
Abstract
Keratoconus is characterised by reduced rigidity of the cornea with distortion and focal thinning that causes blurred vision, however, the pathogenetic mechanisms are unknown. It can lead to severe visual morbidity in children and young adults and is a common indication for corneal transplantation worldwide. Here we report the first large scale genome-wide association study of keratoconus including 4,669 cases and 116,547 controls. We have identified significant association with 36 genomic loci that, for the first time, implicate both dysregulation of corneal collagen matrix integrity and cell differentiation pathways as primary disease-causing mechanisms. The results also suggest pleiotropy, with some disease mechanisms shared with other corneal diseases, such as Fuchs endothelial corneal dystrophy. The common variants associated with keratoconus explain 12.5% of the genetic variance, which shows potential for the future development of a diagnostic test to detect susceptibility to disease.
Similar content being viewed by others


Introduction
Keratoconus is a leading cause for visual impairment in adolescents and young adults which, untreated, can lead to legal blindness1,2,3,4,5,6,7. The prevalence of keratoconus varies between ethnic groups, with figures as high as 1.2% reported in some predominantly European populations8, to 2.3–3.3% in Maori or Iranian populations9,10. A high occurrence rate in first degree relatives, and concordance in twins, suggest that keratoconus has a strong genetic component11,12. Keratoconus can also be a comorbidity of other genetically determined conditions such as Down syndrome13. Several loci and variants for keratoconus have been identified through linkage studies and genome-wide association studies (GWASs) for central corneal thickness (CCT)14,15,16,17,18,19,20. However, although CCT is highly heritable, it is a stable characteristic, in contrast to the acquired and progressive corneal thinning that is a feature of keratoconus. Previous studies have also implicated single nucleotide polymorphism (SNP) alleles upstream of the ZNF469 locus that is associated with a higher CCT but an increased risk for keratoconus16,20,21. Therefore, alternative mechanisms, in addition to those influencing CCT, are likely to be involved. This incomplete knowledge of the genetic predisposition for keratoconus limits our understanding of the mechanisms that drive this disease. In this study we present the largest GWAS for keratoconus performed to date for 4669 cases and 116,547 controls.
Results
Meta-analyses of genome-wide associations with keratoconus
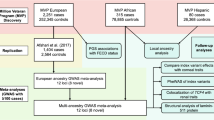
We performed the analyses in three stages (Fig. 1). First, a discovery analysis was conducted in 2116 cases and 24,626 controls of European ancestry. For the second stage, we compared and replicated the discovery results in a meta-analysis of three independent European cohorts (1389 cases and 79,727 controls), and in a separate meta-analysis of two non-European cohorts (759 South Asian cases and 8009 controls, and 405 African cases and 4185 controls). Finally, we combined the discovery and replication cohorts in an overall meta-analysis. Genomic control factors22 were consistent with polygenicity expectations and the absence of uncontrolled population structure in any of the components of this study (Supplementary Data 1).

There were three main phases: a discovery in a European cohort of 2116 cases and 24,626 controls, a replication in a combined meta-analysis of three other European cohorts as well as in two smaller non-European cohorts, and a final meta-analysis involving all the multi-ethnic cases and controls from the previous stages.
The discovery analysis identified 22 GWAS-significant associations (Supplementary Data 2), including six loci previously associated with keratoconus (FOXO117, COL5A117, FNDC3B17, ZNF46917, LOX23 and near PNPLA224), four that were previously associated with CCT16,18 but not keratoconus, and 12 entirely novel loci. Among the novel keratoconus loci, the most significant association was found at a gene-poor region on chr21q2 (p = 1.34 × 10−13 for rs76747345).
At the replication stage, we carried forward the most significant SNPs within each of their regions of association, or other GWAS-significant proxy SNPs if necessary, whenever the index SNPs were missing in the replication data. Nine of the 22 regions (of which 2 are novel) replicated after Bonferroni multiple testing correction (p < 0.05/22 = 0.0022) and another four (of which 2 are novel) at FDR < 0.05 (Supplementary Data 2). All SNPs except three, for which the replication meta-analysis had insufficient power, showed directional consistency (Supplementary Fig. 1). We also observed associations that were highly directionally concordant in non-European samples (Fig. 2). Despite the lower statistical power, there was good replication in the South Asian samples (12 SNPs nominally significant, of which 8 remained significant after correction for multiple testing), but slightly less so in Africans (Supplementary Data 3).

Each dot represents SNPs shown in Supplementary Data 3 and their labels are the respective chromosomal band on which they are located. The shapes of each data point refer to a previous GWAS association with keratoconus (empty circles), with CCT only (empty triangles) or novel associations (solidly filled circular shapes). The colors of both the points and their labels represent the significance (−log10(p-value) of association observed in the European discovery cohort. Polymorphisms identified in the discovery cohort but not shown here were not available for analysis in the replication cohorts.
The final meta-analysis combined data for all 4669 cases and 116,547 controls. Although the genomic inflation factor was nominally large (λ = 1.29), a further LD score regression analysis25 (on European samples only) suggests that these results were in line with expectations of polygenicity (ldsc intercept = 1.09, SE = 0.009). We continue to observe homogeneous effect sizes across all populations (Supplementary Data 4). This meta-analysis yielded significant associations clustering around 36 independent regions (Fig. 3, Table 1), of which 31 are reported for the first time, including six previously associated with CCT but not specifically with keratoconus at GWAS significance.

Analyses were conducted in 4669 cases and 116,547 controls. The log10(p-value) from the final meta-analysis is shown on the y-axis for all the SNPs along the different autosomes (x-axis). Novel associations for keratoconus are in pink. The names of the coding genes nearest to the most significantly associated SNPs are shown, or “no gene” when the association was >250 kb from a coding gene. Different colors are used for genetic loci and genes that to our knowledge, were previously associated with CCT (dark green) keratoconus (dark gray).
Strong associations were found near or within genes that code for fibrillar collagens (types I and V), microfibrillar (VI) and peri-fibrillar (XII) structures26, implicating impaired cohesion of the collagen matrix in the pathogenesis of keratoconus. Association was also found for rs35523808 (p.Glu2160Val, p = 2.90 × 10−25), a missense and potentially deleterious variant within COL12A1 (Table 1, Supplementary Data 5). Collagen XII is localized in Bowman layer and the interfibrillar matrix of the corneal stroma, where it regulates the organization and mechanical properties of collagen fibrils27. We found significant association within the COL6A1 gene (rs142493024, p = 9.07 × 10−12). The collagen VI protein is localized to corneal stroma filaments where it contributes to the integrity of the extracellular matrix28. Other associations were for COL1A1 (rs2075556, p = 3.35 × 10−09) and COL5A1 (rs3118518, p = 1.83 × 10−28). Collagen V is a regulator of collagen fibril formation, matrix assembly and tissue function in the corneal stroma29. Pathogenic variants in COL12A1, COL6A1, COL1A1, and COL5A1 have been described in individuals with different subtypes of the connective tissue disorders Ehlers-Danlos Syndrome and osteogenesis imperfecta30,31,32,33.
We observed a strong association at the previously described23 LOX locus (rs840464 p = 1.72 × 10−20, Table 1). LOX encodes lysyl oxidase, an enzyme that initiates the cross-linking of collagens and elastin34. One of the associated SNPs at this locus, rs840462 (p = 2.08 × 10−17), displayed the most significant expression quantitative trait locus (eQTL) effects over the transcription of the LOX gene (p = 1.61 × 10−13) in a GTEx dataset for sun-exposed skin (Supplementary Data 6). A significant association was found near the integrin gene ITGA2 (rs12515400, p = 3.16 × 10−18). The protein encoded by this gene participates in complexes of integrin α2β1, which are collagen receptors35 and which enhance type I collagen polymerization36.
We found association near the ALDH3A1 gene (rs4646785, p = 9.01 × 10−12) for the same SNP that is also a significant eQTL, controlling transcription (eQTL p = 6.92 × 10−10) in sun-exposed skin (Supplementary Data 6) and other tissues. ALDH3A1 encodes a corneal crystallin, a major component of the corneal stroma and epithelium that is upregulated in keratoconus corneas compared to controls37. It modulates corneal epithelial differentiation and homeostasis and may protect the eye from UV light-induced oxidative stress38,39,40,41,42.
For the first time, we report novel genetic associations implicating corneal cell differentiation and homeostasis in the pathogenesis of keratoconus. A significant association was found for rs17285550 (p = 2.84 × 10−12) near KLF5, a transcription factor that regulates corneal epithelial cell idenitity43,44,45,46. KLF5 knockout mice have an abnormal collagen matrix47 and suppressed levels of FNDC3B, another gene associated with keratoconus in our study (rs4894414, p = 1.21 × 10−26). We found association for SNPs located near genes involved in fibroblast-keratocyte differentiation, such as rs761276 in the intergenic region between CD34 and CD46 (p = 8.02 × 10−09). CD34 is a negative marker of keratocyte differentiation in the cornea, expressed in stromal fibroblasts but not mature keratocytes48. Association was also found for SNPs located at the enhancer region of SMAD3 (rs12912010, p = 1.99 × 10−26), a member of a family of genes involved in fibroblast differentiation49.
Association with several transcription factors involved in cell differentiation was revealed. The NANOS1 and EIF3A genes are located in the same associated region (near rs658352, p = 3.71 × 10−12). NANOS1 controls cell cycle progression and is involved in the SMAD3/TGFβ fibroblast maturation pathway50,51, while EIF3A is a cell differentiation suppressor52. FOXO1 (rs2721051, p = 5.71 × 10−35) is implicated in the maintenance of keratinocyte stem cell identity53, and one associated region (rs12948086, p = 5.33 × 10−09) overlaps a cluster of HOX genes, that participate in early embryonic differentiation and morphogenesis54.
Our results show that two loci previously implicated in Fuchs endothelial corneal dystrophy (FECD)55 are also associated with keratoconus; the PIDD1/SLC25A22 locus (rs7117921, p = 1.09 × 10−26) and ATP1B1 (rs1200108, p = 4.52 × 10−10). The allele increasing the keratoconus risk at both loci also conferred susceptibility to FECD (Supplementary Data 7).
Functional exploration of variants associated with keratoconus
Genes located in regions associated with keratoconus are broadly expressed across all GTEx56 and available eye tissues57 (Supplementary Fig. 2), more so in fetal corneas than in other eye tissues (Supplementary Data 8, Supplementary Fig. 3). In addition, certain genes near the association peaks were shown previously to be differentially expressed in keratoconus compared to control corneas58. Expression of STK35, that is associated in our analyses (rs6106210, p = 2.85 × 10−11), was increased two-fold in keratoconus corneas (FDR = 3.24 × 10−03) and RAB11FIP4 (rs56161228, p = 2.70 × 10−10) found in our study, was increased by 2.4 fold (FDR = 3.24 × 10−03).
Gene-set enrichment analyses found associations with Gene Ontology annotations (Supplementary Data 9), including transcriptional regulation (p = 1.0 × 10−06), embryonic and primary germ layer development (p = 7.0 × 10−06), and embryonic morphogenesis (p = 3.0 × 10−06).
Because of limited availability of corneal eQTL datasets, we conducted a heritability-partitioning analysis to test for enrichment of genes in available eQTLs from other tissues59. The strongest enrichment, albeit not significant after multiple testing, were mainly collagen and fibroblast-rich tissues, such as aortic valve, myometrium, and skin (Supplementary Data 10).
We subsequently investigated the mechanisms through which the DNA variants associated with keratoconus in our meta-analyses alter the susceptibility to disease. We found that many of these variants contribute to epigenetic changes of the genomic regions in which they are located, alter the efficiency of transcription of nearby genes, or both (Supplementary Fig. 4). Mendelian randomization-based (SMR) tests60 which included all the SNPs for which we had summary statistics, regardless of the degree of genetic association with the trait, suggest that methylation is a widespread mechanism mediating the effect of disease-associated SNPs. We found evidence that SNPs associated with keratoconus often alter methylation of many genes, including LOX, PDDC1, SMAD3, HOXB1, KLF5, and BANP (Supplementary Data 11). Interestingly, methylation of KLF4 was significantly (SMR p = 5.10 × 10−08) influenced by SNPs associated with keratoconus. KLF4 is a transcription factor involved in tissue differentiation and development. It induces stem cell pluripotency in fibroblasts61 and regulates corneal epithelial cell cycle progression by suppressing canonical TGFβ signaling62 and is functionally related to KLF5, which specifies corneal epithelial cell identity46. We also found several eQTL mediated effects (Supplementary Data 12), which were less significant than the methylation-mediated effects.
Inter-trait correlation and pleiotropy of keratoconus-associated loci
The strongest genetic correlations between keratoconus and other ocular traits (Supplementary Data 13) were with spherical equivalent (pg = 2.07 × 10−08) and CCT (pg = 2.5 × 10−07). However, as previously noted16, correlation of effect sizes with CCT was not always uniform. SNPs at the ZNF469/BANP locus, but also several loci that, to the best of our knowledge, are newly identified, such as the ATP1B1 locus, diverged considerably from the linear correlation with CCT (Fig. 4, Supplementary Data 14). This supports the view that other mechanisms of loss of corneal integrity and corneal fragility, independent of corneal thickness, contribute to the pathogenesis of keratoconus. There were also nominally significant genetic correlations with asthma and ulcerative colitis (Supplementary Data 13), possibly because of the coincidental high collagen content in tissues involved in these diseases, or reflecting shared inflammatory components63.

Data reported by Choquet et al.20 (also in Supplementary Data 14), with those over keratoconus (4669 cases and 116,547 controls, natural logarithm of the ORs, the y-axis). The alleles increasing susceptibility to keratoconus were selected as reference alleles. The genes located nearest to the most significant SNPs are shown in the labels and their color-codes denote the significance (−log10(p-value) of the association with CCT20. For the sake of clarity, only some of the genes are labeled. Unlabeled points denote SNPs in intragenic regions (distance from the nearest known protein-coding transcript greater than 250kbp). The variants are annotated to the nearest known protein-coding transcript. Because of their proximity with other SNPs, some loci in this figure are not labeled. Polymorphisms identified in the discovery cohort, but not shown in this figure were not available for analysis in the replication cohorts.
Predictive value of common genetic markers associated with keratoconus
An LD score regression analysis revealed that genetic markers associated with keratoconus in our meta-analysis help explain 12.5% of overall keratoconus heritability among the same populations of European ancestry. Next, we assessed the predictive value of the markers we identified. A predictive model tested in a small, but independent panel of mixed British, Dutch and Austrian keratoconus patients and controls of European descent, found that the markers were moderately predictive (AUC = 0.737, SE = 0.017, Fig. 5) of keratoconus. The addition of rare variants, in future studies, is likely to further increase the predictive value of genetic testing.

The area under the receiver operating characteristic curve for the performance of a keratoconus predictive model that used the SNPs identified through the multi-ethnic analysis. The model was trained in the discovery populations of European ancestry and was replicated in a completely independent European panel of 222 keratoconus patients and 2208 controls.
Discussion
In conclusion, we report 36 genetic loci strongly associated with keratoconus, 31 of which we identify for the first time. Their effect sizes are remarkably consistent across different ethnic groups (Supplementary Fig. 5). Larger studies are required to identify the remaining genetic risk for this condition (Supplementary Fig. 6). Our data highlight the importance of the integrity of the corneal collagen matrix in keratoconus. For the first time, we have identified a substantial role for cell differentiation pathways and stem cell regulators such as KLF4 and KLF5 in the pathogenesis of keratoconus, and a role for genes influencing connective tissue maturity.
Our study has several strengths. It is currently the largest of its kind which has allowed the identification of several loci, mostly novel, that predispose to keratoconus. In addition, although dominated by cases and control of European ancestry, the multi-ethnic component of our study allowed an evaluation of the strength of these associations among individuals of African and Asian descent. A limitation of this study is the lack of reliable corneal-specific eQTL and methylation level assessment. This has impeded the functional annotation of the discovered genetic loci, which are currently annotated to the nearest transcript-coding gene. Assuming that the effect of many SNPs is mediated through eQTL or methylation changes, cornea-specific analyses would improve these annotations. Lack of available tissue-specific expression and methylation data also currently limits our ability to further characterize these loci functionally. Our current approach would allow the identification of relationships that transcend tissue specificity and are observed in different cell lines. Also, the genomic inflation factor was moderately high, especially in our non-European cohorts, for which we are not able to fully evaluate the degree to which this is driven by polygenicity, a consequence of the presence of high-effect common variants in a low prevalence disease, or any other potential explanation.
Identification of genetic risk factors and novel disease mechanisms represents a substantial advance of our understanding of the corneal disease and, more broadly, connective tissue homeostasis, highlighting targets for the development of novel therapies. Two patient groups could directly benefit from an improved estimate of their risk of progressive corneal thinning. Firstly, individuals with subclinical keratoconus, in whom corneal collagen cross-linking would be an option to stop disease progression to reduce the need for contact lens wear for visual correction or eventual corneal transplantation. Secondly, genomic screening could be performed before laser refractive surgery for the correction of short sight (laser vision correction), to identify individuals at risk of severe visual loss from secondary progressive corneal thinning. Apart from refractive error, these individuals have normal eyes before surgery and there is currently no reliable mechanism to identify individuals at risk of developing this secondary corneal change, similar to keratoconus. For both groups, genomic data to estimate risk could be incorporated into models based on clinical parameters such as refractive error, corneal thickness, corneal shape, and corneal biomechanics64,65.
Methods
Study overview
This study was approved by the Institutional Review Board (IRB) or equivalent at all participating institutions, and participants provided written informed consent for the use of their genetic information. The study was conducted in concordance with the provisions of the Declaration of Helsinki.
Controls were extracted from a pool of 80,000 randomly selected participants in the UK Biobank cohort. Exclusions included any individual with any ICD9 or ICD10 code for any corneal disease. The cases and controls were ethnically matched.
Multi-ethnic discovery cohort
Phenotyping
The majority of participants were recruited from specialist clinics at Moorfields Eye Hospital, London, UK. The study was approved by the Moorfields Eye Hospital Research Ethics Committee (09/H0721/19). The diagnosis of keratoconus was established based on clinical signs of corneal thinning and corneal distortion, with confirmation by corneal imaging (Orbscan, Bausch & Lomb, Rochester, USA, or Pentacam, Oculus, Wetzlar, Germany). Previous bilateral keratoplasties for keratoconus were also accepted as confirmation of disease status. Patients with keratoconus who had syndromic disease (e.g. Down syndrome, Leber congenital amaurosis, Ehlers Danlos syndrome) were excluded, as were patients with corneal dystrophy, of cornea guttata suggestive of coexisting Fuchs endothelial corneal dystrophy.
Recruitment from specialist corneal clinics at St. James’s University Hospital, Leeds, UK, was approved by Leeds East Research Ethics Committee (reference 10/H1306/63). The diagnosis of keratoconus was determined clinically and confirmed with corneal imaging (Orbscan, Bausch & Lomb, Rochester, USA, or Pentacam, Oculus, Wetzlar, Germany). Patients who had keratoconus associated with syndromes and those without the capacity to consent were excluded.
Participants were recruited from the Corneal and External Eye Disease clinics at Guy’s and St. Thomas’ National Health Service Foundation Trust, London, UK. The diagnosis of keratoconus was established based on a history of uni- or binocular progressive visual disturbance and the presence of some of all of a number of clinical signs seen on slit-lamp biomicroscopic examination including corneal stroma thinning, sub-epithelial iron deposition (Fleischer ring), stromal scarring, deep vertical stromal stress lines (Vogt’s striae) and corneal ectasia, with confirmation by three-dimensional corneal imaging (Pentacam, Oculus, Wetzlar, Germany). A history of previous bilateral keratoplasty for keratoconus was also accepted as confirmation of disease status. Patients with keratoconus who had syndromic disease (e.g. Down syndrome, Leber congenital amaurosis) were excluded.
Czech participants were selected based on the same criteria as used by Moorfields Eye Hospital London. The study protocol for Czech participants was approved by the Ethics Committee of the General Teaching Hospital and Charles University, Prague. Patients were recruited at the Department of Ophthalmology in Prague between 2011 and 2017. The diagnosis of keratoconus was based on the detection of localized steepening on corneal topography maps, together with localized corneal thinning in at least one eye. Only patients with KC grade 1 or higher according to the Pentacam (Oculus Optikgeräte GmbH, Wetzlar, Germany) build in software classification were included. Some eyes had advanced disease with typical signs such as Vogt striae, Fleischer ring, and stromal scarring, but this was not an inclusion requirement for the purposes of this study. Patients with bilateral keratoplasties for KC were also considered as cases. Patients with a known monogenic disorder were excluded.
The Melbourne patients were individuals with keratoconus and European background who were recruited from public clinics at the Royal Victorian Eye and Ear Hospital (RVEEH), private and optometry clinics in Melbourne, Australia. They were required to complete a study questionnaire, clinical examination, and details of family history of disease. A patient information sheet, consent form, privacy statement, and patient rights were provided to all individuals participating in the study. The study protocol was approved by the Royal Victorian Eye and Ear Hospital Human Research and Ethics Committee (Project#10/954H). Written informed consent was obtained from each participant and all protocols followed the tenets of the Declaration of Helsinki. A blood/saliva sample was also collected. DNA was extracted from the blood or saliva sample using NucleoSpin® QuickPure kits66. Keratoconus was diagnosed on the basis of the presence of one or more of the following: (1) an irregular corneal shape, as determined by distortion of keratometric mires and/or corneal tomographic images, (2) scissoring of the retinoscopic reflex; and (3) demonstration of at least one biomicroscopic sign, including Vogt’s striae, Fleischer’s ring or corneal thinning and scarring typical of Keratoconus67. Potential subjects with non-keratoconus ocular disease in both eyes such as keratectasia, corneal degenerations, macular disease, and optic nerve disease (e.g., optic neuritis, optic atrophy) were excluded from the study.
Genotyping
Both cases and controls were genotyped using the Affymetrix UK Biobank Axiom Array. For the discovery cohort cases, DNA was extracted from peripheral blood samples using standard methods (unless otherwise stated). DNA samples (n = 4,032) were quantified and normalized prior to genotyping using the Axiom 2.0 assay for GeneTitan on UK Biobank Axiom arrays (High-Throughput Genomics Centre at the Wellcome Trust Centre for Human Genetics, University of Oxford). 99% of samples reached the SNP QC call rate threshold of ≥97%. The procedures that produced the genotyping information in the UK Biobank participants that were used as controls for our analyses are described elsewhere68.
Intensity (CEL) files from cases and controls were called together using the “apt-probeset-genotype” program from Affymetrix (http://www.affymetrix.com/support/developer/powertools/changelog/apt-probeset-genotype.html, Thermo Fisher Scientific, Waltham, MA). Libraries were downloaded from the UK Biobank Axiom Array product support pages (https://www.thermofisher.com/order/catalog/product/902502). Controls genotyped using the UKBiLEVE SNP chip were removed from the calling process. Genotyping was done in batches of 4,000-5,000 samples each. We subsequently used SNP Polisher functionalities (probeset metrics, classification, OTV caller) following the Affymetrix recommendations (https://www.affymetrix.com/support/developer/powertools/changelog/VIGNETTE-snp-polisher-apt.html#otvcaller).
Post genotyping, UK Biobank sample genotypes were compared (via chi-square association testing) with the genotypes released from the UK Biobank. Only unrelated individuals (PI_HAT < 0.05) were included in the analyses. Subsequently, analyses were run in separate ethnic groups (African, South Asian, and Afro-Caribbean). SNPs with excessive (>0.05) missing genotypes and large (p < 0.01) differences in allele frequency compared with the official UK Biobank genotypes were removed from further analyses. This threshold of significance was adopted because even random missing samples that failed genotyping, either at the UK Biobank processing centre or in our labs, could cause minor and non-significant differences in allele frequencies.
Ancestry information and case-control matching
Cases and controls were subsequently matched for ancestry. First, a Principal Component Analysis (PCA) was run on independent (r2 < 0.3) directly genotyped SNP, then the samples were clustered in either one of the three major ancestral clusters: European, South Asian, or African. Samples that were not part of any of the clusters were removed from further consideration.
To avoid subtler population structure, arising either from ethnic heterogeneity or batch effects, we matched cases and controls based on the information from the first 10 principal components. We sought to match one case with up to 10 controls, if possible.
Only cases and controls that were matched in such a way were taken forward and imputed together.
Imputation
Imputation was run separately for the European and non-European ancestral groups (African and South Asian). The Europeans were split into two groups with an equal number of cases and controls and were imputed up to the HRC r1.1 2016 haplotypes using Minimac4 after Eagle 2.4 pre-phasing. Non-European ancestral groups were imputed up to the 1000 G Phase 3 v5.
Association analyses
Association analyses were conducted based on Firth’s regression models, given that our case-controls were imbalanced. Regression models were built with the keratoconus status as the dependent variable and the allele dosage at each SNP locus as a predictor. Analyses were adjusted for sex and the first 20 principal components and were conducted for each ancestral group separately. The largest case-control samples were of European ancestry and were used for discovery purposes. Samples of African and South Asian ancestries were used for validation purposes.
Replication cohorts
The Los Angeles-affiliated cohort
Enrollment and phenotyping
Clinically affected keratoconus patients were enrolled into the study at three major sites: the longitudinal videokeratography and genetic study at the Cornea Genetic Eye Institute at Cedars-Sinai Medical Center, Los Angeles, CA, USA; the Jules Stein Eye Institute at UCLA, Los Angeles, CA, USA; and the University Hospitals Eye Institute at University Hospitals Case Medical Center and Case Western Reserve University, Cleveland, OH, USA. All patients diagnosed with keratoconus (see below) were offered recruitment into the study.
In addition to keratoconus patients, 126 local controls were recruited at Cedars-Sinai Medical Center. Convenience Caucasian controls from a Cholesterol and Atherosclerosis Pharmacogenetics (CAP) study were also included to make the sample size of controls equivalent to that of cases. CAP sample involved 944 unaffected volunteers, 609 of whom were self-reported white. Participants were aged 30 or above and were recruited from two clinical sites located in Los Angeles and San Francisco, CA, respectively. Additional details of CAP samples are described elsewhere69.
The diagnosis of keratoconus was performed by a corneal specialist ophthalmologist or an experienced research optometrist and based on clinical examination and videokeratography pattern analysis. Clinical examination included slit-lamp biomicroscopy, cycloplegic retinoscopy, and fundus evaluations. Slit-lamp biomicroscopy was used to identify stromal corneal thinning, Vogt’s striae, or a Fleischer ring. Retinoscopy examination was performed with a fully dilated pupil 20 min after phenylephrine 2.5% and cyclopentolate 1% drops had been instilled in the eye to determine the presence or absence of retro-illumination signs of keratoconus, such as the oil droplet sign and scissoring of the red reflex. Videokeratography evaluation was performed on each eye using the Topographic Modeling System (Computed Anatomy, New York, NY, USA), Orbscan II (Bausch & Lomb, Rochester, NY, USA), Oculus Pentacam (Oculus, Inc, Lynnwood, WA, USA) or Keratron (Optikon, Rome, Italy). Subjects were considered to have keratoconus if they had at least one clinical sign of keratoconus and a confirmatory videokeratography map with an asymmetric bowtie with a skewed radial axis above and below the horizontal meridian (AB/SRAX) pattern70. Importantly, topography was screened for mimicking disease such as pellucid marginal degeneration, which was excluded. Subjects that had bilateral keratoplasty for keratoconus were included if the surgical pathology report confirmed the presence of the disease.
Genotyping
DNA was extracted from EBV transformed lymphoblastoid cell lines established from peripheral whole blood of each study participant using NucleoSpin Tissue kit (MACHEREY-NAGEL Inc., Bethlehem, PA, USA) and from saliva samples using QIAsymphony DNA Kit (Qiagen, Germantown, MD).
Genotyping was performed using the Illumina HumanOmni2.5 Beadchip for all keratoconus patients, 126 local controls and 50 quality controls from CAP study. Genome-wide genotyping of half of CAP study subjects was performed using Illumina HumanHap300 BeadChip and of the remaining half of the samples with Illumina HumanCNV610-Quad Beadchip. Additional genotyping with iSelect Beadchip and Metabo-Chip was also available for some CAP samples. Samples with sex mismatches, relatedness (pi-hat>0.1875), low call rate (<95%), and SNPs with low minor allele frequency (MAF < 1%), low genotyping rate (<95%), and Hardy–Weinberg equilibrium p values less than 10−6 were excluded from the analysis. Both cases and controls datasets were imputed using the Michigan Imputation Server71 with the Haplotype Reference Consortium (HRC)72. Post-imputation QC removed SNPs with low imputation quality (rsq < 0.1) and a concordance rate less than 95% among 50 quality control samples. QC was performed using PLINK v1.973. Principal components analysis (PCA) was performed with EIGENSTRAT74 Self-identified Hispanic samples were checked by PCA, and outliers significantly different from self-reported European individuals were excluded. After verifying the participants’ ancestry through a principal component analysis, 662 cases and 676 controls of full European ancestry were included in the analyses.
Association analyses
In total, 662 keratoconus cases and 676 controls (123 local controls and 553 controls from CAP study) with both phenotyping and genotyping data were available for analysis after QC. Under an additive genetic model, we conducted association tests between keratoconus and all autosome SNPs with MAF greater than 5%. The logistic regression model was performed adjusting for gender and three principal components (PCs) with RVTESTS75.
Australian cohort
Phenotyping
Genome-wide association analysis of this cohort has been described previously24. In brief, participants with keratoconus were ascertained through the eye clinic of Flinders Medical Centre, Adelaide; optometry and ophthalmology clinics in Adelaide and Melbourne; or an Australia-wide invitation to members of Keratoconus Australia, a community-based support group for patients. Approval was given by the Southern Adelaide Clinical Human Research Ethics Committee (HREC), the HREC of the Royal Victorian Eye and Ear Hospital and the Health and Medical HREC of the University of Tasmania. All participants gave informed consent and the study conformed to the tenets of the Declaration of Helsinki.
Patients were classified as having keratoconus if they had at least one clinical sign of keratoconus and a confirmatory videokeratography or a penetrating keratoplasty performed because of keratoconus, as described previously.76 The controls included 465 unaffected individuals from the Blue Mountains Eye Study77 and an additional 211 unaffected individuals78 from the Australian cohort previously described as controls in a GWAS for age-related macular degeneration (AMD) from the International AMD Genomics Consortium79.
DNA for cases and controls was extracted from whole blood using the QiaAMP DNA Maxi kit (Qiagen, Hilden, Germany).
Genotyping
Cases were genotyped for 551,839 variants using the HumanCoreExome array (HumanCoreExome-24v1-1_A, Illumina, San Diego, CA, USA) while for the controls, genotypes of 569,645 variants were generated with a customized Illumina HumanCoreExome array (“HumanCoreExome_Goncalo_15038949_A”) as described previously79. Only SNPs common to both arrays were included.
Quality control was carried out according to the protocol described by Anderson et al.80, modified as follows. Reverse and ambiguous strand SNPs were detected using snpflip (https://github.com/biocore-ntnu/snpflip, accessed March 24, 2017) and flipped or excluded. Ancestry outliers identified by principal component analysis (PCA) using EIGENSTRAT74, as well as individuals missing genotype rate > 0.05, heterozygosity more than three standard deviations from the mean, or discordant sex information, were excluded. Related individuals were detected by calculating pairwise identity by descent (IBD), and the individual with the lower genotyping rate in any pair with IBD > 0.185 was removed. Markers were excluded if they had significantly different missing data rates between cases and controls, total missing genotype rate > 3%, minor allele frequency (MAF) < 0.01, or deviated significantly (P < 10−5) from Hardy–Weinberg equilibrium. Following all exclusions, there were 522 cases (mean age 45) and 655 controls (mean age 65) genotyped for 264,115 common platform SNPs.
Association analyses
Genotypes of autosomal SNPs were phased with Eagle (version 2.3.5)81 and imputed to the EUR subset of the 1000 Genomes Project reference panel (Phase III, version 5)82 using Minimac3 (version 2.0.1)71. Indels, SNPs within 5 bp of an indel, rare variants (MAF < 0.01), and variants with poor imputation quality (R2 < 0.8) were excluded. A total of 6,252,612 including 250,964 genotyped variants, passed quality control. Association analysis was performed on most-likely genotypes under a logistic regression model using PLINK (version 1.90)83 using the first three principal components as covariates.
Genetic epidemiology research in adult health and aging cohort (GERA)
The Genetic Epidemiology Research in Adult Health and Aging (GERA) cohort is part of the Kaiser Permanente Research Program on Genes, Environment, and Health (RPGEH) and has been previously described in detail84,85. The GERA cohort comprises 110,266 adult men and women who are consented participants in the RPGEH, an unselected cohort of adult participants who are members of Kaiser Permanente Northern California (KPNC), an integrated health care delivery system, with ongoing longitudinal records from vision examinations. For this analysis, 78,583 adults, who self-reported as non-Hispanic white, were included. Of which 72 cases were males and 85 females, with an average age of 61.9 years (S.D. 12.3). All study procedures were approved by the Institutional Review Board of the Kaiser Foundation Research Institute.
Phenotyping
Keratoconus cases were identified in the KPNC electronic health record system based on the following International Classification of Diseases, Ninth Revision (ICD-9) diagnosis codes: 371.60, 371.61, and 371.62. All selected keratoconus cases (N = 157) had at least one diagnosis of keratoconus made by a Kaiser Permanente ophthalmologist. Our keratoconus control group (N = 78,426) included all the non-cases.
Genotyping
DNA samples from GERA individuals were extracted from Oragene kits (DNA Genotek Inc., Ottawa, ON, Canada) at KPNC and genotyped at the Genomics Core Facility of the University of California, San Francisco (UCSF). DNA samples were genotyped at over 665,000 single nucleotide polymorphisms (SNPs) on Affymetrix Axiom arrays (Affymetrix, Santa Clara, CA, USA)86,87. SNPs with initial genotyping call rate ≥97%, allele frequency difference ≤0.15 between males and females for autosomal markers, and genotype concordance rate >0.75 across duplicate samples were included85. Around 94% of samples and more than 98% of genetic markers assayed passed quality control (QC) procedures. In addition to those QC criteria, SNPs with genotype call rates <90% were removed, as well as SNPs with a minor allele frequency (MAF) < 1%.
Following genotyping QC, we conducted statistical imputation of additional genetic variants. Following the pre-phasing of genotypes with Shape-IT v2.r7271988, variants were imputed from the cosmopolitan 1000 Genomes Project reference panel (phase I integrated release; http://1000genomes.org) using IMPUTE2 v2.3.089,90,91. As a QC metric, we used the info r2 from IMPUTE2, which is an estimate of the correlation of the imputed genotype to the true genotype92. Variants with an imputation r2 < 0.3 were excluded, and we restricted to SNPs that had a minor allele count (MAC) ≥ 20.
Association analyses
We ran a logistic regression of keratoconus and each SNP using PLINK83 v1.9 (www.cog-genomics.org/plink/1.9/) with the following covariates: age, sex, and genetic ancestry principal components (PCs). We modeled data from each genetic marker using additive dosages to account for the uncertainty of imputation93. Eigenstrat74 v4.2 was used to calculate the PCs84. The top 10 ancestry PCs were included as covariates, as well as the percentage of Ashkenazi ancestry to adjust for genetic ancestry, as described previously84.
UK samples of non-European ancestry
Among the subjects recruited at the Moorfield Eye Hospital, there were 759 cases of South Asian ancestry and 405 of African descent. Phenotypes were obtained as described before. These samples were matched from a separate pool of ethnic minorities from the UK Biobank and analyzed using Firth’s regression models, adjusting for sex and the first 20 principal components that were calculated within the ethnically matched case-control group. These samples were primarily used for replication and validation purposes, but also contributed to the final meta-analysis.
Cohorts used for prediction
Prediction models were trained in cases and controls of European descent described above and tested in 222 keratoconus patients and 2208 keratoconus-free controls, pooled from the Erasmus Medical Centre cohort and the UK Biobank.
The Erasmus Medical Centre (EMC) cohort
The Erasmus Medical Centre (EMC) cohort consisted of Caucasian keratoconus patients (n = 156) and controls (n = 1476). Keratoconus samples were collected from both the Erasmus MC and the Rotterdam Eye Hospital. Control samples were collected from 1) the Rotterdam study (n = 448), a population-based study described previously94; 2) the Biomarkers of personalized Medicine (BioPersMed) study (n = 891) which included healthy subjects with one or more risk factors for cardiovascular diseases (Graz, Austria); 3) controls from the Amsterdam Glaucoma Study (n = 137), previously described95. All patients and controls underwent a full ophthalmic examination at the correspondent medical centers as part of the inclusion protocol of each study. The studies were approved by the Institutional Review Board at each institute and informed consents were collected from all participants.
DNA from cases and controls was genotyped using the Illumina bead chip (Infinium Global Screening Array-24 V2; Illumina Inc) at the Human Genetics facility at EMC. Quality control was performed using Genome studio (Illumina-designed software) and PLINK on 683880 genotyped variants from 1632 individuals. In summary, samples with more than 10% missing variants and variants with minor allele frequency less than 5% were excluded. Moreover, a Hardy–Weinberg equilibrium cut-off of 10−7 was used for control samples. The genomic inflation factor (lambda) of 1.08 (based on median chi-square), showing no significant dispersion of test statistics from the expected distribution. A total of 454925 variants (genotyping rate of 0.998) from 1629 subjects (155 cases and 1474 controls) were imputed using Michigan imputation server. Post-imputation quality checks were performed and a total of 57 variants from 1629 subjects were included in the final analysis.
The UK Biobank subset
A subset of 66 cases and 733 controls were extracted from the UK Biobank cohort. In brief cases were individuals who reported keratoconus (ICD code H18.6) and controls were UK Biobank participants who did not report keratoconus but also were negative for other significant corneal disease (ICD10 codes H18.7, H18.8 and H18.9) and without any prior history of eye surgery. UK Biobank participants that were included as controls in the previously described analyses were specifically excluded from this step. All methods, genotyping, imputation, and basic QC were similar to what is described elsewhere before96.
Meta-analyses
We conducted three meta-analyses. For the initial meta-analysis (discovery), we used summary statistic results from the discovery cohorts. These cohorts were genotyped on the same chip and recruited using consistent methodologies. The second meta-analysis aggregated data from summary statistics of three independent cohorts of European ancestry, recruited in addition to the discovery cohort. For the final meta-analysis, we used all available information from all available cohorts. Very rare variants (MAF < 0.01) and rare ones (0.01 < MAF < 0.05) with low imputation quality scores (<0.8) or high meta-analysis heterogeneity I2 > 0.75 were excluded.
For all meta-analyses we applied a fixed-effect inverse variance method as implemented in the software METAL97 and GWAMA98. No genomic control adjustment was applied during the meta-analysis.
Effective population size and power calculations
Power calculations were conducted using the Stata 15 “power”’ package (StataCorp LLC, College Station, TX).
The effective population size was calculated per each locus, aggregating the effective population sizes of each cohort participating in the meta-analyses, using this equation:
as recommended elsewhere97, where N.eff is the effective sample size, N.cases is the number of cases, and N.controls is the number of subjects without keratoconus.
Only SNPs with minor allele frequency of at least 1%, which were available from at least 70% of the maximum number of participants across all studies, and that were not missing in more than one stratum (cohorts), were considered.
Multiple testing correction
Two methods of correcting for multiple testing were used. The first was a classic Bonferroni correction, in which the threshold of significance (0.05) was divided by the number of tests (n):
Given the large number of loci for which replication was needed, we additionally calculated the False Discovery Rates, using the Benjamini–Hochberg method99.
Genomic inflation
To assess the potential inflation of association probabilities, genomic inflation factors22 were calculated and Q-Q plots were drawn using the package “gap” in R (https://cran.r-project.org/).
LDscore regression-based methods
Calculation of genomic inflation attributable to polygenicity vs. stratification
We used LD score regression-based methods to calculate the genomic inflation factors, and measures of stratification within the samples of European ancestry. We followed the LDSC authors’ recommendations (https://groups.google.com/g/ldsc_users/c/yJT-_qSh_44/m/MmKKJYsBAwAJ) to normalize the effective sample sizes and account for the different numbers of cases and controls in the different populations, thereafter using a sample prevalence estimate of 0.5, and a disease prevalence in the population of 0.001.
Calculation of genetic correlation
Bivariate genetic correlations between keratoconus and other complex traits whose summary statistics are publicly available were assessed following previously described methodologies100, using the program LD Score (https://github.com/bulik/ldsc).
Genesis
We used a maximum-likelihood model to estimate the distribution of effect sizes, based on summary statistics of observations and linkage disequilibrium patterns to predict the likely number of SNPs that explain keratoconus heritability as well as explore the relationship between future sample sizes and the number of SNPs identified and variance or heritability explained as described elsewhere101 and implemented in the GENESIS R package (https://github.com/yandorazhang/GENESIS).
SNPs and gene annotations
Polymorphisms associated at a GWAS level (P < 5 × 10−08) were clustered within an “associated genomic region”, defined as a contiguous genomic region where GWAS-significant markers were within 1 million base pairs from each other, as suggested elsewhere102. Significant polymorphisms were annotated with the gene inside whose transcript-coding region they are located, or alternatively, if located between two genes, with the gene nearest to it. The associated genomic regions were collectively annotated with the gene overlapping, or nearest the most significantly associated variant within that region. In addition, the polymorphic sites were functionally annotated using SNPnexus103. CADD scores were generated using Ensembl variant effect predictor (http://grch37.ensembl.org/Homo_sapiens/Tools/VEP).
Previous association with keratoconus and CCT
We collected evidence of previous associations with keratoconus and CCT by querying the GWAS catalog104 and looking up reports in peer-reviewed articles16,17,20,24,105 for genomic markers and regions associated with either. We specifically looked for genome-wide significance level of association.
SMR
SMR (Summary data-based Mendelian randomization) uses GWAS variants as instrumental variables and gene expression levels or methylation levels as mediating traits, in order to test whether the causal effect of a specific variant on the phenotype-of-interest acts via a specific gene60 (but note, in practice, SMR is unable to differentiate between causation and horizontal pleiotropy). SMR incorporates the Heterogeneity in Dependent Instrument (HEIDI) test, which is designed to detect variants with horizontally pleiotropic effects (via their heterogeneity in the SNP-outcome vs. SNP-intermediate trait relationship, in comparison to nearby variants). SMR analyses were repeated after excluding ‘outlier’ variants detected using HEIDI.
Test description
The SMR software helps perform two tests. The first is an SMR test, which correlates GWAS effects with eQTL or methylation effects (or any other intermediate trait)60. This test suggests causation, although it is unable to fully differentiate between it and pleiotropy. The second test is that of Heterogeneity in Dependent Instrument (HEIDI). This test against the null hypothesis that changes in both eQTL (or other intermediary traits) and the phenotype of interest are caused by one single SNP, which is therefore considered as the candidate for the putative causal effect.
Datasets for the SMR analyses: eQTL, cis-mQTL
To perform the above-mentioned tests of causation/pleiotropy, we used three different datasets of association between genetic variants and intermediate traits. The eQTL associations were obtained from the untransformed peripheral blood samples of 5311 subjects106 and methylation data from the analyses of LBC methylation of 1980 subjects described elsewhere107. They were the largest eQTL datasets available.
Gene-set enrichment
To identify pathways or other gene sets that were over or under-represented among our results, we used a Gene-Set Enrichment Analysis (GSEA) as implemented in the Meta-Analysis Gene Set Enrichment of Variant (MAGENTA) software108. This program assigns scores to each gene based on the strength of association with keratoconus, adjusting for potential confounders such as gene length and linkage disequilibrium. Enrichment for any gene set was assessed within genes above the cut-off of the highest 75th centile of significant gene scores. For the current study, the most recent versions of Gene Ontology (GO), Panther, KGG, Biocarta, and MSigDB databases were used. We also carried out a similar enrichment analysis for the presence of transcription factor binding sites. A permutational procedure and false-discovery rates were used to calculate the significance of enrichment and control for multiple testing.
GSEA definitions
For the enrichment analyses, we used updated versions of the GSEA gene sets as described before109. We used the versions from November 2018 which were downloaded from http://software.broadinstitute.org/gsea/login.jsp
Gene expression, GTEx, and other transcription data
We obtained data on tissue expression from several sources for genes located within associated loci. Information about the expression of the genes of interest in systemic (i.e. non-ocular) tissues was obtained from the GTEx Portal for GTEx release v7 (https://gtexportal.org/home/datasets). RNA sequencing data were obtained for both fetal and adult corneal, trabecular meshwork, and ciliary body, as described elsewhere57, which we downloaded from the authors’ supplementary information. In addition, we extracted data from the subset of subjects with presumed healthy adult retinas (AMD=1), described elsewhere110 that obtained from the GTEx Portal (https://gtexportal.org/home/datasets).
Transcription data were processed using different platforms and were available in different units (Transcripts per Million bases, TPM, for the retina and GTEx tissues, and Fragments per Kilobase, FPKM for the other tissues). For purposes of comparing expression across different tissues for which different methodologies may have been used, expression levels for all tissues were rank-transformed. Hierarchical clustering was used to help visualize similarities and differences of patterns of transcript expression across different tissues (“hclust” package in R).
LD score regression applied to specifically expressed genes (LDSC-SEG)
Disease-relevant tissues and cell types were identified by analyzing gene expression data together with summary statistics from the meta-analysis of keratoconus in all cohorts, as described elsewhere59. Briefly, genes were ranked based on the t-statistic of their expression in each tissue and the 10% most expressed genes for each tissue were considered “specifically expressed genes”. A stratified LD score regression was applied to the meta-analysis summary statistics to evaluate the contribution of the focal genome annotation to trait heritability.
Prediction analyses
We built a model which included sex, and the major genetic variants associated with keratoconus. The model included all SNPs from a conditional analysis from a European-only conditional analysis111, as implemented in the program GCTA112 (v1.92.1beta6). The model was trained using the cases and controls of European descent only from the discovery cohort and tested in an independent panel of keratoconus cases and controls. Genotype missingness was < 1% in all cases and controls, and whenever a genotypic value was missing, it was replaced with the population average (calculated on both cases and controls) for each locus. A Receiver Operating Characteristic (ROC) curve was drawn for each case and an Area Under the Curve (AUC) was calculated. R programming language and software environment for statistical computing (https://cran.r-project.org/) was used for both the logistic regression models (‘glm’) and to evaluate the performance of the model (‘ROCR’).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Godefrooij, D. A., de Wit, G. A., Uiterwaal, C. S., Imhof, S. M. & Wisse, R. P. Age-specific incidence and prevalence of keratoconus: a nationwide registration study. Am. J. Ophthalmol. 175, 169–172 (2017).
Nielsen, K., Hjortdal, J., Aagaard Nohr, E. & Ehlers, N. Incidence and prevalence of keratoconus in Denmark. Acta Ophthalmol. Scand. 85, 890–892 (2007).
Kymes, S. M., Walline, J. J., Zadnik, K. & Gordon, M. O. Collaborative longitudinal evaluation of keratoconus study, g. Quality of life in keratoconus. Am. J. Ophthalmol. 138, 527–535 (2004).
Bak-Nielsen, S., Ramlau-Hansen, C. H., Ivarsen, A., Plana-Ripoll, O. & Hjortdal, J. A nationwide population-based study of social demographic factors, associated diseases and mortality of keratoconus patients in Denmark from 1977 to 2015. Acta Ophthalmol. 97, 497–504 (2018).
Mas Tur, V., MacGregor, C., Jayaswal, R., O’Brart, D. & Maycock, N. A review of keratoconus: diagnosis, pathophysiology, and genetics. Surv. Ophthalmol. 62, 770–783 (2017).
Gain, P. et al. Global survey of corneal transplantation and eye banking. JAMA Ophthalmol. 134, 167–173 (2016).
Matthaei, M. et al. Changing indications in penetrating keratoplasty: a systematic review of 34 years of global reporting. Transplantation 101, 1387–1399 (2017).
Chan, E. et al. Prevalence of keratoconus based on Scheimpflug imaging: The Raine Study. Ophthalmology https://doi.org/10.1016/j.ophtha.2020.08.020 (2020).
Hashemi, H., Khabazkhoob, M. & Fotouhi, A. Topographic keratoconus is not rare in an Iranian population: the Tehran Eye Study. Ophthalmic Epidemiol. 20, 385–391 (2013).
Papali’i-Curtin, A. T. et al. Keratoconus prevalence among high school students in New Zealand. Cornea 38, 1382–1389 (2019).
Tuft, S. J. et al. Keratoconus in 18 pairs of twins. Acta Ophthalmol. 90, e482–e486 (2012).
Wang, Y., Rabinowitz, Y. S., Rotter, J. I. & Yang, H. Genetic epidemiological study of keratoconus: evidence for major gene determination. Am. J. Med. Genet. 93, 403–409 (2000).
Woodward, M. A., Blachley, T. S. & Stein, J. D. The association between sociodemographic factors, common systemic diseases, and keratoconus: an analysis of a Nationwide Heath Care Claims Database. Ophthalmology 123, 457–65 e2 (2016).
Bisceglia, L. et al. Linkage analysis in keratoconus: replication of locus 5q21.2 and identification of other suggestive Loci. Invest. Ophthalmol. Vis. Sci. 50, 1081–1086 (2009).
Burdon, K. P. et al. Association of polymorphisms in the hepatocyte growth factor gene promoter with keratoconus. Invest Ophthalmol. Vis. Sci. 52, 8514–8519 (2011).
Iglesias, A. I. et al. Cross-ancestry genome-wide association analysis of corneal thickness strengthens link between complex and Mendelian eye diseases. Nat. Commun. 9, 1864 (2018).
Lu, Y. et al. Genome-wide association analyses identify multiple loci associated with central corneal thickness and keratoconus. Nat. Genet. 45, 155–163 (2013).
Dyrlund, T. et al. Human cornea proteome: identification and quantitation of the proteins of the three main layers including epithelium, stroma and endothelium. Investigative Ophthalmol. Visual Sci. 54 (2013).
Tang, Y. G. et al. Genomewide linkage scan in a multigeneration Caucasian pedigree identifies a novel locus for keratoconus on chromosome 5q14.3-q21.1. Genet. Med. 7, 397–405 (2005).
Choquet, H. et al. A multiethnic genome-wide analysis of 44,039 individuals identifies 41 new loci associated with central corneal thickness. Commun. Biol. 3, 301 (2020).
Liskova, P., Dudakova, L., Krepelova, A., Klema, J. & Hysi, P. G. Replication of SNP associations with keratoconus in a Czech cohort. PLoS ONE 12, e0172365 (2017).
Devlin, B. & Roeder, K. Genomic control for association studies. Biometrics 55, 997–1004 (1999).
Li, X. et al. A genome-wide association study identifies a potential novel gene locus for keratoconus, one of the commonest causes for corneal transplantation in developed countries. Hum. Mol. Genet 21, 421–429 (2012).
McComish, B. J. et al. Association of Genetic Variation With Keratoconus. JAMA Ophthalmol. 38, 174–181 (2019).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Gelse, K., Poschl, E. & Aigner, T. Collagens-structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 55, 1531–1546 (2003).
Young, B. B., Zhang, G., Koch, M. & Birk, D. E. The roles of types XII and XIV collagen in fibrillogenesis and matrix assembly in the developing cornea. J. Cell Biochem. 87, 208–220 (2002).
Feneck, E. M., Lewis, P. N., Ralphs, J. & Meek, K. M. A comparative study of the elastic fibre system within the mouse and human cornea. Exp. Eye Res. 177, 35–44 (2018).
Sun, M. et al. Collagen V is a dominant regulator of collagen fibrillogenesis: dysfunctional regulation of structure and function in a corneal-stroma-specific Col5a1-null mouse model. J. Cell Sci. 124, 4096–4105 (2011).
De Paepe, A., Nuytinck, L., Hausser, I., Anton-Lamprecht, I. & Naeyaert, J. M. Mutations in the COL5A1 gene are causal in the Ehlers-Danlos syndromes I and II. Am. J. Hum. Genet 60, 547–554 (1997).
Klaassens, M. et al. Ehlers-Danlos arthrochalasia type (VIIA-B)-expanding the phenotype: from prenatal life through adulthood. Clin. Genet. 82, 121–130 (2012).
Zou, Y. et al. Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Hum. Mol. Genet. 23, 2339–2352 (2014).
Delbaere, S. et al. Novel defects in collagen XII and VI expand the mixed myopathy/Ehlers-Danlos syndrome spectrum and lead to variant-specific alterations in the extracellular matrix. Genet. Med. 22, 112–123 (2019).
Lucero, H. A. & Kagan, H. M. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol. Life Sci. 63, 2304–2316 (2006).
Kern, A., Eble, J., Golbik, R. & Kuhn, K. Interaction of type IV collagen with the isolated integrins alpha 1 beta 1 and alpha 2 beta 1. Eur. J. Biochem. 215, 151–159 (1993).
Velling, T., Risteli, J., Wennerberg, K., Mosher, D. F. & Johansson, S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins alpha(11)beta(1) and alpha(2)beta(1). J. Biol. Chem. 277, 37377–37381 (2002).
Joseph, R., Srivastava, O. P. & Pfister, R. R. Differential epithelial and stromal protein profiles in keratoconus and normal human corneas. Exp. Eye Res. 92, 282–298 (2011).
Jester, J. V. Corneal crystallins and the development of cellular transparency. Semin Cell Dev. Biol. 19, 82–93 (2008).
Pei, Y., Reins, R. Y. & McDermott, A. M. Aldehyde dehydrogenase (ALDH) 3A1 expression by the human keratocyte and its repair phenotypes. Exp. Eye Res. 83, 1063–1073 (2006).
Stagos, D., Chen, Y., Cantore, M., Jester, J. V. & Vasiliou, V. Corneal aldehyde dehydrogenases: multiple functions and novel nuclear localization. Brain Res. Bull. 81, 211–218 (2010).
Townsend, A. J. et al. Selective protection by stably transfected human ALDH3A1 (but not human ALDH1A1) against toxicity of aliphatic aldehydes in V79 cells. Chem. Biol. Interact. 130-132, 261–273 (2001).
Koppaka, V. et al. ALDH3A1 plays a functional role in maintenance of corneal epithelial homeostasis. PLoS One 11, e0146433 (2016).
Kenchegowda, D., Harvey, S. A., Swamynathan, S., Lathrop, K. L. & Swamynathan, S. K. Critical role of Klf5 in regulating gene expression during post-eyelid opening maturation of mouse corneas. PLoS ONE 7, e44771 (2012).
Diakiw, S. M., D’Andrea, R. J. & Brown, A. L. The double life of KLF5: Opposing roles in regulation of gene-expression, cellular function, and transformation. IUBMB Life 65, 999–1011 (2013).
Dong, J. T. & Chen, C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell Mol. Life Sci. 66, 2691–2706 (2009).
Stephens, D. N. et al. The Ets transcription factor EHF as a regulator of cornea epithelial cell identity. J. Biol. Chem. 288, 34304–34324 (2013).
Swamynathan, S. K. et al. Conditional deletion of the mouse Klf4 gene results in corneal epithelial fragility, stromal edema, and loss of conjunctival goblet cells. Mol. Cell Biol. 27, 182–194 (2007).
Joseph, A. et al. Expression of CD34 and L-selectin on human corneal keratocytes. Invest Ophthalmol. Vis. Sci. 44, 4689–4692 (2003).
Evans, R. A., Tian, Y. C., Steadman, R. & Phillips, A. O. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp. Cell Res. 282, 90–100 (2003).
Li, B. et al. Knockdown of eIF3a ameliorates cardiac fibrosis by inhibiting the TGF-beta1/Smad3 signaling pathway. Cell Mol. Biol. (Noisy-le.-Gd.) 62, 97–101 (2016).
Dong, Z. et al. Role of eIF3a in regulating cell cycle progression. Exp. Cell Res. 315, 1889–1894 (2009).
De Keuckelaere, E., Hulpiau, P., Saeys, Y., Berx, G. & van Roy, F. Nanos genes and their role in development and beyond. Cell Mol. Life Sci. 75, 1929–1946 (2018).
Saoncella, S. et al. Nuclear Akt2 opposes limbal keratinocyte stem cell self-renewal by repressing a FOXO-mTORC1 signaling pathway. Stem Cells 32, 754–769 (2014).
Noordermeer, D. et al. The dynamic architecture of Hox gene clusters. Science 334, 222–225 (2011).
Afshari, N. A. et al. Genome-wide association study identifies three novel loci in Fuchs endothelial corneal dystrophy. Nat. Commun. 8, 14898 (2017).
Consortium, G. T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Carnes, M. U., Allingham, R. R., Ashley-Koch, A. & Hauser, M. A. Transcriptome analysis of adult and fetal trabecular meshwork, cornea, and ciliary body tissues by RNA sequencing. Exp. Eye Res. 167, 91–99 (2018).
Khaled, M. L. et al. Differential expression of coding and long noncoding RNAs in keratoconus-affected corneas. Invest Ophthalmol. Vis. Sci. 59, 2717–2728 (2018).
Finucane, H. K. et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018).
Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 48, 481–487 (2016).
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Tiwari, A. et al. KLF4 regulates corneal epithelial cell cycle progression by suppressing canonical TGF-beta signaling and upregulating CDK inhibitors P16 and P27. Invest Ophthalmol. Vis. Sci. 60, 731–740 (2019).
Galvis, V. et al. Keratoconus: an inflammatory disorder? Eye (Lond.) 29, 843–859 (2015).
Bohac, M. et al. Incidence and clinical characteristics of post LASIK ectasia: a review of over 30,000 LASIK cases. Semin. Ophthalmol. 33, 869–877 (2018).
Ferdi, A. C. et al. Keratoconus natural progression: a systematic review and meta-analysis of 11 529 Eyes. Ophthalmology 126, 935–945 (2019).
Richardson, A. J., Narendran, N., Guymer, R. H., Vu, H. & Baird, P. N. Blood storage at 4 degrees C-factors involved in DNA yield and quality. J. Lab Clin. Med. 147, 290–294 (2006).
Sahebjada, S. et al. Association of the hepatocyte growth factor gene with keratoconus in an Australian population. PLoS ONE 9, e84067 (2014).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Simon, J. A. et al. Phenotypic predictors of response to simvastatin therapy among African-Americans and Caucasians: the Cholesterol and Pharmacogenetics (CAP) Study. Am. J. Cardiol. 97, 843–850 (2006).
Rabinowitz, Y. S. Videokeratographic indices to aid in screening for keratoconus. J. Refract. Surg. 11, 371–379 (1995).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Zhan, X., Hu, Y., Li, B., Abecasis, G. R. & Liu, D. J. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 32, 1423–1426 (2016).
Burdon, K. P. et al. Apparent autosomal dominant keratoconus in a large Australian pedigree accounted for by digenic inheritance of two novel loci. Hum. Genet. 124, 379–386 (2008).
Burdon, K. P. et al. Association of open-angle glaucoma loci with incident glaucoma in the Blue Mountains Eye Study. Am. J. Ophthalmol. 159, 31–36.e1 (2015).
Dimasi, D. P. et al. Genetic investigation into the endophenotypic status of central corneal thickness and optic disc parameters in relation to open-angle glaucoma. Am. J. Ophthalmol. 154, 833–842.e2 (2012).
Fritsche, L. G. et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 48, 134–143 (2016).
Anderson, C. A. et al. Data quality control in genetic case-control association studies. Nat. Protoc. 5, 1564–1573 (2010).
Loh, P.-R. et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Banda, Y. et al. Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the genetic epidemiology research on adult health and aging (GERA) cohort. Genetics 200, 1285–1295 (2015).
Kvale, M. N. et al. Genotyping informatics and quality control for 100,000 subjects in the genetic epidemiology research on adult health and aging (GERA) cohort. Genetics 200, 1051–1060 (2015).
Hoffmann, T. J. et al. Next generation genome-wide association tool: design and coverage of a high-throughput European-optimized SNP array. Genomics 98, 79–89 (2011).
Hoffmann, T. J. et al. Design and coverage of high throughput genotyping arrays optimized for individuals of East Asian, African American, and Latino race/ethnicity using imputation and a novel hybrid SNP selection algorithm. Genomics 98, 422–430 (2011).
Delaneau, O., Marchini, J. & Zagury, J. F. A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2012).
Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. & Abecasis, G. R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955–959 (2012).
Howie, B., Marchini, J. & Stephens, M. Genotype imputation with thousands of genomes. G3 1, 457–470 (2011).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Marchini, J. & Howie, B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 11, 499–511 (2010).
Huang, L., Wang, C. & Rosenberg, N. A. The relationship between imputation error and statistical power in genetic association studies in diverse populations. Am. J. Hum. Genet. 85, 692–698 (2009).
Ikram, M. A. et al. The Rotterdam Study: 2018 update on objectives, design and main results. Eur. J. Epidemiol. 32, 807–850 (2017).
Ramdas, W. D. et al. Common genetic variants associated with open-angle glaucoma. Hum. Mol. Genet. 20, 2464–2471 (2011).
Hysi, P. G. et al. Meta-analysis of 542,934 subjects of European ancestry identifies new genes and mechanisms predisposing to refractive error and myopia. Nat. Genet. 52, 401–407 (2020).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Magi, R. & Morris, A. P. GWAMA: software for genome-wide association meta-analysis. BMC Bioinform. 11, 288 (2010).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statistical Soci. Ser. B (Methodological) 57, 289–300 (1995).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Zhang, Y., Qi, G., Park, J. H. & Chatterjee, N. Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat. Genet. 50, 1318–1326 (2018).
Wood, A. R. et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 46, 1173–1186 (2014).
Dayem Ullah, A. Z., Lemoine, N. R. & Chelala, C. SNPnexus: a web server for functional annotation of novel and publicly known genetic variants (2012 update). Nucleic Acids Res. 40, W65–W70 (2012).
MacArthur, J. et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 45, D896–D901 (2017).
Hao, X. D., Chen, P., Chen, Z. L., Li, S. X. & Wang, Y. Evaluating the association between keratoconus and reported genetic loci in a Han chinese population. Ophthalmic Genet. 36, 132–136 (2015).
Westra, H. J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
McRae, A. F. et al. Identification of 55,000 Replicated DNA Methylation QTL. Sci. Rep. 8, 17605 (2018).
Segre, A. V. et al. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet 6, e1001058 (2010).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Ratnapriya, R. et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 51, 606–610 (2019).
Zhu, Z. et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 9, 224 (2018).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Acknowledgements
The publication of this article has been enabled by a grant from Moorfields Eye Charity (GR001317). UK Biobank: This research has been conducted using the UK Biobank Resource under Application Number 17615. UK Biobank was established by the Wellcome Trust medical charity, Medical Research Council, Department of Health, Scottish Government, and Northwest Regional Development Agency. It also had funding from the Welsh Assembly Government, British Heart Foundation, and Diabetes UK. UCL and Moorfields, London, UK: We are grateful to all the patients at Moorfields Eye Hospital who participated in this study. This study was funded by Moorfields Eye Charity and the Rosetrees Trust. We were supported by infrastructure and funding from the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital and UCL Institute of Ophthalmology. AED is supported by AED is supported by a UK Research and Innovation Future Leadership Fellowship, Moorfields Eye Charity, and Academy of Medical Sciences Springboard award. We thank Beverley Scott and Naushin Waseem for assistance with DNA extraction and DNA banking, and the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (funded by Wellcome Trust grant reference 203141/Z/16/Z) for performing the genotyping on the discovery cohort. APK and NP are supported by Moorfields Eye Charity Career Development Fellowships. GERA, USA: We are grateful to the Kaiser Permanente Northern California members who have generously agreed to participate in the Kaiser Permanente Research Program on Genes, Environment, and Health. Support for participant enrollment, survey completion, and biospecimen collection for the RPGEH was provided by the Robert Wood Johnson Foundation, the Wayne and Gladys Valley Foundation, the Ellison Medical Foundation, and Kaiser Permanente Community Benefit Programs. Genotyping of the GERA cohort was funded by a grant from the National Institute on Aging, National Institute of Mental Health, and National Institute of Health Common Fund (RC2 AG036607). Data analyses were facilitated by National Eye Institute (NEI) grant R01 EY027004 (E.J.), National Institute of Diabetes and Digestive and Kidney Diseases grant R01 DK116738 (E.J.). The Los Angeles-affiliated cohort, USA: The authors gratefully acknowledge the support of The Eye Defects Research Foundation Inc. and NEI R01-09052. CAP study (control cohort) was supported by the National Institutes of Health grant U19 HL069757 from the National Heart, Lung, and Blood Institute; and grant UL1TR000124 from the National Center for Advancing Translational Sciences. In addition, this work was supported in part by the National Institutes of Health, National Eye Institute contract R01 EY009052, and by the National Center for Advancing Translational Sciences, CTSI grant UL1TR001881. Australia: The Australian replication case cohort was funded by a National Health and Medical Research Council (NHMRC) project grant 1104700 and Centre for Research Excellence 1023911. Control genotype data for this cohort were provided by the international AMD Genetics Consortium genotyped under The Center for Inherited Diseases Research (CIDR) Program contract number HHSN268201200008I. SM is supported by an NHMRC Fellowship and NHMRC grants 1150144, 1116360. Dr Burdon was supported by an NHMRC Senior Research Fellowship (1059954). King’s College London and St. Thomas Hospital, UK: M.J. Simcoe is a recipient of a Fight for Sight PhD studentship. K. Patasova is a recipient of a Fight for Sight PhD studentship. P.G. Hysi the recipient of a Fight for Sight ECI fellowship. P.G. Hysi and C.J. Hammond acknowledge the TFC Frost Charitable Trust Support for the KCL Department of Ophthalmology. The statistical analyses were run in King’s College London Rosalind HPC and Department of Twin Research and Genetic Epidemiology Linux clusters and server. The UK Biobank data was accessed as part of the UK Biobank projects 669 and 17615. Leeds University and Leeds Teaching Hospitals NHS Trust, UK: We thank the patients and relatives who participated in this project. This study was supported by grants SAC007 and SAC033 from the National Eye Research Centre, and LAF was supported by a PhD grant award from Fight For Sight. S.Siddiqui was supported by the MRC grant 408757. Thanks to Erica Di Martino for laboratory support. University of Melbourne, Royal Victorian Eye and Ear Hospital, Australia: This study was supported by the Australian National Health and Medical Research Council (NHMRC) project grant 1104700 and Senior Research Fellowship (1138585 to PNB), the Angior Family Foundation and a Lions Eye Foundation Fellowship (SS). The Centre for Eye Research Australia (CERA) receives Operational Infrastructure Support from the Victorian Government. The authors wish to thank all participants who took part in this Keratoconus study and made this work possible. The authors would also like to thank Eye Surgery Associates, Lindsay and Associates and Keratoconus Australia, as well as Mr Tony Ngo for assistance with subject recruitment. Charles University and General University Hospital in Prague, Czech Republic: PL, LD and PS were supported by UNCE/MED/007 and PROGRES1086, Q26/LF1 research programs of the Charles University and by grant GACR 20-19278S. We thank the participating patients and staff of the Cornea service from the Department of Ophthalmology, General University Hospital in Prague for assistance with recruitment. The Erasmus Medical Center: The authors are grateful to Barbara Obermayer-Pietsch, Arthur A Bergen, and Andre G. Uitterlinden for their tremendous help throughout this project. The study was supported by the Combined Ophthalmic Research Rotterdam (CORR) grant.
Author information
Authors and Affiliations
Contributions
A.J.H., P.L., S.J.T., and P.G.H. conceived the project; A.J.H., Y.B., B.J.M., A.E.D., C.F.I., X.L., H.C., M.H., S.E.M.L., S.Sahebjada, N.P., K.E.R.L., M.A., L.D., P.S., B.T.H.V.D., A.J.M.G., C.W.H., V.L.F., A.T., M.J.S., K.P., D.Y., J.Y., S.Sidiqui, A.R., L.A.F., Y-D.I.C.,J.S.R., R.M.K., E.T., J.C.C., L.S-F., C.T., M.A.M-S., A.J.R., P.A.M., K.D.T., R.B.M., A.J.A, R.A.M., K.C., E.C., M.D.D., J.J.W., J.I.R., A.W.H., S.M., C.C.W.K., W.D.R., J.E.C., S.K.I., D.O., E.J., P.N.B., Y.S.R., and K.P.B. were responsible for collecting clinical data and performing genotyping in each study; A.J.H., C.J.H., P.G.H. drafted the paper; A.J.H., Y.B., C.J.H., A.P.K., C.J.H., S.J.T., and P.G.H. critically reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
H.C. is an Editorial Board Member for Communications Biology, but was not involved in the editorial review of, nor the decision to publish, this article. The authors declare the following competing interests: D.O. holds non-commercial research grants with Rayner Inc and Avedro Inc, holds stock in Sparca Inc. All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hardcastle, A.J., Liskova, P., Bykhovskaya, Y. et al. A multi-ethnic genome-wide association study implicates collagen matrix integrity and cell differentiation pathways in keratoconus. Commun Biol 4, 266 (2021). https://doi.org/10.1038/s42003-021-01784-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-021-01784-0
This article is cited by
-
Genetic variants in the FOXO1 and ZNF469 genes are associated with keratoconus in Sweden: a case-control study
BMC Ophthalmology (2024)
-
A multi-ancestry GWAS of Fuchs corneal dystrophy highlights the contributions of laminins, collagen, and endothelial cell regulation
Communications Biology (2024)
-
Keratoconus International Consortium (KIC)- advancing keratoconus research
BMC Ophthalmology (2023)
-
Contribution of common and rare variants to Asian neovascular age-related macular degeneration subtypes
Nature Communications (2023)
-
Evaluating the association between MPDZ-NF1B rs1324183 and keratoconus in an independent northwestern Chinese population
BMC Ophthalmology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
