
Abstract
Transporters play vital roles in acquiring antimicrobial resistance among pathogenic bacteria. In this study, we report the X-ray structure of NorC, a 14-transmembrane major facilitator superfamily member that is implicated in fluoroquinolone resistance in drug-resistant Staphylococcus aureus strains, at a resolution of 3.6 Å. The NorC structure was determined in complex with a single-domain camelid antibody that interacts at the extracellular face of the transporter and stabilizes it in an outward-open conformation. The complementarity determining regions of the antibody enter and block solvent access to the interior of the vestibule, thereby inhibiting alternating-access. NorC specifically interacts with an organic cation, tetraphenylphosphonium, although it does not demonstrate an ability to transport it. The interaction is compromised in the presence of NorC-antibody complex, consequently establishing a strategy to detect and block NorC and related transporters through the use of single-domain camelid antibodies.
Similar content being viewed by others
Introduction
Integral membrane transporters involved in multidrug efflux render pathogenic bacteria resistant to antimicrobial compounds through the reduced accumulation of drugs within cells1. Structurally and mechanistically diverse classes of primary and secondary active transporters facilitate the survival of pathogens against antibacterial compounds, either through direct efflux or through enhanced fitness or persistence2,3. The Gram-positive pathogen, Staphylococcus aureus, is known to cause mild skin and soft-tissue infections to severe infections like endocarditis, bacteremia, sepsis, and pneumonia4. A major factor that aids in the gain of multidrug resistance is the enhanced expression and activity of efflux transporters5. Among multidrug efflux transporters, the major facilitator superfamily (MFS) constitutes an extensive array of transporters with a promiscuous ability to transport a diverse array of substrates in both Gram-positive and Gram-negative pathogens6,7,8. Drug-resistant strains of S. aureus employ a diverse set of chromosomal and plasmid-encoded MFS transporters to gain antibiotic resistance9. Transporters like NorA, NorB, and NorC are chromosomally encoded and protect S. aureus against fluoroquinolones9,10,11. QacA and QacB are plasmid-encoded and provide resistance to monovalent and divalent quaternary ammonium compounds12,13.
MFS transporters involved in multidrug efflux primarily act by coupling efflux to proton gradients across the bacterial membrane14. The drug:H+ stoichiometry can differ, and substrate efflux can happen through electroneutral exchange or via electrogenic transport. MFS transporters are multi-pass integral membrane proteins comprising 12 or 14 transmembrane (TM) helices, and drug:H+ antiporters (DHA) are classified as DHA1 and DHA2 depending on the presence of 12 or 14 TM helices, respectively6. Multiple structures of DHA1 members, including MdfA, LmrP, and EmrD, have been solved in different conformational states that facilitate an understanding of the alternating-access in DHA members through the rocker-switch mechanism15,16,17. However, there is no representative structure for the DHA2 members that comprise well-studied transporters, including QacA/B, Tet38, NorB, and NorC. Besides S. aureus, numerous pathogens express DHA2 members to effectively counter antibacterial stress. Efflux transporters provide a potential therapeutic strategy to counter antimicrobial resistance through efflux pump inhibitors18. Blockers of antimicrobial efflux can enhance the efficacy of existing antibiotics, thereby serving as antibiotic adjuvants19.
In this study, we report the X-ray structure of NorC, a 14-TM putative efflux transporter representing a unique subset among MFS transporters in complex with a Zn2+-bound single-domain Indian camelid antibody (ICab). The structure of the ICab–NorC complex was solved at a resolution of 3.6 Å. Isolated initially as a crystallization chaperone for NorC, the ICab interacts with NorC through the insertion of CDR loops into the vestibule to effectively lock the transporter in an outward-open state. The transporter does not display a natural ability to transport fluoroquinolone or monovalent cations but can specifically interact with tetraphenylphosphonium, a monovalent cationic antibacterial. This interaction is blocked by the presence of the ICab. We anticipate that the structure of the DHA2 member, NorC, would facilitate the investigation of other DHA2 members in diverse pathogens, and the identification of single-domain antibodies that block efflux can be explored as a novel paradigm for efflux pump detection and inhibition.
Results
NorC represents a unique subset among DHA2 members. Phylogenetic analysis reveals that NorC and related sequences form a separate clade among MFS transporters implicated in multidrug efflux (Fig.1a). Despite a clear prediction of 14-TM helices, NorC/NorB-like transporters differ substantially in comparison to the DHA2 members of the QacA-like transporters and the 14-TM proton-coupled oligopeptide symporters (POTs). Among the members of this subset, a high degree of sequence conservation is observed (~58–88%) (Supplementary Fig. S1). NorC and NorB display a high sequence identity of nearly 70%. It is observed that NorB can aid in the survival of S. aureus in an abscess environment and is also overexpressed in persister populations of antibiotic-resistant S. aureus strains10,20,21. Interestingly, the NorC/NorB-like transporters lack the typical protonation and conserved motif C residues characteristic of DHA22. While conventional DHA2 members retain one or more negatively charged residues for protonation-driven efflux, NorB and NorC lack negative charges facing the transport vestibule. For instance, D34 (TM1), a conserved acidic residue meant for substrate recognition and protonation among DHA2 members like QacA, is replaced by glutamine in the NorB/NorC clade. These changes may have significant consequences on the transport properties of NorC. We screened a wide array of potential substrates that could be transported by NorC, including fluoroquinolones. We employed capillary-based differential scanning fluorimetry (DSF) to identify compounds that can enhance the stability of NorC, thereby suggesting a potential substrate23. The stability increments were observed primarily for tetraphenylphosphonium (TPP+) (Supplementary Fig. S2). An analysis of the binding propensity for TPP+ revealed an ability to interact with NorC (described later). We also evaluated the ability of NorC to facilitate survival of an E. coli strain, deficient in acrB, mdfA, and ydhE, in the presence of antibacterial substrates like norfloxacin and tetraphenylphosphonium (TPP+) (Supplementary Fig. S3). The assay revealed no visible ability of NorC to protect against TPP+ and norfloxacin when overexpressed in E. coli.

a Radial phenogram of DHA1 and DHA2 transporters. Characterized DHA2 transporters and their close homologs have been categorized with QacA/EmrB and their homologs for their sequentially coherent residues. b Side view of NorC bound to the ICab in an outward-open conformation. The transmembrane helices have been grouped in repeats of three helices to depict symmetric organization; vestibule lining TM helices 1, 4, 9, and 12 are in blue, 2, 5, 10, and 13 lining the domain interface in orange, 3, 6, 11, and 14 in green, and helices 7 and 8 are in gray. c APBS electrostatic representation showing positively charged region of the vestibule near the cytoplasmic end of NorC, owing to K398 (shown as sticks). d, e Note that TM7 and TM8 lie outside the primary pseudo-two-fold symmetric helical bundles that surround the vestibule. f Structural superposition of LmrP (blue, RMSD = 3.0 Å), MdfA (brown, RMSD = 2.5 Å), GkPOT (olive, RMSD = 4.4 Å) and XcPOT (cyan, RMSD = 4.2 Å) with NorC; angular differences between TMs 7 and 8 of NorC and those of POTs are shown in the same superposition. MdfA and LmrP are members of the DHA family while GkPOT and XcPOT are proton-dependent oligopeptide symporters from Geobacillus kaustophilus and Xanthomonas campestris, respectively. g Solid section of NorC showing vestibular environment.
Crystallization and structure determination
The NorC WT (wild-type) protein was heterologously expressed and purified from E. coli membranes and crystallized in complex with a Zn2+-bound ICab that was identified and isolated in an earlier study24 (Supplementary Fig. S4). Despite obtaining single crystals, only a minor fraction would diffract and were susceptible to radiation damage. Multiple datasets were merged and scaled together to obtain a complete dataset to a resolution of 3.7 Å. The phases were estimated through Se-SAD phasing (described in methods). The resulting electron density was subjected to density modification, followed by manual model building and refinement. All the 14 TM helices could be modeled, and the main chain was traced with the help of selenium peaks resulting in the correct assignment of amino acids into the density. Multiple rounds of model building and refinement iteratively led to the convergence of refinement and generated a significantly improved electron density map (Supplementary Fig. S5). A mutation in TM13, K398A, allowed a marginal improvement of data quality to 3.6 Å with improved side-chain densities in the molecule leading to the refinement of the structure to acceptable R-factors (Table 1).
X-ray structure of NorC bound to single-domain camelid antibody
The structure of NorC retains a typical MFS fold and all the 14 TM helices of NorC could be modeled unambiguously in the electron density, although loop regions connecting TM6 with TM7 (189–198), TM7 with TM8 (223–226), and TM8 with TM9 (247–257) were missing due to inherent disorder (Fig. 1b, Supplementary Fig. S5). The TM helices are organized in two six-helix bundles, a conserved feature of MFS transporters, with the linker between the helical domains forming two additional TM helices leading to a 6 + 2 + 6 arrangement (Fig. 1d). The helices 1 to 6 and 9 to 14 are related by a pseudo twofold symmetry that facilitates conformational transitions through the rocker-switch mechanism25. Among the symmetrically arranged helices, TMs 1, 4, 9, 12 line the solvent-exposed vestibule forming cavity helices (blue), TMs 2, 5, 10, 13 are organized as rocker helices (orange) and 3, 6, 11, 14 are on the exterior facing the lipid interface forming the support helices (green) (Fig. 1e)26. The arrangement of the helical bundles is consistent with the organization in MFS transporter (MdfA and LmrP) structures in the outward-open state15,16. The MFS members, proton-coupled oligopeptide transporters (POTs) that are involved in the symport of di or tri-peptides, also comprise 14 TM helices27,28,29. However, all the POT structures known thus far were elucidated in the inward-open conformation unlike NorC, which is in the outward-open conformation. The relative positions of the additional TM helices 7 and 8 differ substantially in their orientation in NorC, in comparison to equivalent helices in POTs (Fig. 1f). Despite these differences, the presence of two additional helices as an insertion in the linker connecting the six-helix bundles among distantly related NorC and POTs makes this a consistent organization among 14-TM MFS members. The TMs 7 and 8, in NorC, are observed to occlude a wide opening between the helical bundles towards the outer leaflet of the membrane lined by TMs 2 and 13. When compared to the Hoechst bound LmrP structure16, the position of TMs 7 and 8 clashes with Hoechst, potentially alluding to the ability of TMs 7 and 8 to influence substrate interactions among NorC-like transporters (Supplementary Fig. S6).
The structure of NorC is solved in an outward-open conformation with entry into the vestibule blocked by the ICab that was used as a crystallization chaperone (Fig. 2a). Both aromatic and non-polar residues W23 (TM1), W139 (TM5), W144 (TM5), M116 (TM4) line the vestibule creating hydrophobic pockets as well as regions that can allow interactions with charged substrates (Fig. 1g). Unlike QacA, NorC lacks any negatively charged residues within the vestibule although a single cationic charge at K398 is observed in the cytosolic half of TM13 whose side chain is positioned facing the vestibule (Fig. 1c). Theoretical estimation of pKa of K398 yielded a value of 8.030. This value is lower than the pKa of solvent-exposed lysine, which is usually around 10.431. The reduction in pKa of lysine to a physiological range facilitates proton exchange during substrate transport. While acidic residues like glutamate and aspartate remain the predominant protonatable residues for substrate transport, lysines also couple protonation-driven transport as observed in Na+/H+ antiporters and ApcT family of amino acid transporters32,33. The location of K398, in NorC, coincides with the position of E407 in TM13 of QacA homology model where it was observed to be vital as both a general protonation site and substrate recognition site for certain substrates34. The vestibule also has a substitution of Q27 instead of acidic residues (D34) commonly observed in DHA members including QacA and MdfA that transport cations.

a Solid section of NorC (light blue) in complex with ICab (purple). CDRs 1–3 are shown in red, green, and cyan loops respectively, with Zn2+ as a blue sphere. The region of interest in dashed border is magnified in b with interchain H-bonds (dashed lines, lengths labeled) and participating residues (in sticks; lines shown for those not participating via their side chains, for clarity). NorC helices interacting with ICab through H-bonds are shown as cartoon. c Y36 (CDR1) and F111 and L112 (CDR3) involved in extensive hydrophobic interactions at the interface of the N- and C- terminal domain of NorC are shown with participating residues (sticks) and complementary NorC contour (transparent surface). d CDR3 (cyan cartoon and surface) forming a near-perfect shape complementarity with interacting residues from NorC (white surface patches) involved in polar and VdW interactions. e Structural alignment of ICab in native state (gray) and when bound to NorC (purple). CDR1 and CDR3 display the conformational changes that residues (sticks and transparent spheres) undergo during complexation (arrows).
Most MFS transporters are identified by the presence of conserved motifs that are characteristic of the superfamily despite weak sequence identities. In NorC, the motif A with a consensus sequence of G66X3D70(K/R)XGR74X(K/R) is well-conserved and consistent with other MFS transporters (Supplementary Fig. S1). The D70 is involved in a salt bridge interaction with R74 of the same motif (Supplementary Fig. S7). R105 in TM4 forms the motif B and interacts with the main chain carbonyl groups of A26 (TM1) and occupies a similar position in comparison to its equivalent residue R112 in MdfA. The TM5 helix comprises motif C, which is characteristically present in antiporters and comprises a stretch of glycine residues interspersed with a GP dipeptide (GX8GX3GPX2GG)22. This motif is consistently present across numerous antiporter sequences and is absent in the case of symporters and uniporters22. While the glycine-rich sequence provides for the necessary freedom during conformational transitions from outward to inward-facing states, the main-chain carbonyl group of proline in the motif mediates interaction with the helices of the other domain via ordered water molecules in the inward-facing state, thus stabilizing that conformation, as seen in case of MdfA15. Incidentally, NorC retains a bulk of the glycine residues although the -GP- dipeptide is substituted by -CS- dipeptide (C150, S151) (Supplementary Fig. S1).
ICab interacts with NorC in the outward-open state
The vestibule of NorC is open to the extracellular side although solvent access is limited through the interactions with ICab (Figs. 2a and 4a). Nanobodies derived from llamas have proven to be powerful tools in studying integral membrane protein structures35. While a large number of GPCR structures have been studied using nanobodies, a few transporters like LacY have been crystallized in complex with nanobodies that bind to extracellular face of the transporter36. Despite interacting with LacY with high affinity, the nanobodies allow the interaction of sugar in the binding pocket36. The ICab that we isolated has a longer CDR1 loop compared to other camelid and llama antibodies and the CDR3 loop harbors a unique Zn2+-binding site. Destabilizing the site leads to a loss of interactions with NorC24. The CDR1 loop has an additional -STYS- motif that forms a β-turn and inserts deep into the vestibule of NorC, to nearly half its depth (Fig. 2a) forming multiple polar and hydrophobic interactions with NorC (Fig. 2b). The motif wedges between symmetry-related helices TM5 from the N-terminal domain and TM9 and TM10 from the C-terminal domain. TM5 of MdfA undergoes angular shifts of 15° and a clockwise twist of 45° while changing from inward-open to outward-open conformation to facilitate alternating-access15. The TM5 of NorC is blocked by the presence of ICab whose CDR1 as well as CDR3 interacts with it. The presence of ICab CDR1 loop in the NorC vestibule further prevents the TM9 from curving as observed in the equivalent helix in MdfA(TM7) and restrains it as a linear helix (Supplementary Fig. S8). The CDR3 also interacts closely with residues in TMs 1, 2 and 5 towards the extracellular face (Fig. 2d), which undergo substantial shifts during the rocker-switch motion. While the epitope residues do not display any interaction with the Zn2+ ion directly, it is evident from the NorC-ICab complex that Zn2+-binding allows the CDR3 to have a conformation that facilitates high-affinity interactions with NorC. The residues F111 and L112 of ICab also wedge in the gap between TMs 5 and 10 that are part of the rocker helices (TMs 2, 5, 10, 13) that facilitate conformational changes in MFS transporters (Fig. 2c).
NorC interaction induces conformational changes within ICab
The ICab used in this study displays a unique mode of interaction with the NorC. It has a high affinity of 38 nM and interacts specifically with NorC without any cross-reactivity to NorB, despite their close similarity, as observed in fluorescence-detection size-exclusion chromatography (FSEC) and microscale thermophoresis (MST) experiments (Fig. 3a, b). A comparison of the ICab crystal structures in the NorC-bound and free forms reveals subtle conformational changes in the CDR1 loop when it interacts with the antigen. The β-turn undergoes a straightening in its position and the T35 undergoes a 3.0 Å shift in its Cα position. Similarly, the Y36 in the β-turn undergoes a displacement of its phenol side chain by 3.3 Å to facilitate its wedging between TMs 5 and 10 (Fig. 2e). The CDR3 largely retains a conformation in the bound state similar to that of the free ICab with a minor displacement of about 1.5 Å in the region between 109 and 115. However, the side chain of F111 undergoes a massive shift in the χ1 torsion angle with 148° rotation to interact with NorC residues L152 and L298 (Supplementary movie SM1).

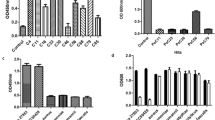
a Tryptophan fluorescence trace of NorC in FSEC, shifts to a slightly higher molecular mass when complexed with ICab. b NorB does not display a shift indicating a lack of interaction with ICab (inset). c, d Corresponding MST profiles of NorC and NorB respectively with Kd = 37.5 ± 16 nM for ICab and NorC but no detectable affinity for NorB. n = 2 for NorB vs ICab titration and n = 3 for NorC vs ICab for independent replicates. Source data are provided in the source data file.
ICab blocks access to antibacterial compounds
The ability of ICab to interact with the vestibule results in a substantial reduction of solvent accessibility within the vestibule of NorC, likely compromising substrate interactions (Fig. 4a). The natural substrates of NorB/NorC-like transporters are unknown. Most members related to NorC however retain an amidohydrolase in an open reading frame alongside the transporter gene in the di/tri-cistronic mRNA, which could be involved in processing the substrates transported by NorC. While we do not observe a direct efflux of fluoroquinolones with NorC (Supplementary Fig. S3b), we do however observe specific interactions with TPP+ with a Kd value of 4 μM despite lacking a propensity to transport cationic antibacterials (Fig. 4b). TPP+ interaction is observed in both NorB and NorC K398A leading to a nominal enhancement of stability (ΔTm = +1.2 °C and +2.7 °C, respectively) (Supplementary Fig. S3a). This is entirely plausible in the context of other MFS transporter structures like LmrP, which has a detergent molecule, a phospholipid and a cationic dye (Hoechst) observed in distinct subsites within the vestibule16. We suggest that TPP+, given its multiple phenyl groups could stack and interact with the aromatic side chains within the NorC vestibule, thereby acting as an allosteric inhibitor of NorC instead of being a substrate. This presents two potential modes of TPP+ affecting ICab interactions with NorC. If TPP+ binding induces a conformational shift of NorC to occluded or inward-open states or binds competitively to the ICab binding site, the ICab interactions with NorC would weaken leading to reduced affinity (mode A, Fig. 4c). On the contrary, if TPP+ interacts with the outward-open state of the transporter and does not block ICab binding to the transporter the affinity would remain unaltered (mode B, Fig. 4c). We observe that while ICab binding blocks TPP+ interactions with NorC, the binding of TPP+ with NorC, on the contrary, does not influence the binding of ICab. In the presence of 1 mM TPP+, ICab interacts with similar affinity of 35.6 ± 16 nM with NorC (Fig. 4c). We could however observe a reduction in the enthalpy of ICab binding to NorC-TPP+ complex and suggest that TPP+ interaction likely pre-stabilizes a conformation favored for ICab binding (Fig. 4c, Supplementary Table ST1). However, the free energy of binding remains unaltered, hinting at the possibility that TPP+ too, like ICab, binds to the same conformation of NorC without impeding the interaction of the ICab.

a Water accessible surface (green spheres) in NorC’s vestibule starting from W139/K398 (black spheres), in the absence (left) and presence (right) of ICab. The outward-open conformation completely blocks accessibility from the cytosolic side of NorC. b Microscale thermophoresis profiles for NorC and tetraphenylphosphonium (TPP+) in the presence and absence of ICab. Kd = 4 ± 0.8 μM for NorC and TPP+. n = 4 for NorC-TPP+ titration for independent replicates. The value provided is a concurrent representative from one. c ITC traces of ICab titrated against NorC WT in the presence and absence of TPP+ (offset corrected differential power, top panel; baseline subtracted binding isotherm with integrated peaks normalized against moles of injectant, bottom panel) concurs with mode “B” of TPP+ and ICab binding. n = 2 for independent replicates. Source data is provided in the source data file.
ICab and TPP+ binding induce a conformational shift of NorC
In order to evaluate if NorC is capable of conformational transitions in the membrane in response to ICab or TPP+ binding, we devised a biochemical strategy. A polyethylene glycol 5 K maleimide (PEG-Mal) was used as a crosslinker to form a covalent adduct with exposed cysteines in NorC and observed for a 5 kDa shift in the migration of NorC in overexpressed E. coli membranes through western blots. Despite having an exposed cysteine (C150) in the outward-open conformation of NorC, in the absence of bound ICab, we failed to observe any PEG-Mal crosslinking with NorC WT suggesting the lack of PEG-Mal exposure of C150 in NorC. In order to evaluate whether NorC is in a likely cytosol-facing conformation, we further engineered a cysteine at T378 position (TM12), which likely does not change the conformation of NorC as the mutant protein retains interactions with the ICab (Fig. 5a). Interestingly, the T378C mutant displays a clear PEG-Mal adduct formation suggesting that NorC in the membranes exists in an inward-open conformation. We further investigated the conformation by incubating the membranes with an excess of ICab and TPP+ and observed that in both cases the extent of PEG-Mal crosslink is visibly reduced (Fig. 5b, Supplementary Fig. S9). A combination of ICab and TPP+ displays the least extent of crosslinking suggesting that both the ICab and TPP+ force the conformation of NorC into an outward-open state (Fig. 5b).

a Schematic showing the accessibility of C150 and T378 of NorC in different conformations in native membranes. FSEC profile with detergent purified NorC T378C incubated with ICab shows that the mutation does not alter NorC’s ability to interact with ICab. b Methoxypolyethylene glycol maleimide (or PEG5k Mal; avg mol wt = 5 kDa) accessibility assay in NorC’s vestibule in E. coli membranes. PEG5k Mal does not react with NorC WT in native membranes or in detergent purified form, while it readily interacts with NorC T378C, as seen in the western blot (probed with Anti-Histag antibody), but the reactivity decreases in presence of excess ICab, TPP+ or both. n = 8 for independent replicates. Source data are provided in the source data file.
Discussion
NorC structure is the first representative structure of the DHA2 family and reveals the architecture of this distinct class of transporters within the MFS fold. While the true physiological substrate(s) of NorC is not evident, both NorC and its closely related homolog, NorB are observed to protect S. aureus against fluoroquinolones, which are broad-spectrum antibiotics. In this study, we have identified that TPP+, a cationic substrate of antibacterial efflux pumps, interacts with the outward-open conformation of NorC. TPP+ interacts despite mutating the protonatable lysine in the vestibule (K398) suggesting that the compound, given its multiple phenyl groups, could interact at an allosteric site stabilizing the outward-open conformation of NorC. This is very different from competitive binding displayed by TPP+ in antibacterial efflux transporters, MdfA and QacA, wherein substituting the primary protonation sites leads to a clear loss of TPP+ interaction and transport propensity34,37. In the presence of the ICab, entry into the vestibule of NorC is clearly blocked causing a complete loss of interactions of the antibacterial compound TPP+ with NorC (Fig. 4a, b). The ICab–NorC interactions happen through the insertion of the CDR loops deep into the vestibule of NorC with the transporter stabilized in an outward-open conformation. In doing so, the ICab–NorC interaction, resembles a “bottle-cork” that prevents antibacterial compound interactions and enforces a lock on the conformational changes within the transporter.
The ability to detect and block efflux transporters is proposed as a promising strategy to aid and improve the efficacy of existing antibiotics. The use of nanobodies to target plasma membrane primary and secondary active transporters for therapeutic outcomes has been conceptually proposed38. Nanobodies that antagonize purinergic receptor activity have been developed39,40. Nanobodies have also been developed as blockers of both primary and secondary active eukaryotic transporters including the Zn-transporting P-type ATPase41 and the vesicular glutamate transporter (VGlut)42. Nanobodies have been developed as blockers of bacterial transporters like the cyanocobalamine (Vitamin B12) transporter BtuF43 and used as chaperones to study the structure of a peptide-oligopeptide transporter-like DptA29 and NorC. The ICab characterized in this study provides a high-fidelity detection tool to observe the presence of NorC in S. aureus populations. Its ability to wedge deeply in the NorC vestibule and lock it in an outward-open conformation serves as a proof-of-principle to use ICabs or nanobodies as efflux pump inhibitors and a potential strategy to overcome antimicrobial resistance.
Methods
Statistics and reproducibility
All experiments described were performed in independent replicates (and technical replicates as well in the case of DSF screening). All comparative statistical analyses were done on complete sets of experiments. To ensure no variability arising due to batch-to-batch variations of proteins, statistical analyses for DSF experiments were performed within both technical replicates as well as independent replicates. However, no conflation on technical and biological variability has been performed.
Sequence alignment and phylogenetic analysis
NorC (Uniprot ID A0A0E1ACG1) was used to search for closely related homologous sequences in a non-redundant sequence database. To search for more homologous sequences amongst the known DHA family members, a transporter classification database was used. The alignment was carried out using clustalw program with iterative HMM clustering. The output properly aligned the TMs of all the query proteins—a benchmark we used to check the alignment accuracy for this highly divergent dataset. The alignment was used further for functional and phylogenetic analysis. The evolutionary history was inferred by using the maximum likelihood method and JTT matrix-based model44. The tree with the highest log likelihood (−35519.34) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor–Join and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model, and then selecting the topology with a superior log-likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (2 categories (+G, parameter = 3.0280)). This analysis involved 39 amino acid sequences. There were a total of 611 positions in the final dataset. Evolutionary analyses were conducted in MEGA X45.
Survival assays
To check for phenotype (resistance against antibacterials), C41 cells (transformed with IPTG inducible pET16b-norC or pET16b-norA) and JD838 cells (transformed with L-arabinose inducible pBAD-norC, pBAD-empty or pBAD-qacA) were grown as primary culture overnight. A secondary culture was inoculated using this and split into two parts: one treated for induction of protein expression (using 0.2 mM IPTG or 0.05% (w/v) L-arabinose) and the other left untreated. The induction was done when the O.D.s600nm reached 0.4 A.U. The cultures were further grown till all of them had an O.D.600nm between 1.5 and 2.0 A.U.; the cultures were then diluted accordingly to bring their O.D.s to 1.0 and spotted in tenfold serial dilutions on 1.5% (w/v) agar plate containing 2% (w/v) luria bertani (LB) broth and 100 μg/ml ampicillin with/without inducer in the media. Control plates containing none of the antibacterials but ampicillin were spotted similarly with/without inducer. The cells were allowed to grow overnight at 37 °C. These assays were replicated independently with n = 5.
Cloning, expression, and purification of NorC
Full length norC gene was cloned into pET-16b vector between restriction sites NcoI and KpnI with 8X-His tag at its C-terminus. The E. coli C41(DE3) cells were transformed with the cloned vector containing an ampicillin resistance cassette. A primary culture was grown in LB-broth (HiMedia) for 10–12 h at 37 °C. Primary culture was used to inoculate large scale culture (2–3 l) that were grown at 37 °C until the OD600 reached 0.4–0.5 following which protein expression was induced with 0.2 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG). The cells were further grown for 12–15 h at 20 °C. Cells were harvested by centrifugation and flash-frozen in liquid N2 for storage or processed immediately. The cell pellet was dissolved in HBS (20 mM Hepes pH 7.0, and 200 mM NaCl). The cell suspension was lysed at high pressure of about 800 bar using a homogenizer (GEA Niro Soavi PandaPlus 1000 Homogenizer) and spun at ~100,000 g for 1 h at 4 °C. The pelleted membranes were resuspended in HBS and 1 mM PMSF using a rotor-stator homogenizer, following which DDM (n-Dodecyl-β-D-maltopyranoside, Anatrace) was added to a final concentration of 20 mM. The solution was gently mixed for 2 h at 4 °C to extract NorC into micelles, followed by ultracentrifugation at 100,000 g for 1 h to remove the insoluble debris. The supernatant was incubated with Ni-NTA beads pre-equilibrated with buffer A (1 mM DDM in HBS) at 4 °C for 1 h. The solution containing beads was transferred into a gravity column (Bio-rad) and washed with 50 column volumes of buffer A having 30 mM imidazole. The protein was eluted in buffer A containing 300 mM imidazole and concentrated using a 30 kDa cut-off centrifugal filter (Amicon, Merck Millipore). It was further purified by size exclusion chromatography using Superdex S-200 increase 10/300 GL column (GE Healthcare) in HBS containing 4 mM n-Decyl-β-D-maltopyranoside (DM, Anatrace). The purity of the protein was analyzed by SDS-PAGE. The mutant NorC K398A protein was purified in a similar manner.
NorC WT was labeled with L-selenomethionine by growing E. coli C41(DE3) cells, transformed with NorC-containing vector, in M9 medium supplemented with 50 mg/L of L-selenomethionine. The purification of SeMet-labeled NorC WT was similar to that of the native protein.
ICab generation and purification
Generation and purification of ICab were carried out in a manner described previously24. The ICab isolated through yeast-display screening was subcloned into the pET-22b vector. E. coli Rosetta cells were transformed with a vhh-containing vector. Large scale cultures were grown in LB broth at 37 °C until OD600 reached 0.6 and protein expression was induced by adding IPTG to a final concentration of 0.5 mM; cells were further grown at 37 °C for 5–6 h. The cells were harvested and the protein was extracted from inclusion bodies using urea denaturation followed by refolding. The refolding was done using step-wise dialysis at 4 °C. The protein was further purified by size-exclusion chromatography using Superdex S-75 10/300 GL column (GE Healthcare) in HBS. The purity and integrity of the protein were checked by SDS-PAGE and MALDI-TOF.
Site-directed mutagenesis
To perform site-directed mutagenesis of NorC, primers were designed to amplify the entire plasmid carrying norc gene in a single step with desired point mutation. The amplicons were treated with Dpn1 at 37 °C for 2–3 h to digest the wild-type plasmid. E. coli Top10 cells were transformed with the mutant amplicon and plated on an LB-agar plate containing 100 µg/ml ampicillin and grown overnight at 37 °C. The positive clones were confirmed by DNA sequencing. The mutant proteins were purified in a manner similar to that of the wild type protein.
Fluorescence-detection size exclusion chromatography (FSEC)
FSEC was done using a high-performance liquid chromatography system attached to an autosampler and a multi-wavelength fluorescence detector (Shimadzu). In total, 10 μM each of NorC WT, NorC-K398A and NorB purified in DM buffer were incubated with 1:1.2 molar ratios of ICab and shifts in their elutions (measured at λex = 295 nm, λem = 340 nm) in the same DM buffer were used to qualitatively determine whether or not ICab binds to them. A Superdex 200 increase 10/300 GL column was used for analyzing elution times.
Crystallization and structure determination
Purified NorC WT (native and SeMet-derivative) and NorC K398A (native only) proteins were concentrated to 3 mg/ml using a 50 kDa cut-off centrifugal filter (Amicon, Merck Millipore) and mixed with ICab in a molar ration of 1:1.2. The crystallization was carried out by hanging drop vapor diffusion method at 20 °C where protein and crystallization conditions were mixed in a ratio of 3:2 v/v. NorC WT and NorC-K398A crystallized in a condition containing 0.1 M MES pH 6.0, 50 mM NaCl, 35.7 % PEG 600, 57 mM MgCl2, 10 mM YCl3 and 6.0 mM CHAPSO. The crystals appeared within 3–4 days but took about 2 weeks to attain full size. The crystals were washed in the crystallization condition and flash-frozen in liquid N2. The X-ray diffraction datasets were collected at the advanced photon source (APS) synchrotron, USA. The crystals diffracted X-rays to a resolution of about 3.6–4 Å.
All the data sets were indexed and integrated with XDS46. Eight data sets were merged and scaled for SeMet-derivative NorC WT using AIMLESS47 resulting in the highest resolution of 3.65 Å. The space group was determined to be P21 with unit cell dimensions of a = 71.2 Å, b = 139.9 Å, c = 118.0 Å and α = γ = 90° and β = 106° (Table 1). The Matthews coefficient suggested two NorC-ICab complexes in the asymmetric unit with a solvent content of about 70%. SAD phasing was carried out using CRANK248 in CCP4i249. For the substructure determination, the highest resolution was set at 5.0 Å as the anomalous signal was very weak at a higher resolution. SHELX C and D50 in CRANK2 were used for substructure determination and refinement. The resulting phases were extended to a resolution of 3.65 Å and the electron density was subjected to density modification using PARROT51. Manual model building was carried out in the density modified map using Coot52. Selenium positions were used as markers to trace the main chain and place amino acids.
Multiple X-ray diffraction data sets were collected for the native NorCK398A crystals. Six best data sets were merged and scaled using AIMLESS to a resolution of 3.6 Å. The structure was determined by molecular replacement using PHASER53 with NorC WT structure as a model. The refinement of both the structures was carried out using REFMAC54 until the refinement converged. The resulting structures were validated using MOLPROBITY55. The data collection and refinement statistics are presented in Table 1.
Structural analysis and visualization
All structural analysis and visual representations of atomic coordinates were done using the PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. The pKa of K398 was determined on the PROPKA server used through its extension in PyMOL(TM) 2.4.030.
Microscale thermophoresis for protein ligand interaction quantitation
Quantification of NorC’s interaction with TPP+ was carried out using microscale thermophoresis (Nanotemper)56. The protein was labeled with red Tris-NTA dye (Nanotemper) by mixing both in an equimolar proportion. The labeled protein was mixed with TPP+ such that the concentration of the labeled protein and TPP+ were 10 nM and 0.1 mM, respectively. A total of 16 twofold serial dilutions of TPP+ were prepared to keep the protein concentration constant at 10 nM. The experiment was carried out with Monolith™ NT.115 MST premium-coated capillaries. For NorC-ICab complex titrations against TPP+, 10 nM of NorC was mixed with ICab in a 1:3 molar ratio and the starting concentration of TPP+ was kept at 0.1 mM. For ICab titrations against NorC, 10 nM of NorC was used with a starting concentration of 5 µM for the ICab. The curves were fitted with the single-site binding model. To assess the same with NorB and ICab, identical concentrations were taken, with NorB in place of NorC.
Differential scanning fluorimetry for substrate screening
The propensity of TPP+ to interact with NorC WT, NorB WT, and NorC-K398A was assessed using differential scanning fluorimetry (DSF) using Prometheus NT.48 instrument (NanoTemper Technologies)57. A temperature scan was carried out between 20 and 80–90 °C with a scan rate of 1 °C/min. All measurements were carried out at least in duplicates. The first derivative of the ratio of Femission350 nm/330 nm was plotted against temperature, and shift in Tm was used to determine whether or not TPP+ binds to the proteins. Similar protocol was employed for initial substrate screening where 5 μM of NorC WT or NorC K398A was taken with a 1:10 molar ratio of each substrate and assessed on a temperature range of 20–80 °C. n ≥ 4 for every substrate tested.
Isothermal titration calorimetry
The binding affinity of NorC with ICab was determined by ITC using Microcal PEAQ-ITC (Malvern Panalytical). Both NorC and ICab were purified in a buffer containing 20 mM HEPES, pH 7.0, 200 mM NaCl, and 4 mM n-decyl-β-D-maltopyranoside detergent. The titration consisted of 20 injections of 4 s duration each, with the first injection of 0.4 µl and all the subsequent injections of 2 µl. The time between two consecutive injections was kept at 120 s, and the sample in the cell was stirred at 750 rpm during the entire run. For the run involving TPP+, each injection volume was increased to 2.5 µl to accommodate a low signal-to-noise ratio, while the number of injections were reduced to 16 keeping the overall volume unchanged. The concentration of NorC in the cell was kept at 20 µM and the concentration of ICab in the syringe was kept at 200 µM. The titration was carried out at a constant temperature of 25 °C. For blank, ICab was titrated against buffer, and the heat of dilution was subtracted and offset corrected from the titration data against NorC. All the data were fit using Microcal PEAQ-ITC analysis software with one set of sites model24.
PEG maleimide accessibility assay
E. coli JD838 cells were transformed with pBAD norc-wt and pBAD norc-t378c vectors and grown in 2% (w/v) LB broth overnight. These cells were used to overexpress NorC WT and its T378C mutant, lysed, and the membranes were isolated, using the protocol described earlier. Each of the membrane fractions was resuspended in 2 ml of phosphate buffer saline, pH 7.4, snap-frozen in Liq. N2 and stored in −80 °C in 30–50 μL aliquots for further use. For the PEG5k maleimide adduction reaction, the 5 μl of each membrane was diluted in HEPES buffer saline (HBS, pH 7.0, containing 20 mM HEPES and 200 mM NaCl) 10×. In a 20 μl reaction, 5 μl of the diluted membrane was added. In total, 60 μM ICab and/or 1 mM TPP+ was added in some reaction mixes prior to the addition of 1 mM PEG5k mal. The final mix was incubated for 10 min in dark at 25 °C before quenching the reaction with 100 mM β-mercaptoethanol. These membranes were solubilized in 3% SDS and centrifuged at 17,000 g for 30 min before loading the supernatant on a 10% resolving SDS-PAGE. The gel was then western transferred onto PVDF membrane and probed with 1:4000 of anti-His8 tag mouse primary antibody (Invitrogen) and 1:10000 HRP tagged goat anti-mouse secondary antibody (Invitrogen) and analyzed for band shifts after the adduction reactions. The blots were independently repeated eight times with multiple batches of membranes. A similar protocol was also used for SEC purified proteins, with 10 mM PEG5k mal was used for the assay.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data supporting the manuscript would be available upon reasonable request from the corresponding author. A reporting summary is available as a Supplementary Information file. The atomic coordinates and structure factors of NorC WT and NorC-K398A are deposited in the Protein Data Bank with PDB IDs 7D5P (https://doi.org/10.2210/pdb7D5P/pdb) and 7D5Q (https://doi.org/10.2210/pdb7D5Q/pdb), respectively. Source data are provided with this manuscript.
References
Poole, K. Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 56, 20–51 (2005).
Blair, J. M., Webber, M. A., Baylay, A. J., Ogbolu, D. O. & Piddock, L. J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51 (2015).
Piddock, L. J. Multidrug-resistance efflux pumps—not just for resistance. Nat. Rev. Microbiol. 4, 629–636 (2006).
Tong, S. Y., Davis, J. S., Eichenberger, E., Holland, T. L. & Fowler, V. G. Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 28, 603–661 (2015).
Pu, Y. et al. Enhanced efflux activity facilitates drug tolerance in dormant bacterial cells. Mol. Cell 62, 284–294 (2016).
Reddy, V. S., Shlykov, M. A., Castillo, R., Sun, E. I. & Saier, M. H. Jr. The major facilitator superfamily (MFS) revisited. FEBS J. 279, 2022–2035 (2012).
Kumar, S. et al. Bacterial multidrug efflux pumps of the major facilitator superfamily as targets for modulation. Infect. Disord. Drug Targets 16, 28–43 (2016).
Henderson, P. J. F. et al. Physiological functions of bacterial “multidrug” efflux pumps. Chem. Rev. 121, 5417–5478 (2021).
Costa, S. S., Viveiros, M., Amaral, L. & Couto, I. Multidrug efflux pumps in staphylococcus aureus: an update. Open Microbiol J. 7, 59–71 (2013).
Truong-Bolduc, Q. C., Strahilevitz, J. & Hooper, D. C. NorC, a new efflux pump regulated by MgrA of Staphylococcus aureus. Antimicrob. Agents Chemother. 50, 1104–1107 (2006).
Truong-Bolduc, Q. C., Dunman, P. M., Strahilevitz, J., Projan, S. J. & Hooper, D. C. MgrA is a multiple regulator of two new efflux pumps in Staphylococcus aureus. J. Bacteriol. 187, 2395–2405 (2005).
Chitsaz, M. & Brown, M. H. The role played by drug efflux pumps in bacterial multidrug resistance. Essays Biochem 61, 127–139 (2017).
Paulsen, I. T., Brown, M. H., Littlejohn, T. G., Mitchell, B. A. & Skurray, R. A. Multidrug resistance proteins QacA and QacB from Staphylococcus aureus: membrane topology and identification of residues involved in substrate specificity. Proc. Natl Acad. Sci. USA 93, 3630–3635 (1996).
Paulsen, I. T., Brown, M. H. & Skurray, R. A. Proton-dependent multidrug efflux systems. Microbiol Rev. 60, 575–608 (1996).
Nagarathinam, K. et al. Outward open conformation of a major facilitator superfamily multidrug/H(+) antiporter provides insights into switching mechanism. Nat. Commun. 9, 4005 (2018).
Debruycker, V. et al. An embedded lipid in the multidrug transporter LmrP suggests a mechanism for polyspecificity. Nat. Struct. Mol. Biol. 27 829–835 (2020).
Yin, Y., He, X., Szewczyk, P., Nguyen, T. & Chang, G. Structure of the multidrug transporter EmrD from Escherichia coli. Science 312, 741–744 (2006).
Stavri, M., Piddock, L. J. & Gibbons, S. Bacterial efflux pump inhibitors from natural sources. J. Antimicrob. Chemother. 59, 1247–1260 (2007).
Wright, G. D. Antibiotic adjuvants: rescuing antibiotics from resistance. Trends Microbiol 24, 862–871 (2016).
Ding, Y., Onodera, Y., Lee, J. C. & Hooper, D. C. NorB, an efflux pump in Staphylococcus aureus strain MW2, contributes to bacterial fitness in abscesses. J. Bacteriol. 190, 7123–7129 (2008).
Dawan, J., Wei, S. & Ahn, J. Role of antibiotic stress in phenotypic switching to persister cells of antibiotic-resistant Staphylococcus aureus. Ann. Microbiol. 70, 1 (2020).
Varela, M. F., Sansom, C. E. & Griffith, J. K. Mutational analysis and molecular modelling of an amino acid sequence motif conserved in antiporters but not symporters in a transporter superfamily. Mol. Membr. Biol. 12, 313–319 (1995).
Majd, H. et al. Screening of candidate substrates and coupling ions of transporters by thermostability shift assays. Elife 7, e38821 (2018).
Kumar, S., Mahendran, I., Athreya, A., Ranjan, R. & Penmatsa, A. Isolation and structural characterization of a Zn(2+)-bound single-domain antibody against NorC, a putative multidrug efflux transporter in bacteria. J. Biol. Chem. 295, 55–68 (2020).
Drew, D. & Boudker, O. Shared molecular mechanisms of membrane transporters. Annu. Rev. Biochem. 85, 543–572 (2016).
Zhang, X. C., Zhao, Y., Heng, J. & Jiang, D. Energy coupling mechanisms of MFS transporters. Protein Sci. 24, 1560–1579 (2015).
Solcan, N. et al. Alternating access mechanism in the POT family of oligopeptide transporters. EMBO J. 31, 3411–3421 (2012).
Doki, S. et al. Structural basis for dynamic mechanism of proton-coupled symport by the peptide transporter POT. Proc. Natl Acad. Sci. USA 110, 11343–11348 (2013).
Ural-Blimke, Y. et al. Structure of prototypic peptide transporter DtpA from E. coli in complex with valganciclovir provides insights into drug binding of human PepT1. J. Am. Chem. Soc. 141, 2404–2412 (2019).
Li, H., Robertson, A. D. & Jensen, J. H. Very fast empirical prediction and rationalization of protein pKa values. Proteins 61, 704–721 (2005).
Panahi, A. & Brooks, C. L. 3rd Membrane environment modulates the pKa values of transmembrane helices. J. Phys. Chem. B 119, 4601–4607 (2015).
Shaffer, P. L., Goehring, A., Shankaranarayanan, A. & Gouaux, E. Structure and mechanism of a Na+-independent amino acid transporter. Science 325, 1010–1014 (2009).
Uzdavinys, P. et al. Dissecting the proton transport pathway in electrogenic Na(+)/H(+) antiporters. Proc. Natl Acad. Sci. USA 114, E1101–E1110 (2017).
Majumder, P. et al. Dissection of protonation sites for antibacterial recognition and transport in QacA, a Multi-Drug Efflux Transporter. J. Mol. Biol. 431, 2163–2179 (2019).
Manglik, A., Kobilka, B. K. & Steyaert, J. Nanobodies to study G protein-coupled receptor structure and function. Annu Rev. Pharm. Toxicol. 57, 19–37 (2017).
Kumar, H. et al. Crystal structure of a ligand-bound LacY-nanobody complex. Proc. Natl Acad. Sci. USA 115, 8769–8774 (2018).
Fluman, N., Ryan, C. M., Whitelegge, J. P. & Bibi, E. Dissection of mechanistic principles of a secondary multidrug efflux protein. Mol. Cell 47, 777–787 (2012).
Van Campenhout, R., Muyldermans, S., Vinken, M., Devoogdt, N. & De Groof, T. W. M. Therapeutic nanobodies targeting cell plasma membrane transport proteins: a high-risk/high-gain endeavor. Biomolecules 11, 63 (2021).
Bergmann, P. et al. Generation and characterization of specific monoclonal antibodies and nanobodies directed against the ATP-gated channel P2X4. Front. Cell Neurosci. 13, 498 (2019).
Danquah, W. et al. Nanobodies that block gating of the P2X7 ion channel ameliorate inflammation. Sci. Transl. Med 8, 366ra162 (2016).
Longhin, E. et al. Isolation and characterization of nanobodies against a zinc-transporting P-Type ATPase. Antibodies 7, 39 (2018).
Schenck, S. et al. Generation and characterization of anti-VGLUT nanobodies acting as inhibitors of transport. Biochemistry 56, 3962–3971 (2017).
Mireku, S. A., Sauer, M. M., Glockshuber, R. & Locher, K. P. Structural basis of nanobody-mediated blocking of BtuF, the cognate substrate-binding protein of the Escherichia coli vitamin B12 transporter BtuCD. Sci. Rep. 7, 14296 (2017).
Jones, D. T., Taylor, W. R. & Thornton, J. M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282 (1992).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Kabsch, W. Xds. Acta Crystallogr. D. Biol. Crystallogr. 66, 125–132 (2010).
Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 (2006).
Skubak, P. & Pannu, N. S. Automatic protein structure solution from weak X-ray data. Nat. Commun. 4, 2777 (2013).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011).
Schneider, T. R. & Sheldrick, G. M. Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 58, 1772–1779 (2002).
Cowtan, K. Recent developments in classical density modification. Acta Crystallogr. D. Biol. Crystallogr. 66, 470–478 (2010).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 (2004).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr. 67, 355–367 (2011).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Seidel, S. A. et al. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 59, 301–315 (2013).
Kotov, V. et al. High-throughput stability screening for detergent-solubilized membrane proteins. Sci. Rep. 9, 10379 (2019).
Acknowledgements
Research in this manuscript was supported by the DBT/Wellcome Trust India Alliance Intermediate Fellowship (IA/1/15/2/502063) awarded to A.P. and a Department of Biotechnology, India grant (BT/PR31976/MED/29/1421/2019) awarded to A.P. and R.R. A.P. is also the recipient of the DBT-IYBA grant-2015 (BT/09/IYBA/2015/13) and is an EMBO Global Investigator from India. S.K. was supported through the DBT-IISc research associate program and A.A. is a graduate student of the integrated Ph.D. program of the Indian Institute of Science. M.I. was supported by a DST-NPDF fellowship (DSTO1605). The authors acknowledge the DBT-IISc partnership program phase-I and phase-II support and DST-FIST program support to carry out this work. The authors would like to thank Prof. B. Gopal for sharing the genomic DNA of S. aureus strains and access to PEAQ-ITC. The authors would like to thank the staff of the northeastern collaborative access team (NECAT), Advanced Photon Source (APS), particularly Dr. Surajit Banerjee for access and help with data collection. This work is based upon research conducted at the NECAT beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DoE) Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank the beamline staff at the Elettra XRD2 particularly Dr. Babu Manjashetty, Dr. Annie Heroux, and Dr. Raghurama P. Hegde for beamline support. Access to the XRD2 beamline at the Elettra synchrotron, Trieste was made possible through grant-in-aid from the Department of Science and Technology (DST), India, vide grant number DSTO-1668. The X-Ray diffraction facility for macromolecular crystallography at the Indian Institute of Science is supported by the Department of Science and Technology—Science and Engineering Research Board (DST-SERB) grant IR/SO/LU/0003/2010-PHASE-II.
Author information
Authors and Affiliations
Contributions
S.K. and A.A. purified NorC and ICab. S.K. and A.A. optimized the crystallization of NorC-ICab complex and selmet derivatization of NorC. S.K. solved and built the structure of NorC with inputs from A.P. A.A., S.K. performed the microbial growth and biochemical assays with NorC. A.G. and R.M.N. cloned, isolated and optimized the behavior of NorC. M.I. performed the screening and identification of the ICab used in the study and R.R. performed camel immunization and aided in PBMC isolation. A.P. designed the project. S.K. and A.P. wrote the manuscript with inputs from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Joanna Timmins and Anam Akhtar. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kumar, S., Athreya, A., Gulati, A. et al. Structural basis of inhibition of a transporter from Staphylococcus aureus, NorC, through a single-domain camelid antibody. Commun Biol 4, 836 (2021). https://doi.org/10.1038/s42003-021-02357-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-02357-x
This article is cited by
-
Structural basis for triacylglyceride extraction from mycobacterial inner membrane by MFS transporter Rv1410
Nature Communications (2023)
-
A (Fab)ulous tool to block efflux
Nature Chemical Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
