consistent estimates under both conditions. The coefficients of determination, R2, ranging from −∞ to 1.00, are utilized to evaluate the consistency. h Overlay of FLIM images in red and blue channels (left: low-medium-photon-count; right: medium-high-photon-count).

a Schematic of CFP-g-YFP FRET pair interacting with glucose and normalized excitation and emission spectra of CFP and YFP. b Intensity contrast and FLIM images of CFP generated by TD_LSE, TD_MLE, DFD_LSE and flimGANE before and after adding 2 mM glucose. c Energy transfer efficiency, E, versus glucose concentration (error bars, standard deviation errors on the parameter estimate, n = 1507~6824 pixels). The flimGANE FRET data can be well fitted by a sigmoidal curve (R2 = 0.92).

a Intensity contrast images of FAD and NAD(P)H. b FLIM images of FAD and NAD(P)H generated by TD_LSE, TD_MLE, DFD_LSE, and flimGANE. c Pre-exponential factors (α1 and α2 = 1-α1, where α1 is the fraction of short lifetime component) of FAD and NAD(P)H obtained by different methods (error bars, standard deviation, n = 160,000 pixels). d FLIRR images show that flimGANE result best matches with TD_MLE result. e Intensity contrast images from a are normalized and segmented for mitochondria, cytoplasm, and nuclei. f Comparison of FLIRR results obtained from TD_LSE, TD_MLE, DFD_LSE, and flimGANE (solid line: based on the mitochondria images; dashed line: based on the whole-cell images without nuclei; n = 5 cells).
To obtain accurate FLIM images, the IRF of the imaging system, which is mainly decided by the width of the laser pulse and the timing dispersion of detector, should be carefully considered during lifetime estimation. While the full width at half maximum (FWHM) of IRF is stable in most of the commercial FLIM imaging systems (detector time jittering within 35–500 ps44), users often observe that the delay between the single-photon detector output and the photon-counting electronics input varies from day to day, possibly due to the instability of the photon-counting electronics caused by radio-frequency interference, laser lock instability, and temperature fluctuation. Such delay changes cause the onsets of the decays to drift, deteriorating the flimGANE analysis results. A preprocessing step, termed Center of Mass Evaluation (CoME), is thus introduced to adjust (or standardize) the temporal location of the onset of experimental decays (Supplementary Figs. 7–9). After preprocessing, the apparent lifetimes estimated by flimGANE are found free of onset-delay bias.
To prove the reliability of flimGANE in estimating an apparent fluorescence lifetime from a mixture, two fluorophores, Cy5-NHS ester (τ1 = 0.60 ns) and Atto633 (τ2 = 3.30 ns), were mixed at different ratios, creating ten samples of distinct apparent fluorescence lifetimes (τα) ranging from 0.60 to 3.30 ns. Here τ1 and τ2 were measured from the pure dye solutions and estimated by TD_MLE, whereas the theoretical apparent lifetime ταT was predicted by the equation ταT = τ1α1 + τ2(1 − α1). α1, the pre-exponential factor45, was derived from the relative brightness of the two dyes and their molar ratio41 (see Supplementary Methods). When analyzing 256 × 256-pixel images with emission rates fluctuating between 80 and 200 photons per pixel, flimGANE and TD_MLE produced the most accurate τα estimates among the four methods (Fig. 1f, and Supplementary Table 8). TD_LSE and DFD_LSE performed poorly in this low-light, two-dye mixture experiment.
Discriminating fluorescence lifetime barcode beads
We then tested flimGANE in discriminating the fluorescence lifetime barcodes. To create fluorescence lifetime barcodes, biotinylated Cy5- and Atto633-labeled DNA probes were mixed at three different ratios, Cy5-DNA:Atto633-DNA = 1:0 (barcode_1, expecting lifetime 1.90 ns); 1:1 (barcode_2, 2.40 ns) and 0:1 (barcode_3, 3.50 ns), and separately conjugated to streptavidin-coated polystyrene beads (3–4 μm in size). It was noted that the lifetime of Cy5-DNA (1.90 ns) is different from that of Cy5-NHS ester (0.60 ns). Similarly, the lifetime of Atto633-DNA (3.50 ns) is different from that of Atto633 (3.30 ns). A coverslip coated with the three barcode beads (at equal molar concentration) was scanned by the ISS Alba v5 confocal microscopic system (equipped with a 20 MHz 635 nm diode laser for excitation and a FastFLIM module for DFD acquisition41) for 31 s, generating 512 × 512-pixel DFD data with photon counts ranging from 50-300 per pixel on the beads (Fig. 2a). The acquired DFD data (i.e., cross-correlation phase histograms41) were converted into time decays for flimGANE, TD_LSE, and TD_MLE analysis (Fig. 2b). Each barcode bead was registered by ImageJ ROI manager and assigned an ID number (Supplementary Fig. 10). An apparent lifetime was assigned to each pixel on the bead (~292 pixels) and lifetimes of all pixels were plotted in a histogram. The mean lifetime for the bead was determined by the Gaussian fitting of the histogram. After examining 97 beads, we chose the midpoint lifetimes (2.15 and 2.95 ns) to be the cutoffs for barcode identification (Fig. 2c) and assigned pseudocolors to the beads (Fig. 2d). It was clear to see that flimGANE is the only method that can correctly identify the three barcodes and restore the 1:1:1 barcode ratio, while other methods often misidentified the barcodes (Fig. 2e). Whereas it was a general trend that beads with more Atto633-DNA are dimmer, possibly due to stronger self-quenching, brightness alone could not classify the three barcodes (Fig. 2c, Supplementary Fig. 11, Supplementary Table 9). It was noted that the brightness of barcode_1 beads could vary by sixfold, but the coefficient of variance (CV) of the barcode_1 lifetimes was only 0.06, making lifetime a better metric to differentiate barcodes. Barcode discrimination by other MLE-based approaches were provided in Supplementary Fig. 12. Whereas the analysis speed of MLE-pattern matching46 was much faster than that of flimGANE (0.08 vs. 0.32 ms/pixel), flimGANE still had the best barcode discrimination accuracy.
Visualizing cellular structures of Convallaria and HeLa cells
The DFD fluorescence data of Convallaria (lily of the valley) and live HeLa cells, acquired under the low- and medium-photon-count conditions (Fig. 3a), were analyzed by DFD_LSE and flimGANE (Fig. 3b), where TD_MLE (in the medium-photon-count condition, ~243 photons per pixel) served as the standard for comparison. The histogram clearly showed two characteristic lifetimes (0.90 ± 0.13 ns; 4.84 ± 1.20 ns) in the Convallaria sample (Fig. 3c). As TD_MLE with medium-photon counts had all lifetime estimates within 0.0–6.0 ns range, we limited the upper bound of the lifetime estimates to be 6.0 ns. Those 6.0 ns pixels were given the white pseudocolor and regarded as failed pixels in the FLIM images (Fig. 3d). A large number of failed pixels were seen in the DFD_LSE images (37% and 25% for the low- and medium-count images, respectively; Fig. 3b), deteriorating the visualization of structure details in the Convallaria sample. In contrast, there were very few failed pixels in the flimGANE images under the ultra-low-light condition (~83 photons per pixel), making them most resemble the TD_MLE images under the medium-light condition and provide better visualization of the structure details (Fig. 3c, d). The structure similarity index (SSIM)47 indicated that the flimGANE images were 73% more similar to the gold standard TD_MLE images than those generated by DFD_LSE (flimGANE −0.88, DFD_LSE −0.51; Supplementary Table 10), and visual information fidelity (VIF)48 showed that the flimGANE images were 1.44-fold higher than those reconstructed by DFD_LSE (flimGANE – 0.22, DFD_LSE – 0.09; Supplementary Table 10).
In the live HeLa cell sample, nuclei and membranes were stained with Hoechst and CellMaskTM Red and excited by 405 nm and 635 nm diode lasers, respectively. The contours of nuclei and cell membranes could not be clearly defined by the intensity-based images even under the low-light condition (Fig. 3e, f). Although FLIM overlay images allowed us to better visualize structural details in HeLa cells, the lifetime estimates could be biased when there were ~180 photons per pixel (low-light condition; TD_LSE and DFD_LSE images in Fig. 3h). Using the medium-high-count TD_MLE images (~600 photons per pixel) as the standard for comparison, flimGANE clearly outperformed TD_LSE and DFD_LSE in producing images that resemble the standard under the medium-light condition (Fig. 3h; Supplementary Table 11). Interestingly, when scrutinizing the assigned lifetime at each pixel, we found not only TD_LSE and DFD_LSE but also TD_MLE gave inconsistent lifetime estimates at the two excitation powers (e.g., R2 in red channel were 0.35, −1.77, and −2.32 for TD_MLE, TD_LSE, and DFD_LSE, respectively). In contrast, flimGANE provided much more consistent lifetime estimates regardless the excitation power (R2 was 0.75 in red channel; Fig. 3g).
Quantifying Förster resonance energy transfer (FRET) efficiency in live MDA-MB-231 cells
Combined with the glucose FRET sensor, FLIM has been employed to image the glucose concentration in live cells10,49. However, depending on the lifetime analysis methods, the trend of FRET change can be skewed, especially when the donor lifetime change is very small (e.g., only 0.1–0.2 ns). Our glucose FRET sensor, termed CFP-g-YFP50,51, consisted of a glucose binding domain flanked by a cyan fluorescent protein (CFP) donor and a yellow fluorescent protein (YFP) acceptor (Fig. 4a). The overlap between CFP emission and YFP absorption leads to efficient dipole-dipole interactions. The CFP-g-YFP sensor-expressed MDA-MB-231 cancer cells were starved for 24 h before adding different amount of glucose to the cell culture (final concentrations: 0, 0.5, 1.0, 2.0, 5.0, 10.0, 15.0 mM). The DFD data were collected by the confocal scanning system from a 256 × 256-pixel area before and after the addition of glucose and then analyzed by TD_LSE, TD_MLE, DFD_LSE, and flimGANE methods to generate FLIM images based on the CFP donor decays (Fig. 4b). By proper selection of regions of interest (ROI) in imaging analysis, single cells were separated from each other and from the background noise (Supplementary Fig. 13; Supplementary Table 12). Thousands of lifetime data points (apparent lifetimes, τa) were plotted in a histogram and the mean was extracted by Gaussian fitting, giving one representative donor lifetime for each glucose concentration (Supplementary Fig. 14). The energy transfer efficiency (E) was calculated based on the equation: E = 1 − (τDA/τD), where τD and τDA were the representative CFP lifetimes before and after addition of glucose, respectively (Fig. 4c). Whereas only subtle lifetime changes were seen in the mean CFP donor lifetime (0.04–0.20 ns in Supplementary Table 12, which led to low FRET efficiencies around 0.02–0.07), flimGANE-derived FRET efficiencies were not only highly reproducible but also showing a general increasing trend at higher glucose concentrations. On the other hand, the lifetime of the acceptor (YFP) did not change upon the addition of glucose (Supplementary Fig. 15). Among the four methods, DFD_LSE failed to provide a FRET efficiency response curve due to its poor lifetime estimation in this experiment, thus being excluded from Fig. 4c.
When the intensity-based method, E = 1 − (FDA/FD), was used to estimate E, the resulting response curve clearly deviated from the reasonable trend, possibly due to the artifacts such as photobleaching. At 2 mM glucose concentration, we could clearly see that the flimGANE image of CFP is the most similar to the TD_MLE image, but quite different from the TD_LSE and DFD_LSE images, in which there were many failed pixels (Fig. 4b; Supplementary Table 13). Although the TD_MLE images were similar to the flimGANE images, TD_MLE-derived FRET efficiencies had higher variations and showed an unrealistic, decreasing trend at higher glucose concentrations. In this demonstration, flimGANE not only gave a correct sensor response curve but also provided an analysis speed up to 2,800-fold faster than TD_MLE in reconstructing a FLIM image.
Quantifying metabolic states in live HeLa cells
Autofluorescence of endogenous fluorophores, such as nicotinamide adenine dinucleotide (NADH), nicotinamide adenine dinucleotide phosphate (NADPH), and flavin adenine dinucleotide (FAD), are often used to characterize the metabolic states of cancer cells through metrics such as optical redox ratio (ORR)52, optical metabolic imaging index (OMI index)53 and fluorescence lifetime redox ratio (FLIRR)54. Since the fluorescence signatures of NADH and NADPH overlap, they are often referred to as NAD(P)H fluorescence in literature. NAD(P)H (an electron donor) and FAD (an electron acceptor) are metabolic coenzymes in live cells, whose autofluorescence intensity ratio reflects the redox states of the cells and the shifts in the metabolic pathways. However, intensity-based metrics (e.g., ORR) often suffer from wavelength- and depth-dependent light scattering and absorption issues when they are used to characterize the metabolic states of tumor tissues. In contrast, lifetime-based metrics (e.g., FLIRR) bypass these issues, revealing bias-free protein-binding activities of NAD(P)H and FAD54. As ORR and fluorescence lifetimes of NAD(P)H and FAD provide complementary information, they have been combined into the OMI index that can distinguish drug-resistant cells from drug-responsive cells in tumor organoids55.
Here we demonstrate that flimGANE provides rapid and accurate autofluorescence FLIM images of live HeLa cells. DFD data at two emission channels (NAD(P)H: 425–465 nm and FAD:511–551 nm) were collected by the ISS confocal scanning system (with 405 nm excitation) and the acquired data were analyzed by the four methods, generating both intensity and FLIM images (Fig. 5a, b). We adopted FLIRR (α2_NAD(p)H/α1_FAD) as a metric to assess the metabolic response of cancer cells to an intervention. It was found that flimGANE-derived FLIRR was highly correlated with its counterpart derived by TD_MLE (Fig. 5c, d; Supplementary Table 14). Since the NAD(P)H signals came from both the mitochondrial oxidative phosphorylation and cytosolic glycolysis and the FAD signals mainly originated from the mitochondria, image segmentation was often needed to deduce the relative contributions of oxidative phosphorylation and glycolysis to the cellular redox states and help quantify the heterogeneity of cell responses54. In our analysis, an intensity threshold was selected to isolate the mitochondrial regions from the rest of the cell area, where the nuclei were manually zeroed (Fig. 5e). Again, flimGANE outperformed the other three methods, generating results most similar to those reported in literature54,56,57,58, with FLIRR values of cancer cells around 0.2–0.4 (Fig. 5f). TD_LSE and DFD_LSE provided an incorrect representation, where the former was largely skewed by the low FLIRR values and the latter showed two unrealistic peaks. TD_MLE gave a distribution similar to that of flimGANE, but with a larger FLIRR peak value, due to the inaccurate estimate of NAD(P)H lifetime under the photon-starved conditions.
Discussion
flimGANE addresses an unmet need for FLIM analysis—a computationally efficient, high-throughput and high-quality method for fluorescence lifetime estimation that works reliably even in the ultra-low-photon-count conditions (e.g., 50–100 photon counts per pixel; Fig. 1d). Among the cases studied in this report, flimGANE generated FLIM images with quality similar to those produced by the gold standard TD_MLE, but flimGANE clearly outperforms TD_MLE in barcode identification (Fig. 2), FRET characterization (Fig. 4), and metabolic state analysis (Fig. 5). We emphasize that here we intentionally acquired fluorescence data under the low- to medium-light conditions in order to compare the performance of the four methods. We found even the gold standard TD_MLE may not necessarily give consistent lifetime estimates under different excitation powers (Fig. 3g). It is thus critically important for users to understand the limitations of their lifetime analysis methods, especially when handling the low-count decays. Here we provide users with an alternative FLIM analysis approach, where the low-laser power requirement can reduce photobleaching and phototoxicity issues in delicate samples.
As FLIM finds more clinical applications such as in retinal imaging59 and tumor margin identification60 in recent years, it is necessary that we have an accurate and fit-free method to perform the lifetime imaging analysis. Through the use of convolutional and residual blocks, our model generates high-quality decays from low-photon-count inputs without introducing bias. The inference of lifetime is non-iterative and does not require parameter search to optimize the network performance. In this work, we evaluated the network performance using in-silico data, demonstrating that flimGANE can generate reasonable lifetime estimates with photon counts as low as 50 per pixel.
To the best of our knowledge, this is the first demonstration of a GAN model applied to reconstruct FLIM images. Since the use of Jensen-Shannon divergence as the objective function can cause problems such as vanishing gradients and mode collapse during GAN training, we incorporated Wasserstein metric in our model, which bypasses these issues by providing much smoother value spaces (Supplementary Figs. 16 and 17)34. We are continuing to explore the incorporation of other frameworks in our model, including the gradient penalty (WGAN-GP)61, the sequence generation framework (SeqGAN)62, and the context-aware learning63, that may in some instances provide more suitable approximate inference.
While flimGANE provides rapid, accurate, and fit-free FLIM analysis, its cost lies in the network training. In other words, flimGANE is particularly valuable for the FLIM applications where retraining is not frequently required. For instance, samples have similar fluorophore compositions (i.e., autofluorescence from metabolites in patient-derived organoids) and the IRF of the imaging system seldom changes. Different training datasets were employed to train the model separately that eventually led to the reliable results shown in Figs. 1–5 (Supplementary Table 1). A primary reason to retrain the model is due to the change of IRF (Supplementary Fig. 18). Whenever a different laser source is chosen for excitation, the filters are replaced, or the optics system is realigned, the IRF can also change, and the network should be retrained. For an entirely new imaging system, it can take up to 500 h to fully train the network with a lifetime range of 0.1–10.0 ns (two components, τ1 and τ2) and a pre-exponential factor range of 0.0–1.0 (α1). However, if we know the range of lifetime on our samples (e.g., 1.3–4.0 ns for the two lifetime components in barcode identification and 0.5–5.0 ns in live HeLa cell studies), a smaller training dataset can be employed to speed up the training process (e.g., 19 h in Supplementary Table 1).
Transfer learning64 from a previously trained network for handling a new sample can speed up the convergence of the learning process, and even potentially enhance overall performance. However, this is neither a replacement nor a required step for the entire training process. After running a sufficiently large number of training iterations for the generator (>2000), the optimal network can be selected when the validation loss no longer decreases. An essential step in generating a reliable FLIM image by our network is the accurate alignment of the fluorescence decay with respect to its corresponding IRF. A multistage preprocessing step, termed CoME (Supplementary Fig. 9), is employed to guarantee such an alignment. When running an IRF fluctuation test, a quality of estimate (G-quality score) based on known samples, and a discriminability test, we clearly show the advantages of flimGANE over other methods (Supplementary Figs. 19–21).
Taken together, our work represents an important step forward towards real-time and super-resolution65,66 FLIM. In fundamental biological research, further development of flimGANE will enable monitoring of fast binding kinetics and molecular transport dynamics inside live cells67. In medicine, flimGANE can provide rapid identification of tumor-free margin during tumor surgery60 and investigation of disease progression in the retina59. We envision that our method will soon replace TD_MLE and TD_LSE analysis packages in some commercial FLIM systems.
Methods
Structure of dataset
The dataset is composed of training data and testing data. Both training and testing data can be obtained either by a Monte Carlo (MC) simulation with the parameters or by acquiring experimental data from the ISS Alba v5 confocal microscopic system. The experimental data is the fluorescence decay histogram matrix of dimension n by n by b (n is the image size, which is either 256 or 512; b is the number of histogram bin size, which is 256 in this work).
Structure of flimGANE
The flimGANE consists of a generator, a discriminator, and an estimator. The generator is composed of a convolutional block, a multi-task layer associated with the rectified linear unit (ReLU) activation function, a decoding layer associated with the tanh activation function, and a residual block implicitly. The discriminator is composed of four layers of neural networks. All the layers are fully connected layers composed of 128, 64, 8, and 1 node. The first three layers are associated with the sigmoid activation function, and the last one used a linear function. The estimator begins with two fully connected neural networks with 128 and 64 nodes, respectively, for incoming inputs, followed by the concatenation layer and a multitask layer associated with ReLU activation function (for further details, see Supplementary Methods).
We trained flimGANE with three-stage processes: generative model training, estimative model training, and flimGANE combination training. In generative model training, we adopt the Wasserstein GAN algorithm, where the generator and the discriminator are trained with Wasserstein loss. In estimative model training, the estimator is trained with the mean-squared error cost function. In flimGANE combination training, a well-trained generator and estimator are combined and trained with the mean-squared error cost function. The Adam optimizer is applied to the generator by setting the learning rate as 1 × 10−4. The RMSprop optimizer is applied to the discriminator by setting the learning rate as 5 × 10−5. The Adam optimizer is applied to the estimator by setting the learning rate as 0.001 (for further details, see Supplementary Methods).
Evaluation metrics
To compare the proposed methods with other existing algorithms, we utilized several metrics, including execution time, mean-squared error for pixel-wise comparison, peak signal-to-noise ratio, structural similarity index, and visual information fidelity for the quality of the FLIM image with respect to the reference FLIM image (for further details, see Supplementary Methods).
Statistics and reproducibility
Paired t tests and Kolmogorov-Smirnov tests were used for statistical comparisons of data. All data presented in the text are shown as mean ± standard deviation or mean ± standard deviation error, which are specified in each section. The significance level was set to 0.05 (* <0.05, **<0.001). Sample sizes and numbers are indicated in detail in each figure caption and main text. Exclusion criteria, if applied, are specified in each corresponding section.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Code availability
All source codes, which can be obtained from https://github.com/NinaYIC/flimGANE, are available from the corresponding author on reasonable request.
References
Berezin, M. Y. & Achilefu, S. Fluorescence lifetime measurements and biological imaging. Chem. Rev. 110, 2641–2684 (2010).
Suhling, K. et al. Fluorescence lifetime imaging (FLIM): Basic concepts and some recent developments. Med. Photonics 27, 3–40 (2015).
Datta, R., Heaster, T. M., Sharick, J. T., Gillette, A. A. & Skala, M. C. Fluorescence lifetime imaging microscopy: fundamentals and advances in instrumentation, analysis, and applications. J. Biomed. Opt. 25, 071203 (2020).
Ogikubo, S. et al. Intracellular pH sensing using autofluorescence lifetime microscopy. J. Phys. Chem. B 115, 10385–10390 (2011).
Kuimova, M. K., Yahioglu, G., Levitt, J. A. & Suhling, K. Molecular rotor measures viscosity of live cells via fluorescence lifetime imaging. J. Am. Chem. Soc. 130, 6672–6673 (2008).
Okabe, K. et al. Intracellular temperature mapping with a fluorescent polymeric thermometer and fluorescence lifetime imaging microscopy. Nat. Commun. 3, 1–9 (2012).
Gerritsen, H. C., Sanders, R., Draaijer, A., Ince, C. & Levine, Y. Fluorescence lifetime imaging of oxygen in living cells. J. Fluorescence 7, 11–15 (1997).
Skala, M. C. et al. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc. Natl Acad. Sci. 104, 19494–19499 (2007).
Unger, J. et al. Method for accurate registration of tissue autofluorescence imaging data with corresponding histology: a means for enhanced tumor margin assessment. J. Biomed. Opt. 23, 015001 (2018).
Marx, V. Probes: FRET sensor design and optimization. Nat. Methods 14, 949–953 (2017).
Grant, D. M. et al. Multiplexed FRET to image multiple signaling events in live cells. Biophysical J. 95, L69–L71 (2008).
Lakowicz, J. R. & Szmacinski, H. Fluorescence lifetime-based sensing of pH, Ca2+, K+ and glucose. Sens. Actuators B: Chem. 11, 133–143 (1993).
Sun, Y., Day, R. N. & Periasamy, A. Investigating protein-protein interactions in living cells using fluorescence lifetime imaging microscopy. Nat. Protoc. 6, 1324 (2011).
Bastiaens, P. I. & Squire, A. Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell. Trends Cell Biol. 9, 48–52 (1999).
Wallrabe, H. & Periasamy, A. Imaging protein molecules using FRET and FLIM microscopy. Curr. Opin. Biotechnol. 16, 19–27 (2005).
Schrimpf, W. et al. Chemical diversity in a metal–organic framework revealed by fluorescence lifetime imaging. Nat. Commun. 9, 1647 (2018).
Straume, M., Frasier-Cadoret, S. G. & Johnson, M. L. Least-squares analysis of fluorescence data. In Topics in Fluorescence Spectroscopy. (Springer, 2002).
Laurence, T. A. & Chromy, B. A. Efficient maximum likelihood estimator fitting of histograms. Nat. Methods 7, 338 (2010).
Pelet, S., Previte, M., Laiho, L. & So, P. A fast global fitting algorithm for fluorescence lifetime imaging microscopy based on image segmentation. Biophysical J. 87, 2807–2817 (2004).
Rowley, M. I., Barber, P. R., Coolen, A. C. & Vojnovic, B. Bayesian analysis of fluorescence lifetime imaging data. In Proceedings of SPIE Conference on Multiphoton Microscopy in the Biomedical Sciences XXI). (International Society for Optics and Photonics, 2011).
Redford, G. I. & Clegg, R. M. Polar plot representation for frequency-domain analysis of fluorescence lifetimes. J. Fluorescence 15, 805 (2005).
Digman, M. A., Caiolfa, V. R., Zamai, M. & Gratton, E. The phasor approach to fluorescence lifetime imaging analysis. Biophysical J. 94, L14–L16 (2008).
Lee, K. B. et al. Application of the stretched exponential function to fluorescence lifetime imaging. Biophysical J. 81, 1265–1274 (2001).
Jo, J. A., Fang, Q., Papaioannou, T. & Marcu, L. Fast model-free deconvolution of fluorescence decay for analysis of biological systems. J. Biomed. Opt. 9, 743–753 (2004).
Wu, G., Nowotny, T., Zhang, Y., Yu, H.-Q. & Li, D. D.-U. Artificial neural network approaches for fluorescence lifetime imaging techniques. Opt. Lett. 41, 2561–2564 (2016).
Smith, J. T. et al. Fast fit-free analysis of fluorescence lifetime imaging via deep learning. Proc. Natl Acad. Sci. 116, 24019–24030 (2019).
Yao, R., Ochoa, M., Yan, P. & Intes, X. Net-FLICS: fast quantitative wide-field fluorescence lifetime imaging with compressed sensing–a deep learning approach. Light.: Sci. Appl. 8, 26 (2019).
Goodfellow, I. et al. Generative adversarial nets. In Advances in Neural Information Processing Systems (2014).
Rivenson, Y. et al. Virtual histological staining of unlabelled tissue-autofluorescence images via deep learning. Nat. Biomed. Eng. 3, 466 (2019).
Schawinski, K., Zhang, C., Zhang, H., Fowler, L. & Santhanam, G. K. Generative adversarial networks recover features in astrophysical images of galaxies beyond the deconvolution limit. Monthly Not. R. Astronomical Soc.: Lett. 467, L110–L114 (2017).
Wang, H. et al. Deep learning enables cross-modality super-resolution in fluorescence microscopy. Nat. Methods 16, 103–110 (2019).
Guimaraes, G. L., Sanchez-Lengeling, B., Outeiral, C., Farias, P. L. C. & Aspuru-Guzik, A. Objective-reinforced generative adversarial networks (organ) for sequence generation models. arXiv preprint at https://arxivorg/abs/170510843 (2017).
Ledig, C. et al. Photo-realistic single image super-resolution using a generative adversarial network. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (2017).
Arjovsky, M., Chintala, S. & Bottou, L. Wasserstein generative adversarial networks. In International conference on machine learning, PMLR. 214–223 (2017).
Ware, W. R., Doemeny, L. J. & Nemzek, T. L. Deconvolution of fluorescence and phosphorescence decay curves. Least-squares method. J. Phys. Chem. 77, 2038–2048 (1973).
Gratton, E., Breusegem, S., Sutin, J. D., Ruan, Q. & Barry, N. P. Fluorescence lifetime imaging for the two-photon microscope: time-domain and frequency-domain methods. J. Biomed. Opt. 8, 381–391 (2003).
Chen, Y.-I. et al. Measuring DNA hybridization kinetics in live cells using a time-resolved 3D single-molecule tracking method. J. Am. Chem. Soc. 141, 15747–15750 (2019).
Liu, C. et al. 3D single-molecule tracking enables direct hybridization kinetics measurement in solution. Nanoscale 9, 5664–5670 (2017).
Turton, D. A., Reid, G. D. & Beddard, G. S. Accurate analysis of fluorescence decays from single molecules in photon counting experiments. Anal. Chem. 75, 4182–4187 (2003).
Lieske, T. et al. Embedded Fluorescence Lifetime Determination for High-Throughput, Low-Photon-Number Applications. J. Signal Process. Syst. 91, 819–831 (2019).
Colyer, R. A., Lee, C. & Gratton, E. A novel fluorescence lifetime imaging system that optimizes photon efficiency. Microsc. Res. Tech. 71, 201–213 (2008).
Yang, H. et al. Protein conformational dynamics probed by single-molecule electron transfer. Science 302, 262–266 (2003).
Elson, D. et al. Real-time time-domain fluorescence lifetime imaging including single-shot acquisition with a segmented optical image intensifier. N. J. Phys. 6, 180 (2004).
Buller, G. & Collins, R. Single-photon generation and detection. Meas. Sci. Technol. 21, 012002 (2009).
Lakowicz, J. R. Fluorescence spectroscopic investigations of the dynamic properties of proteins, membranes and nucleic acids. J. Biochemical Biophysical Methods 2, 91–119 (1980).
Enderlein, J. & Sauer, M. Optimal algorithm for single-molecule identification with time-correlated single-photon counting. J. Phys. Chem. A 105, 48–53 (2001).
Wang, Z., Bovik, A. C., Sheikh, H. R. & Simoncelli, E. P. Image quality assessment: from error visibility to structural similarity. IEEE Trans. Image Process. 13, 600–612 (2004).
Sheikh, H. R., Bovik, A. C. A visual information fidelity approach to video quality assessment. In International Workshop on Video Processing and Quality Metrics for Consumer Electronics (2005).
Veetil, J. V., Jin, S. & Ye, K. Fluorescence Lifetime Imaging Microscopy of Intracellular Glucose Dynamics. J. Diabetes Sci. Technol. 6, 1276–1285 (2012).
Takanaga, H., Chaudhuri, B. & Frommer, W. B. GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochimica et. Biophysica Acta (BBA)-Biomembranes 1778, 1091–1099 (2008).
Yang, J. et al. Longitudinal FRET Imaging of Glucose and Lactate Dynamics and Response to Therapy in Breast Cancer Cells. Molecular Imaging and Biology, 1–12 (2021).
Chance, B., Schoener, B., Oshino, R., Itshak, F. & Nakase, Y. Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. J. Biol. Chem. 254, 4764–4771 (1979).
Walsh, A. J. et al. Quantitative optical imaging of primary tumor organoid metabolism predicts drug response in breast cancer. Cancer Res. 74, 5184–5194 (2014).
Wallrabe, H. et al. Segmented cell analyses to measure redox states of autofluorescent NAD (P) H, FAD & Trp in cancer cells by FLIM. Sci. Rep. 8, 1–11 (2018).
Walsh, A. J., Castellanos, J. A., Nagathihalli, N. S., Merchant, N. B. & Skala, M. C. Optical imaging of drug-induced metabolism changes in murine and human pancreatic cancer organoids reveals heterogeneous drug response. Pancreas 45, 863 (2016).
Alam, S. R. et al. Investigation of mitochondrial metabolic response to doxorubicin in prostate cancer cells: an NADH, FAD and tryptophan FLIM assay. Sci. Rep. 7, 1–10 (2017).
Cao, R., Wallrabe, H., Siller, K., Rehman Alam, S. & Periasamy, A. Single‐cell redox states analyzed by fluorescence lifetime metrics and tryptophan FRET interaction with NAD (P) H. Cytom. Part A 95, 110–121 (2019).
Penjweini, R. et al. Single cell-based fluorescence lifetime imaging of intracellular oxygenation and metabolism. Redox Biology, 101549 (2020).
Dysli, C. et al. Fluorescence lifetime imaging ophthalmoscopy. Prog. Retinal Eye Res. 60, 120–143 (2017).
Alfonso‐Garcia, A. et al. Real‐time augmented reality for delineation of surgical margins during neurosurgery using autofluorescence lifetime contrast. J. Biophotonics 13, e201900108 (2020).
Gulrajani, I., Ahmed, F., Arjovsky, M., Dumoulin, V., Courville, A. C. Improved training of wasserstein gans. In Advances in Neural Information Processing Systems (2017).
Yu, L., Zhang, W., Wang, J. & Yu, Y. Seqgan: Sequence generative adversarial nets with policy gradient. In AAAI conference on artificial intelligence (2017).
Perdikis, S., Leeb, R., Chavarriaga, R., Millan, J. D. R. Context–aware Learning for Generative Models. IEEE Transactions on Neural Networks and Learning Systems (2020).
Pan, S. J. & Yang, Q. A survey on transfer learning. IEEE Trans. Knowl. Data Eng. 22, 1345–1359 (2009).
Castello, M. et al. A robust and versatile platform for image scanning microscopy enabling super-resolution FLIM. Nat. Methods 16, 175–178 (2019).
Niehörster, T. et al. Multi-target spectrally resolved fluorescence lifetime imaging microscopy. Nat. Methods 13, 257–262 (2016).
Chen, Y.-I. et al. Recent developments in the characterization of nucleic acid hybridization kinetics. Curr. Opinion Biomed. Eng. 19, 100305 (2021).
Acknowledgements
The authors thank Mr. Li-Heng Chen from Prof. Alan C. Bovik’s Image & Video Engineering (LIVE) lab at University of Texas at Austin, Dr. Beniamino Barbieri from ISS Inc. and Mr. Jason Smith from Rensselaer Polytechnic Institute for valuable inputs and discussions. This work is supported by NSF (2041345), the Welch Foundation (F-1833), NIH (GM129617 and EY033106), Texas 4000 Foundation, Texas Health Catalyst Program at Dell Medical School and CPRIT grant RR160005 (to T.E.Y.). T.E.Y. is a CPRIT Scholar in Cancer Research. Y.-I.C. is supported by the University Graduate Continuing Fellowship at UT Austin.
Author information
Authors and Affiliations
Contributions
Y.-I.C., Y.-J.C. and H.-C.Y. conceived the project and wrote the article. Y.-I.C., Y.-J.C., H.-C.Y. and S.-C.L. developed the image processing and analysis software. Y.-I.C., J.Y. and H.-C.Y. designed the experiments. Y.-I.C., J.Y., Y.-A.K. and S.H. prepared samples, performed cell culture, and collected images. Y.-I.C., S.-C.L. and T.D.N. supported the special instrumental setup for all experiments. H.G.R., S.R.S., T.E.Y. and Y.-L.L. advised the experimental designs and data analysis. H.-C.Y. supervised the project.
Corresponding author
Ethics declarations
Competing interests
H.-C.Y., Y.-I.C., Y.-J.C., S.-C.L., T.D.N., S.H. and Y.-A.K. have a patent application on the contents of the presented results. The remaining authors declare no competing interests.
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Natalie Elia and Christina Karlsson Rosenthal.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, YI., Chang, YJ., Liao, SC. et al. Generative adversarial network enables rapid and robust fluorescence lifetime image analysis in live cells. Commun Biol 5, 18 (2022). https://doi.org/10.1038/s42003-021-02938-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02938-w
This article is cited by
-
Assessing the impact of extracellular matrix fiber orientation on breast cancer cellular metabolism
Cancer Cell International (2024)
-
Coupling a recurrent neural network to SPAD TCSPC systems for real-time fluorescence lifetime imaging
Scientific Reports (2024)
-
CASPI: collaborative photon processing for active single-photon imaging
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
